Back to Journals » Drug Design, Development and Therapy » Volume 11
Association of genetic variations with pharmacokinetics and lipid-lowering response to atorvastatin in healthy Korean subjects
Authors Woo HI, Kim SR, Huh W, Ko JW ![]() , Lee SY
, Lee SY ![]()
Received 2 January 2017
Accepted for publication 21 February 2017
Published 4 April 2017 Volume 2017:11 Pages 1135—1146
DOI https://doi.org/10.2147/DDDT.S131487
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Hye In Woo,1 Suk Ran Kim,2 Wooseong Huh,3,4 Jae-Wook Ko,4 Soo-Youn Lee4,5
1Department of Laboratory Medicine, Samsung Changwon Hospital, Sungkyunkwan University School of Medicine, Changwon, Korea; 2Clinical Research and Development, Hanmi Pharm. Co., Ltd., Seoul, Korea; 3Department of Medicine, 4Department of Clinical Pharmacology and Therapeutics, 5Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea
Background: Statins are effective agents in the primary and secondary prevention of cardiovascular disease, but treatment response to statins varies among individuals. We analyzed multiple genetic polymorphisms and assessed pharmacokinetic and lipid-lowering responses after atorvastatin 80 mg treatment in healthy Korean individuals.
Methods: Atorvastatin 80 mg was given to 50 healthy Korean male volunteers. Blood samples were collected to measure plasma atorvastatin and lipid concentrations up to 48 hours after atorvastatin administration. Subjects were genotyped for 1,936 drug metabolism and transporter genetic polymorphisms using the Affymetrix DMET plus array.
Results: The pharmacokinetics and lipid-lowering effect of atorvastatin showed remarkable interindividual variation. Three polymorphisms in the SLCO1B1, SLCO1B3, and ABCC2 genes were associated with either the maximum concentration (Cmax) of atorvastatin or changes in total cholesterol or low-density lipoprotein cholesterol (LDL-C). Minor homozygotes (76.5 ng/mL) of SLCO1B1 c.-910G>A showed higher Cmax than heterozygotes (34.0 ng/mL) and major homozygotes (33.5 ng/mL, false discovery rate P=0.040). Cmax and the area under the plasma concentration curve from hour 0 to infinity (AUC∞) were higher in carriers of the SLCO1B1*17 haplotype that included c.-910G>A than in noncarriers (46.1 vs 32.8 ng/mL for Cmax; 221.5 vs 154.2 ng/mL for AUC∞). SLCO1B3 c.334G>T homozygotes (63.0 ng/mL) also showed higher Cmax than heterozygotes (34.7 ng/mL) and major homozygotes (31.4 ng/mL, FDR P=0.037). A nonsynonymous ABCC2 c.1249G>A was associated with small total cholesterol and LDL-C responses (0.23% and -0.70% for G/A vs -11.9% and -17.4% for G/G). The Cmax tended to increase according to the increase in the number of minor allele of SLCO1B1 c.-910G>A and SLCO1B3 c.334G>T.
Conclusion: Genetic polymorphisms in transporter genes, including SLCO1B1, SLCO1B3, and ABCC2, may influence the pharmacokinetics and lipid-lowering response to atorvastatin administration.
Keywords: atorvastatin, pharmacokinetics, pharmacogenomics, SLCO1B1, SLCO1B3, ABCC2
Introduction
As therapeutic agents administered to reduce the risk of cardiovascular disease and manage hypercholesterolemia,1 statins upregulate low-density lipoprotein (LDL) receptors, increase plasma clearance of LDL, and reduce hepatic secretion of apolipoprotein B (ApoB)-containing lipoproteins, very low-density lipoprotein (VLDL), and LDL. Statins can reduce the plasma concentration of low-density lipoprotein cholesterol (LDL-C) by as much as 50% as well as triglycerides.2
Atorvastatin is a potent competitive inhibitor of 3-hydroxy-3-ethylglutaryl-coenzyme A (HMG-CoA) reductase, an enzyme that catalyzes conversion of HMG-CoA to mevalonate, an early rate-limiting step in cholesterol synthesis. In spite of the beneficial effects of statin treatment in cardiovascular disease prevention,1 responses to statin therapy show considerable interindividual variation,3,4 and some patients may not achieve sufficient LDL-C reduction even with the most efficacious statins.5
Genetic factors are expected to be part of the interindividual variation in the pharmacokinetic and pharmacodynamic response to statins.6,7 Various genes encoding for enzymes and transporters that influence pharmacokinetics and the targets of pathways on which a drug acts, as well as those involved in related disease conditions, have been evaluated in candidate gene studies and hypothesis-free genome-wide investigations.6–12 Genetic variations on drug transporter genes, ABCB1 and SLCO1B1; P450 system genes, CYP3A4, CYP3A5, and CYP2D6; and other genes encoding lipoproteins and enzymes of lipid metabolic pathways such as APOE and HMACR, have been suggested to have associations with statin responsiveness.6,8,9 Genetic variations that affect pharmacokinetics of statins may modify atorvastatin disposition and hence its efficacy and toxicity.8,13 However, the effects of genetic variations have been inconsistently replicated, and there are relatively few data available in Asian populations.14 In addition, there are lots of variations in metabolic processes according to statin type, and in the frequency of genetic variations and responsiveness to statins according to ethnic background.6,15
Accordingly, we investigated pharmacokinetic and pharmacodynamic changes in healthy Korean individuals after high-dose atorvastatin administration through serial plasma measurements of drug and lipid concentrations. We assessed associations between genetic variations and pharmacokinetics or lipid-lowering effects of atorvastatin using a predesigned gene panel including genes related to absorption, distribution, metabolism, and elimination.
Materials and methods
Subjects and study design
This study enrolled 50 healthy Korean male subjects. All subjects were from unrelated families and were ascertained to be healthy by medical history, physical examination, vital signs, electrocardiography, and routine clinical laboratory tests. Subjects were given a single oral dose of 80 mg atorvastatin calcium at 08:00 am with 240 mL of water in the overnight fasting state. Subjects fasted for 4 hours after atorvastatin administration, then lunch and dinner were served. Venous blood samples for pharmacokinetic analysis were collected via an intravenous catheter at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24, 36, and 48 hours after dosing. Plasma concentrations of total cholesterol, LDL-C, and triglycerides were measured before and at 24 and 48 hours after atorvastatin administration. Blood sampling for genotyping was performed before drug administration. The study protocol was approved by the Institutional Review Board of Dankook University and Samsung Medical Center, Korea. Written informed consent was obtained from each participant.
Pharmacokinetic and pharmacodynamic measurements
Plasma concentrations of atorvastatin were determined by liquid chromatography–tandem mass spectrometry (LC–MS/MS) using a TSQ Quantum Discovery mass spectrometer (Thermo Electron, San Jose, CA, USA). The ion transitions monitored were m/z 559.2 → 440. Pharmacokinetic parameters were determined by BA-Calc software (Korea Food and Drug Administration, Korea) using actual sampling times. Plasma concentrations of the terminal phase were fitted to a log-linear line by the least squares method to obtain the elimination rate constant. The area under the plasma concentration curve from hour 0 to infinity (AUC∞) was calculated using a combination of the trapezoidal rule and extrapolation to infinity by the elimination rate constant. The maximum drug concentration in plasma (Cmax) and time to Cmax (tmax) were determined from observed values. Clearance (CL) of atorvastatin was adjusted according to the body weight of each subject. Plasma lipid concentrations were measured with an Hitachi 7600-110 chemistry analyzer (Hitachi, Tokyo, Japan).
Genotyping
Genomic DNA was isolated from peripheral blood samples using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA). Genotyping was performed using the Affymetrix drug-metabolizing enzyme and transporter (DMET) Plus array (Affymetrix, Santa Clara, CA, USA), which gauges 1,936 polymorphisms from 225 genes encoding phase I and phase II drug metabolism enzymes as well as drug transporters.16,17 Briefly, we determined the yields of pure double-stranded genomic DNA samples before genotyping. Samples were adjusted to concentrations of 60 ng/μL. Normalized genomic DNA (17 μL) was used as a template for DMET arrays. For loci that had pseudogenes and close homologs, initial genomic amplification using locus-specific primers in a multiplex polymerase chain reaction (mPCR) was performed. By hybridization of highly selective molecular inversion probes (MIPs) to their complementary genomic templates, sequences containing polymorphisms of interest were amplified and then fragmented to improve hybridization onto DMET arrays. Hybridized DMET arrays were scanned with an Affymetrix GeneChip Scanner 3000 7G. Genotyping was performed according to the predefined software algorithms of the manufacturer using DMET Console version 1.0.16,18
Statistical analyses
Of 1,936 polymorphisms in 225 genes screened, 519 nonmonomorphic polymorphisms in 181 genes were identified with a ≥90% call rate, ≥5% minor allele frequency, and nonsignificant deviation from Hardy–Weinberg equilibrium (P≥0.001). Analysis of variance (ANOVA) or Kruskal–Wallis tests were used to test for associations between genotypes and pharmacokinetic parameters or lipid concentration changes from baseline to 48 hours after atorvastatin administration. Pharmacokinetic parameters included AUC∞, Cmax, and clearance adjusted with body weight (CLadj). An ANOVA test was applied for polymorphisms that satisfied the assumptions of normality and homogeneity of variances in phenotype distribution. A P-value less than 0.050 was considered statistically significant. For statistically significant associations, the Jonckheere–Terpstra test was performed to test for ordered differences among genotypes. Corrected P-values were obtained using the Benjamini–Hochberg false discovery rate (FDR) approach. Statistical analyses were conducted using R, version 2.9.1 (R Foundation for Statistical Computing, Vienna, Austria), and IBM SPSS Statistics version 18.0 (SPSS Inc., Chicago, IL, USA).
Results
Participant demographics
A total of 50 healthy individuals were included in this study. Baseline characteristics are presented in Table 1. All subjects were male with a mean age of 24 years (range, 20–27 years) and a mean body weight of 69 kg (50–98 kg). Mean plasma concentrations of total cholesterol, LDL-C, and triglycerides were 150, 72.6, and 110 mg/dL, retrospectively.
 | Table 1 Demographics and baseline characteristics (n=50) |
Atorvastatin pharmacokinetics and pharmacodynamics
Pharmacokinetic properties of atorvastatin are summarized in Table 2. The mean AUC∞, Cmax, tmax, CL, CLadj, and half-life (t1/2) were 172 ng·h/mL, 36.2 ng/mL, 1.07 h, 603 L/h, 8.85 L/(h·kg), and 7.75 h, respectively. Pharmacokinetic parameters showed marked interindividual variability, with a coefficient of variation (CV) ranging from 33.3% to 81.8%. The mean and standard deviation (SD) of the plasma concentration–time profile for atorvastatin after a single oral administration in all participants are shown in Figure 1A. The mean dose-per-body weight normalized AUC∞ and Cmax were 148 ng·h/mL per mg/kg and 31.0 ng/mL per mg/kg, respectively. The CVs of dose-per-body weight normalized AUC∞ and Cmax were 59.0% and 54.4%.
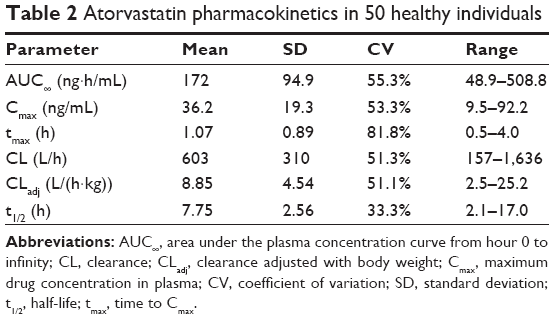 | Table 2 Atorvastatin pharmacokinetics in 50 healthy individuals |
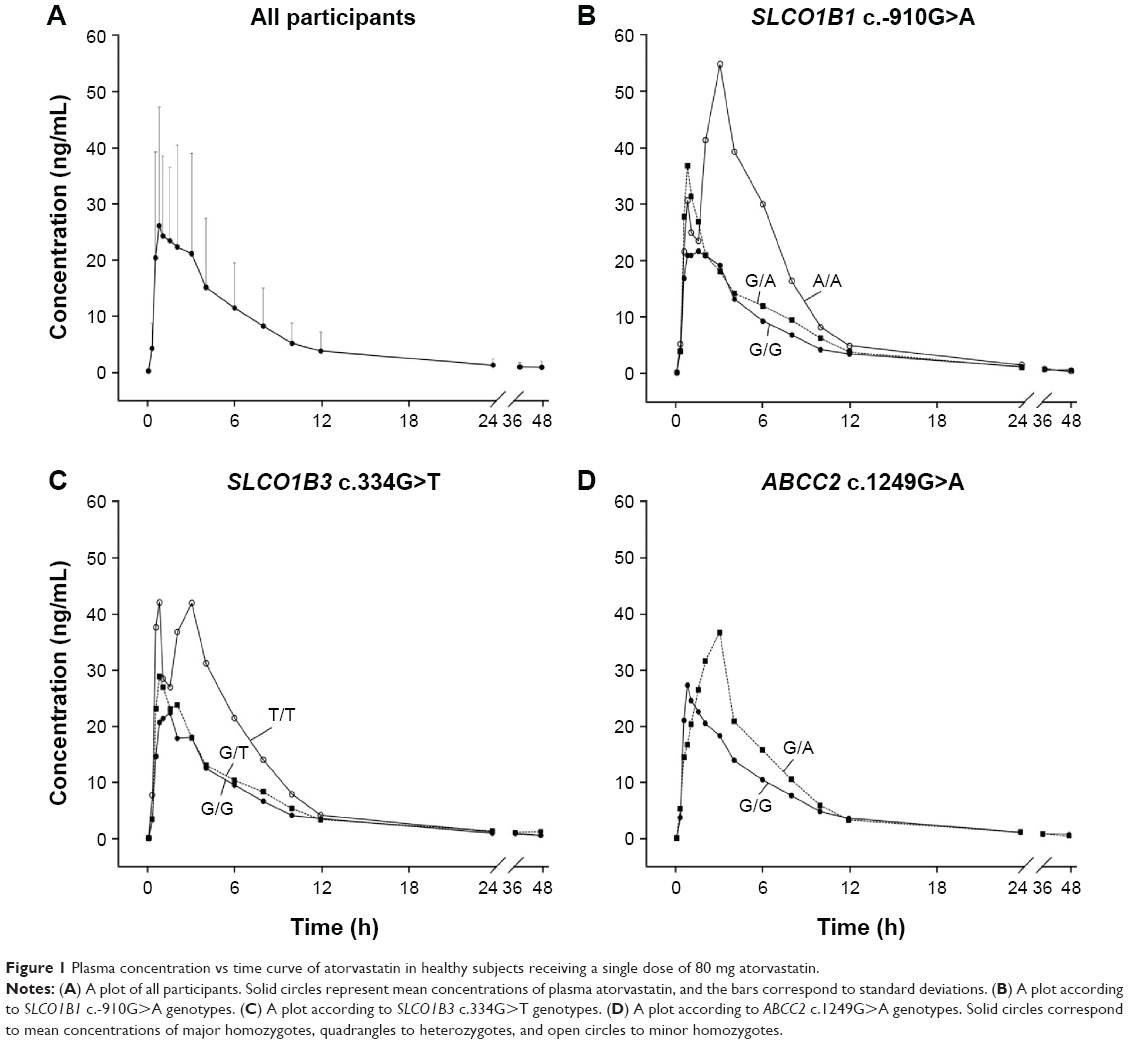 | Figure 1 Plasma concentration vs time curve of atorvastatin in healthy subjects receiving a single dose of 80 mg atorvastatin. |
Lipid concentration changes at 24 and 48 hours from baseline are summarized in Table 3. Mean total cholesterol and LDL-C concentrations at 48 hours after single atorvastatin administration were decreased by 10.2% and 15.1%, respectively. Triglyceride concentrations did not show any statistically significant change. There was no correlation between baseline plasma concentrations or pharmacokinetic parameters of atorvastatin and changes in lipid concentrations.
 | Table 3 Lipid concentrations (mg/dL) and percentage changes (%Δ) from baseline to 24 and 48 hours after atorvastatin administration |
Genetic polymorphisms associated with pharmacokinetic/pharmacodynamic variables
Sixty-four polymorphisms from 47 genes were associated with pharmacokinetic variables or lipid concentration changes according to genotype (P<0.050 in ANOVA or Kruskal–Wallis tests; Table S1). Seventeen polymorphisms were associated with AUC∞, 26 polymorphisms with Cmax, and 16 polymorphisms with CLadj. In terms of a lipid-lowering effect, 16 polymorphisms were associated with LDL-C, and 12 polymorphisms with total cholesterol. The 13 genes related to variance in LDL-C lowering included ABCB11, ABCC1, ABCC2, AHR, CBR1, CYP19A1, CYP1B1, CYP4F11, NAT2, SLC10A2, SLC5A6, SLC7A8, and SULT1B1.
Among these 64 polymorphisms, 3 in the SLCO1B1, SLCO1B3, and ABCC2 genes showed ordinal associations with Cmax, or changes in total cholesterol or LDL-C (Tables 4 and S2). The mean of the plasma concentration–time profile for atorvastatin according to each genotype is shown in Figure 1B–D. For c.-910G>A (rs4149015) in the SLCO1B1 gene, 33 subjects were G/G homozygotes, 14 were G/A heterozygotes, and 3 were A/A homozygotes. The mean Cmax was 76.5 ng/mL for A/A, 34.0 ng/mL for G/A, and 33.5 ng/mL for G/G (Figure 2). In the analysis of haplotypes that included rs4149015, carriers possessing the SLCO1B1*17 variant allele (rs2306283, rs4149056, and rs4149015) showed higher Cmax and AUC∞ compared to noncarriers (46.1 vs 32.8 ng/mL, P=0.032 for Cmax; 222 vs 154 ng/mL, P=0.026 for AUC∞). The SLCO1B3 c.334G>T (p.Ala112Ser, rs4149117) also influenced the Cmax of atorvastatin. Mean Cmax for 6 subjects with T/T (63.0 ng/mL) was higher than that in 16 with G/T (34.7 ng/mL) and 28 with G/G (31.4 ng/mL, FDR P=0.037). In genotype combination analysis of SLCO1B1 c.-910G>A and SLCO1B3 c.334G>T, major homozygous individuals of both polymorphisms showed the lowest mean Cmax (30.7 ng/mL), and the Cmax tended to increase according to the increase in the number of minor alleles (P=0.011, Figure 3); 39.3 ng/mL for 1 minor allele, 29.0 ng/mL for 2 minor alleles, 56.0 ng/mL for 3 minor alleles, and 76.5 for 4 minor alleles. ABCC2 c.1249G>A (p.Val417Ile, rs2273697) was associated with changes in total cholesterol and LDL-C at 48 hours after atorvastatin administration. In particular, the decrease in total cholesterol and LDL-C was smaller in those with G/A (n=7) than in the 43 subjects with G/G. There was no A/A homozygote identified. The mean percentage changes in total cholesterol and LDL-C in subjects with G/A were 0.23% and −0.70%, compared to −11.9% and −17.4% for those with G/G.
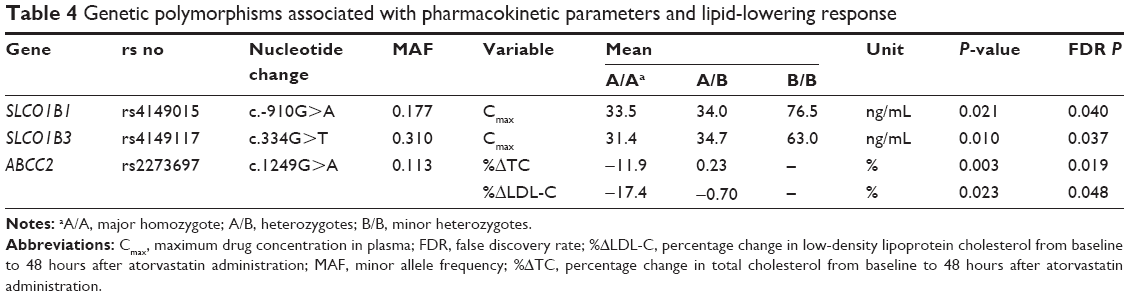 | Table 4 Genetic polymorphisms associated with pharmacokinetic parameters and lipid-lowering response |
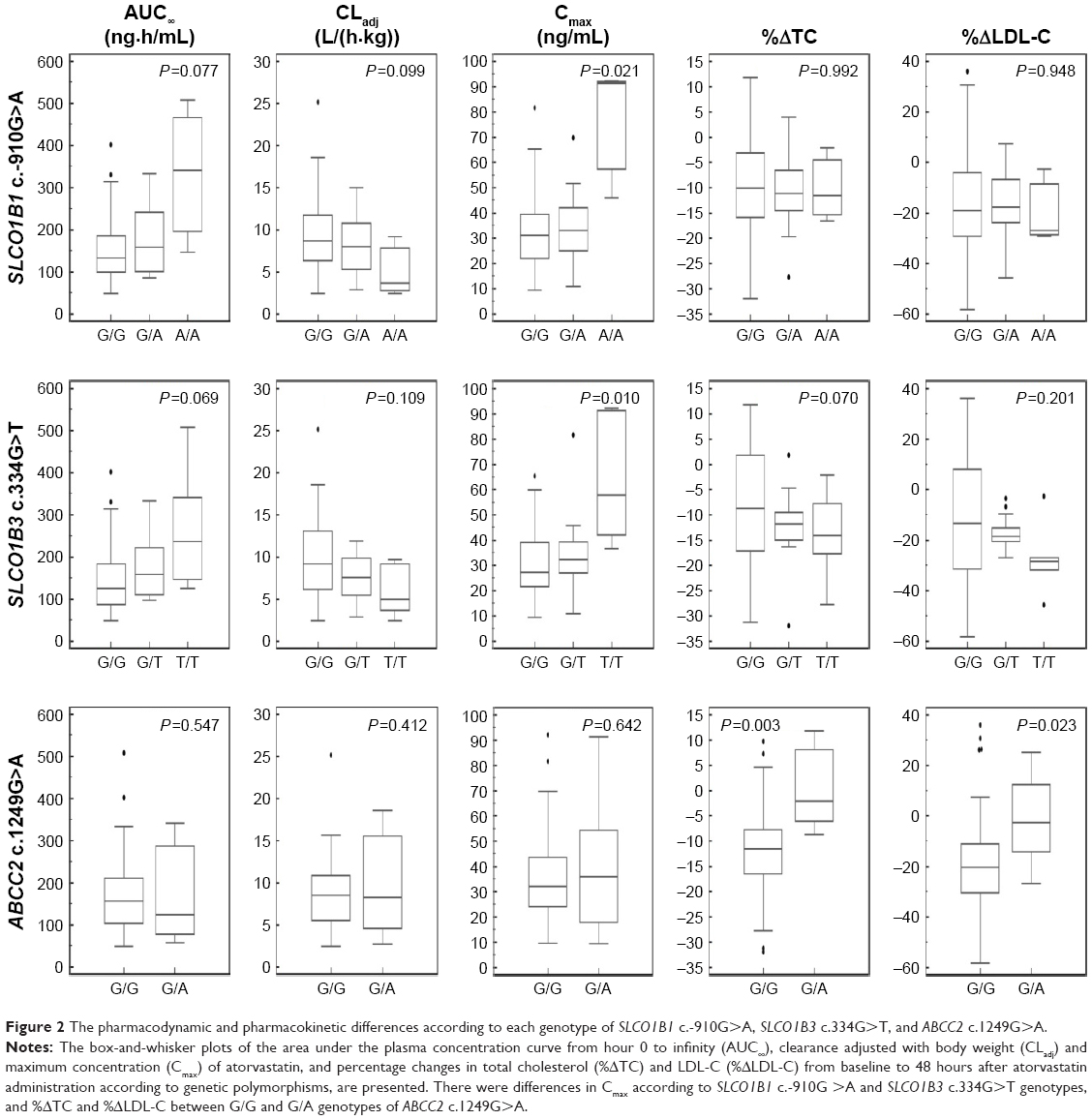 | Figure 2 The pharmacodynamic and pharmacokinetic differences according to each genotype of SLCO1B1 c.-910G>A, SLCO1B3 c.334G>T, and ABCC2 c.1249G>A. |
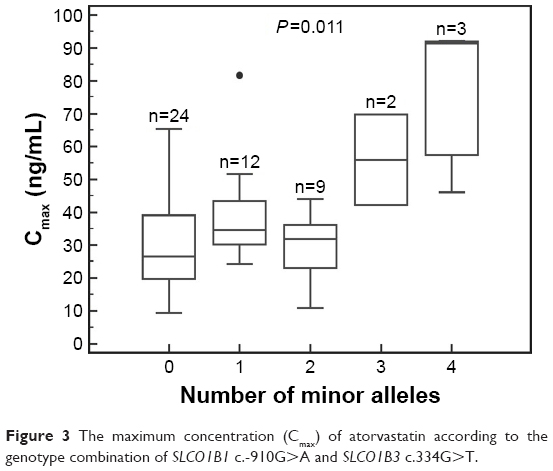 | Figure 3 The maximum concentration (Cmax) of atorvastatin according to the genotype combination of SLCO1B1 c.-910G>A and SLCO1B3 c.334G>T. |
Discussion
This study investigated pharmacokinetic characteristics and lipid-lowering response following high-dose atorvastatin treatment in young, healthy Korean males in association with genotypes in genes related to absorption, distribution, metabolism, and elimination of drugs.
Statins are known to produce immediate biochemical changes.19,20 We showed that atorvastatin achieved Cmax at around 1.07 hours. Dose-per-body weight normalized AUC∞ and Cmax were comparable to the results from previous studies in Asians and Caucasians.14 Interindividual variability of pharmacokinetic parameters was observed in spite of the uniformity of the enrolled subjects, who were all young and healthy males, and controlled conditions. This finding suggests that much of the pharmacokinetic variability is caused by innate or underlying conditions such as genetic factors and the gut microbiome, instead of controllable environmental factors such as concomitant medicines and compliance. The lipid-lowering effect of atorvastain also showed interindividual variation. In agreement with previous studies, there were no pharmacokinetic parameters associated with the lipid-lowering effect of atorvastatin.19,20
Because the pharmacokinetic and pharmacodynamic changes in this study were as expected, we next inspected their association with multiple genetic polymorphisms. Polymorphisms in the SLCO1B1, SLCO1B3, and ABCC2 genes were ordinally associated with pharmacokinetic properties or lipid-lowering responses. SLCO1B1 c.-910G>A, identified in 17 subjects, was associated with Cmax, and the SLCO1B1*17 haplotype including this polymorphism was also related to Cmax and AUC∞. The SLCO1B1 gene encodes the organic anion transporting polypeptide (OATP) 1B1, which facilitates hepatic uptake of statins on the sinusoidal membrane of hepatocytes.21,22 Variations in SLCO1B1, c.-910G>A and c.521T>C (rs4149056), and haplotypes, *5, *15, and *17, have been reported to be associated with pharmacokinetic and lipid-lowering responses in previous studies.6–8,23 In addition, a loss-of-function variation, c.521T>C, which reduced liver influx of the statins, has a potent effect on myalgia, one adverse effect of statins.8 Similar findings have been reported in individuals who received atorvastatin, including Asians.8,24,25
SLCO1B3 c.334G>T was associated with a higher Cmax in this study. The OATP 1B3 encoded by the SLCO1B3 gene is one of the major hepatic OATPs and has a potent function as an active transporter of atorvastatin, following the OATP 1B1.26,27 Several genetic polymorphisms in the SLCO1B3 gene have been investigated in previous in vitro studies.28,29 A preclinical study showed no effect of c.334G>T on cellular uptake of atorvastatin,28 which suggests the minor effect of the SLCO1B3 gene on the distribution of atorvastatin. As previous studies suggested the aggregate effect of top-associated polymorphisms,30,31 we evaluated the genotype combination effect. We observed the genotype combination effect of SLCO1B1 c.-910G>A and SLCO1B3 c.334G>T; thus, further in vivo analysis of the role of transporter enzymes on the metabolism of statins is needed to clarify the interaction.
c.1249G>A in the ABCC2 gene was associated with a small lipid-lowering response in this study. Multidrug resistance-associated protein 2 (MRP2/ABCC2) is an efflux transporter expressed in various types of cells, including hepatocytes, enterocytes, and proximal renal tubular cells,32 and plays an important role in reducing gastrointestinal absorption and facilitating the biliary and urinary excretion of its substrates, including pravastatin and fluvastatin.32–34 A polymorphism in the ABCC2 gene has been related to low plasma concentrations of pravastatin,33 as well as dose decreases or switches to other cholesterol-lowering agents during simvastatin and atorvastatin therapy.35 In addition, after atorvastatin administration, mRNA levels of transporters, including MRP2/ABCC2, are downregulated and positively correlated with the percentage of reduction in LDL-C.36 Collectively, these data indicate that the ABCC2 gene might affect the lipid-lowering response to atorvastatin treatment.
In this prospective study, we performed a pharmacokinetic and pharmacodynamic analysis in healthy Korean individuals following high-dose atorvastatin administration. However, we should acknowledge the limitation of our study. Because of the relatively small sample size, some associations may have been missed or noticed only by chance, and also the aggregate effect of polymorphisms was partially evaluated. Our study findings should be confirmed through future large prospective studies in various ethnic populations. The strength of our study is that we provided prospective data about pharmacokinetics and the lipid-lowering response after atorvastatin 80 mg administration for a hypothesis-free genetic association study in healthy, young male Asian subjects; a population that has not been studied in this context before.25 Our findings support the value of further studies investigating factors that affect interindividual atorvastatin treatment variability, such as genes related to pharmacodynamics, and the contribution to the risk of adverse effects of polymorphisms identified as associated with the Cmax of atorvastatin.
Conclusion
In conclusion, we genotyped multiple polymorphisms in genes related to phase I and II drug metabolism enzymes and drug transporters, and evaluated the association of these with pharmacokinetic properties and lipid-lowering response following atorvastatin administration. Our findings describe pharmacokinetic and pharmacodynamic changes with variations among individuals after high-dose atorvastatin treatment in healthy Korean subjects. We also identified various genetic polymorphisms related to the response to atorvastatin treatment, including the association between polymorphisms in the transporter genes, SLCO1B1, SLCO1B3, and ABCC2, and either Cmax of atorvastatin or lipid-lowering response. These findings contribute to the understanding of interindividual variation in atorvastatin treatment.
Acknowledgments
We thank Hyung-Gun Kim (Department of Pharmacology, Dankook University) for his contribution in performing the clinical experiments and pharmacokinetic analysis. This research was supported by a grant (HI13C2098) from Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea.
Disclosure
The authors report no conflicts of interest in this work.
References
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2889–2934. | ||
Mangravite LM, Thorn CF, Krauss RM. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenomics J. 2006;6(6):360–374. | ||
Bousoula E, Kolovou V, Perrea D, Kolovou G. Pharmacogenetics and statin treatment: reality or theory? Curr Vasc Pharmacol. 2015;13(5):616–623. | ||
Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685–696. | ||
LaRosa JC. Low-density lipoprotein cholesterol reduction: the end is more important than the means. Am J Cardiol. 2007;100(2):240–242. | ||
Kajinami K, Akao H, Polisecki E, Schaefer EJ. Pharmacogenomics of statin responsiveness. Am J Cardiol. 2005;96(9A):65K–70K; discussion 34K–35K. | ||
Chasman DI, Giulianini F, MacFadyen J, Barratt BJ, Nyberg F, Ridker PM. Genetic determinants of statin-induced low-density lipoprotein cholesterol reduction: the Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER) trial. Circ Cardiovasc Genet. 2012;5(2):257–264. | ||
Sirtori CR, Mombelli G, Triolo M, Laaksonen R. Clinical response to statins: mechanism(s) of variable activity and adverse effects. Ann Med. 2012;44(5):419–432. | ||
Hopewell JC, Parish S, Offer A, et al. Impact of common genetic variation on response to simvastatin therapy among 18 705 participants in the Heart Protection Study. Eur Heart J. 2013;34(13):982–992. | ||
Deshmukh HA, Colhoun HM, Johnson T, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: importance of Lp(a). J Lipid Res. 2012;53(5):1000–1011. | ||
Barber MJ, Mangravite LM, Hyde CL, et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS One. 2010;5(3):e9763. | ||
Thompson JF, Hyde CL, Wood LS, et al. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ Cardiovasc Genet. 2009;2(2):173–181. | ||
Rodrigues AC, Hirata MH, Hirata RD. The genetic determinants of atorvastatin response. Curr Opin Mol Ther. 2007;9(6):545–553. | ||
Gandelman K, Fung GL, Messig M, Laskey R. Systemic exposure to atorvastatin between Asian and Caucasian subjects: a combined analysis of 22 studies. Am J Ther. 2012;19(3):164–173. | ||
Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99(3):410–414. | ||
Burmester JK, Sedova M, Shapero MH, Mansfield E. DMET microarray technology for pharmacogenomics-based personalized medicine. Methods Mol Biol. 2010;632:99–124. | ||
Hu Y, Ehli EA, Nelson K, et al. Genotyping performance between saliva and blood-derived genomic DNAs on the DMET array: a comparison. PLoS One. 2012;7(3):e33968. | ||
Daly TM, Dumaual CM, Miao X, et al. Multiplex assay for comprehensive genotyping of genes involved in drug metabolism, excretion, and transport. Clin Chem. 2007;53(7):1222–1230. | ||
Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–1160. | ||
Stern RH, Yang BB, Hounslow NJ, MacMahon M, Abel RB, Olson SC. Pharmacodynamics and pharmacokinetic-pharmacodynamic relationships of atorvastatin, an HMG-CoA reductase inhibitor. J Clin Pharmacol. 2000;40(6):616–623. | ||
Ho RH, Kim RB. Transporters and drug therapy: implications for drug disposition and disease. Clin Pharmacol Ther. 2005;78(3):260–277. | ||
Hsiang B, Zhu Y, Wang Z, et al. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J Biol Chem. 1999;274(52):37161–37168. | ||
Niemi M, Schaeffeler E, Lang T, et al. High plasma pravastatin concentrations are associated with single nucleotide polymorphisms and haplotypes of organic anion transporting polypeptide-C (OATP-C, SLCO1B1). Pharmacogenetics. 2004;14(7):429–440. | ||
Morimoto K, Oishi T, Ueda S, Ueda M, Hosokawa M, Chiba K. A novel variant allele of OATP-C (SLCO1B1) found in a Japanese patient with pravastatin-induced myopathy. Drug Metab Pharmacokinet. 2004;19(6):453–455. | ||
Lee YJ, Lee MG, Lim LA, Jang SB, Chung JY. Effects of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of atorvastatin and 2-hydroxyatorvastatin in healthy Korean subjects. Int J Clin Pharmacol Ther. 2010;48(1):36–45. | ||
Vildhede A, Karlgren M, Svedberg EK, et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug-drug interactions. Drug Metab Dispos. 2014;42(7):1210–1218. | ||
Kunze A, Huwyler J, Camenisch G, Poller B. Prediction of organic anion-transporting polypeptide 1B1- and 1B3-mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab Dispos. 2014;42(9):1514–1521. | ||
Schwarz UI, Meyer zu Schwabedissen HE, Tirona RG, et al. Identification of novel functional organic anion-transporting polypeptide 1B3 polymorphisms and assessment of substrate specificity. Pharmacogenet Genomics. 2011;21(3):103–114. | ||
DeGorter MK, Ho RH, Leake BF, Tirona RG, Kim RB. Interaction of three regiospecific amino acid residues is required for OATP1B1 gain of OATP1B3 substrate specificity. Mol Pharm. 2012;9(4):986–995. | ||
Derringer J, Krueger RF, Dick DM, et al. The aggregate effect of dopamine genes on dependence symptoms among cocaine users: cross-validation of a candidate system scoring approach. Behav Genet. 2012;42(4):626–635. | ||
Ferrari M, Guasti L, Maresca A, et al. Association between statin-induced creatine kinase elevation and genetic polymorphisms in SLCO1B1, ABCB1 and ABCG2. Eur J Clin Pharmacol. 2014;70(5):539–547. | ||
Gerk PM, Vore M. Regulation of expression of the multidrug resistance-associated protein 2 (MRP2) and its role in drug disposition. J Pharmacol Exp Ther. 2002;302(2):407–415. | ||
Niemi M, Arnold KA, Backman JT, et al. Association of genetic polymorphism in ABCC2 with hepatic multidrug resistance-associated protein 2 expression and pravastatin pharmacokinetics. Pharmacogenet Genomics. 2006;16(11):801–808. | ||
Lindahl A, Sjoberg A, Bredberg U, Toreson H, Ungell AL, Lennernas H. Regional intestinal absorption and biliary excretion of fluvastatin in the rat: possible involvement of mrp2. Mol Pharm. 2004;1(5):347–356. | ||
Becker ML, Elens LL, Visser LE, et al. Genetic variation in the ABCC2 gene is associated with dose decreases or switches to other cholesterol-lowering drugs during simvastatin and atorvastatin therapy. Pharmacogenomics J. 2013;13(3):251–256. | ||
Rodrigues AC, Hirata MH, Hirata RD. Impact of cholesterol on ABC and SLC transporters expression and function and its role in disposition variability to lipid-lowering drugs. Pharmacogenomics. 2009;10(6):1007–1016. |
Supplementary materials
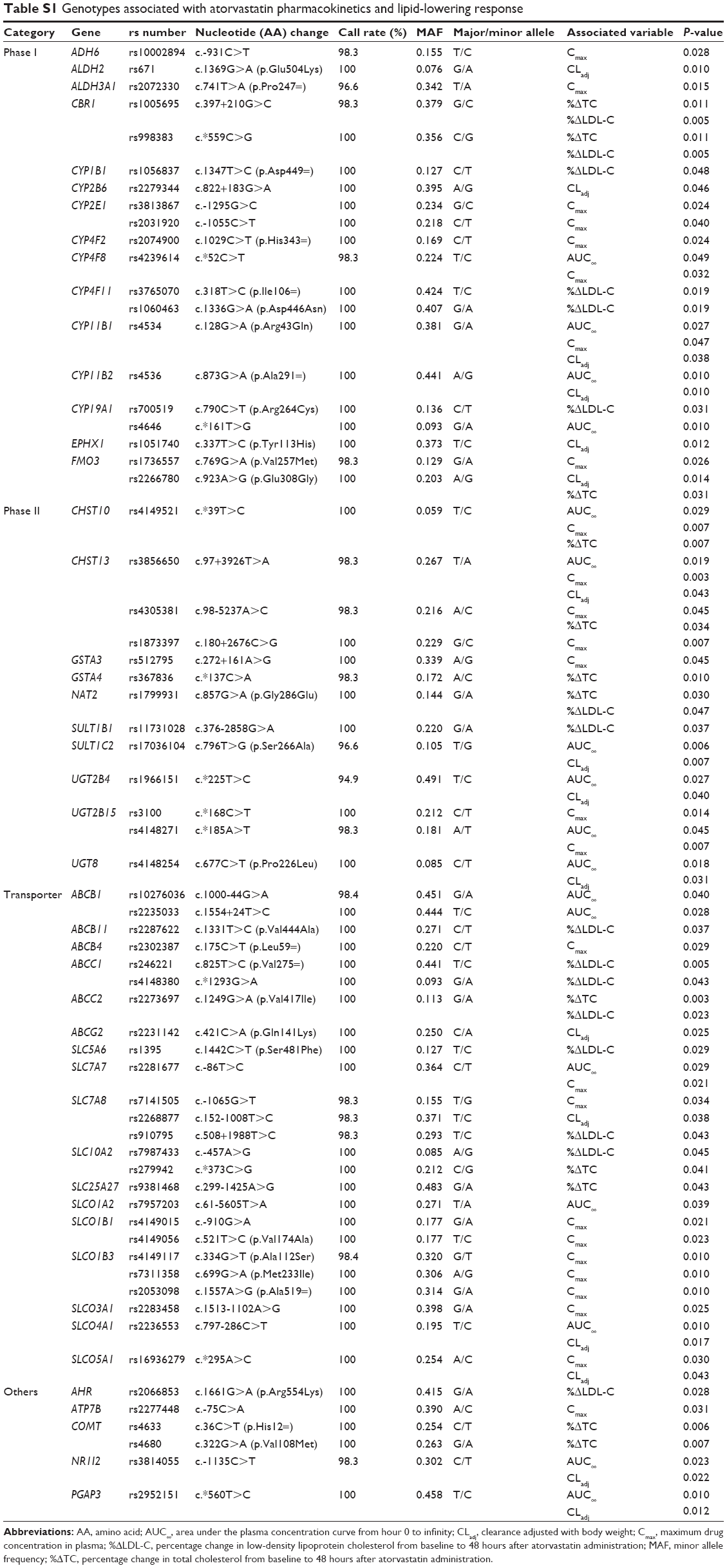 | Table S1 Genotypes associated with atorvastatin pharmacokinetics and lipid-lowering response |
 | Table S2 Pharmacokinetic parameters and lipid-lowering response of SLCO1B1 c.-910G>A, SLCO1B3 c.334G>T, and ABCC2 c.1249G>A |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.