")
Back to Journals » Cancer Management and Research » Volume 11
Assessment of the presence and anti-tumor potential of tumor-infiltrating lymphocytes in patients with acute myeloid leukemia
Authors Wei L, Wang Z, Zhang Z, Li Y, Fan S, Zhao Y, Liu Z, Ye X, Zhang F, Yu Y, Liu X, Cao F, Zhou J
Received 29 December 2018
Accepted for publication 18 March 2019
Published 12 April 2019 Volume 2019:11 Pages 3187—3196
DOI https://doi.org/10.2147/CMAR.S199817
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Alexandra R. Fernandes
Liya Wei,1 Zhenkun Wang,2 Zhuo Zhang,1 Yinghua Li,1 Shengjin Fan,1 Yanqiu Zhao,1 Zhiyu Liu,2 Xiangmei Ye,2 Fan Zhang,2 Yingying Yu,2 Xiaolong Liu,2 Fenglin Cao,2 Jin Zhou1,2
1Department of Hematology, The First Affiliated Clinical Hospital of Harbin Medical University, Harbin, Heilongjiang 150001, People’s Republic of China; 2Central Laboratory of Hematology and Oncology, First Affiliated Hospital, Harbin Medical University, Harbin, Heilongjiang 150001, People’s Republic of China
Purpose: Assessing the possibility of finding tumor-infiltrating lymphocytes (TIL) in bone marrow of acute myeloid leukemia (AML) patients and evaluating the anti-tumor activity of these TIL against autologous AML cells.
Patients and methods: TIL were immunomagnetically isolated by using anti-CD3 from bone marrow samples of 20 patients at the presentation of AML or four weeks upon completion of chemotherapy. TIL were ex vivo expanded for two weeks and immunophenotyped. Functionality in terms of cytokine secretion and cytotoxicity was assessed by γ-interferon quantitation and Elispot assay, respectively.
Results: TIL were detected in bone marrow samples of 50% (10/20) of the patient cohort. They were noted to highly express CD137 and PD-1 and display a significantly higher anti-tumor reactivity compared to that of autologous peripheral blood lymphocytes. TIL could be expanded in co-cultures with irradiated feeder cells supplemented with interleukin (IL)-7 and IL-15.
Conclusion: Data suggested the presence of reactive γ-interferon-secreting TIL in AML patients. They are expandable and possess anti-tumor activity, which might have a great potential in the development of adoptive cellular therapy for AML.
Keywords: tumor-infiltrating lymphocytes, acute myeloid leukemia, adoptive cell therapy, immunotherapy
Introduction
Acute myeloid leukemia (AML) is the most common hematological malignancies in adults, with an incidence rate ranging from three to five cases per 1,00,000 population.1,2 AML is characterized by monoclonal expansion of abnormal myeloid precursors, resulting in impaired hematopoiesis and bone marrow failure.3 With the contemporary chemotherapy, the mortality rate is high especially among patients above 60 years of age, being approximately 70% within one year of diagnosis.4,5 Besides, less than 50% of AML patients achieved a complete response, and in most cases, the disease relapsed quickly and became refractory to standard treatment.6 Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the effective therapy available to refractory and relapsed AML patients.7–9 However, serious adverse events and graft-versus-host disease are often associated with allo-HSCT.10 Many AML patients are also not eligible for allo-HSCT. Undoubtedly, new treatment modalities are urgently needed.
Immunotherapies might be good alternatives, but reports of the clinical outcomes available in the literature were not very encouraging.11–15 The human immune system can recognize and eliminate autologous tumor cells expressing “nonself” antigens.16 Malignant tumors are known to have genetic mutations, some of which mediate neo-epitopes,provoking an immune response of host defense.17 Neo-epitopes are “non-self” so they may trigger T cell responses. Indeed, neoantigen-reactive T cells which infiltrated human malignant tumors were detected.18–21 Adoptive cell therapy (ACT) with tumor-infiltrating lymphocytes (TIL) was noted to be effective in the treatment of metastatic melanoma patients, leading to response rates to 50%.22,23 In addition, it was reported that adoptive transfer of mutation-reactive CD4+ T cells mediated a durable and ongoing regression of metastatic diseases of more than two years in a patient with metastatic cholangiocarcinoma.24 However, it is unclear whether TIL are present in AML patients and how the naturally occurring and mutation-specific T cells interact with autologous leukemic counterparts.
Like solid tumors, AML cells may have gene mutations. Some of which may possess neo-antigens and become “non-self” that can trigger T cell responses. In this study, we assessed TIL in AML patients and examined the potential significance in the ACT of AML.
Patients and methods
Patient samples
The study was approved by the Institutional Review Board of Harbin Medical University and conducted according to the Declaration of Helsinki. Patients were at least 18 years of age at presentation of AML or four weeks upon completion of chemotherapy. Patients with a life expectancy of less than six months, impaired organ function, having undergone allo-HSCT were excluded. Bone marrow and peripheral blood samples were taken with written informed consents of the patients. All patients had disease-relevant gene mutations examined by next-generation sequencing at the time of enrollment. Mutations in the following 127 genes were detected in the indicated number of patients (Supplementary materials, Methods).
Enrichment and expansion of TIL
Bone marrow mononuclear cells (BMMC) and peripheral blood mononuclear cells (PBMC) were enriched by density-gradient centrifugation using Ficoll-Hypaque (Supplier, Country). Cells were then labeled with CD3 microbeads (Miltenyi Biotech, Germany) for 15 mins at 4°C–6°C. Immunomagnetic device VarioMACS (Miltenyi Biotech, Germany) was used for cell selection according to the manufacturer’s protocol. CD3+ cells were considered AML TIL, whereas CD3-cells were considered autologous AML cells.
BMMC and PBMC were seeded at 1×106 cells/mL in X-VIVO 15 medium (Lonza, Switzerland) supplemented with 5% heat-inactivated human serum, 100 ng/mL anti-CD3, 1000 IU/mL of interleukin (IL)-2, 10 ng/mL of IL-7 and 10 ng/mL of IL-15 (PeproTech, USA) and co-cultured with 50 Gy-irradiated PBMC derived from unrelated donors as feeder cells at the ratio (10:1) of feeder cells to BMMC or PBMC for 14 days at 37°C. Fresh X-VIVO 15 medium with cytokines was added whenever the medium turned yellow.
AML TIL were cultured with autologous AML cells at the ratio of 1:1 in X-VIVO 15 medium with 5% heat-inactivated human serum, 10 ng/mL of IL-7 and 10 ng/mL of IL-15 overnight at 37°C. Cultured cells were washed with phosphate-buffered saline (PBS) and labeled with CD137 microbeads. Immunomagnetic sorting was conducted using the selection kit according to the manufacturer’s instructions (Miltenyi Biotech, Germany).
Immunophenotyping
Cells were stained with anti-human CD3-APC, anti-human CD4-PE, anti-human CD8-FITC, anti-human CD56-PE, anti-human CD279-APC, and anti-human CD137-PE (BD Biosciences, USA) for 15 mins at 4°C. They were then washed with and re-suspended in PBS. Events were acquired using a flow cytometer (Calibur, BD Bioscience) and analyzed by Cell Quest software (BD Biosciences).
Elispot assay
Assays were performed according to the manufacturer’s protocol (Mabtech, Nacka, Sweden). In brief, AML TIL of 1×105 were mixed with equal numbers of autologous AML cells in 200 μL of X-VIVO 15 medium (Lonza, Switzerland) and seeded onto wells of Interferon-γ (IFN-γ) Elispot plate pre-coated with monoclonal antibody IFN-γ. Having cultured overnight at 37°C in a 5% CO2 incubator, the plate was washed. Spots were detected and visualized by sequential incubation with the antibody 7-B6-1-biotin, streptavidin-ALP and the substrate BCIP/NBT-plus. Spots were counted using an ELISPOT Plate Reader (ImmunoSpot, CTL-Europe GmbH). Assays were run in duplicate.
IFN-γ release assay
AML TIL or AML activated peripheral blood lymphocytes (aPBL) of 1×105 were co-cultured with equal numbers of autologous AML cells in 200 μL of X-VIVO 15 medium on 96-well plates overnight at 37°C. Cultures in triplicate were harvested and centrifuged. Supernatant was collected. Secreted IFN-γ were assayed in triplicate by enzyme-linked immunosorbent assay according to the manufacturer’s instructions (R&D, USA).
Cell-mediated cytotoxicity assay
Leukemic cell line K562 and NB4 were obtained from America Type Culture Collection (Manassas, VA, USA). Cytosine arabinoside (Actavis S.P.A, Italia) was used as a positive control. AML TIL or AML aPBL of 2×105 and K562 cells or NB4 cells of 2×104 at the ratio of 10:1 were co-cultured for 4 hrs at 37°C in 200 μL of X-VIVO 15 medium. Cultures were harvested. K562 cells and NB4 cells (at a density of 2×105cells/mL) were treated with cytosine arabinoside (Ara-C) at a final concentration of 20 uM in 100 μL of X-VIVO 15 medium. Supernatant was collected upon centrifugation. The cytosolic enzyme, lactate dehydrogenase, was assayed in triplicate using CytoTox 96R kit according to the manufacturer’s instructions (Promega, USA).
Statistical analysis
Data are presented as mean and SD, unless stated otherwise. The non-parametric two-tailed Student’s t-test was used to analyze variables. Differences in values between tested groups were regarded as significant at a value of p≤0.05.
Results
Patient characteristics
Twenty AML patients (13 patients with newly diagnosed AML and seven patients with relapsed AML) of a median year of age of 38 (range: 19–56) were enrolled to the study between March 2017 and July 2018 (Table 1). Blast counts of bone marrow samples at disease presentation ranged from 20% to 97% of nucleated cells. Cytogenetic analysis of bone marrow of nine patients exhibited a normal karyotype accounting to 45% (9/20) of the patient cohort and gene mutations were detected in 80% (16/20) of the patients. None had undergone transplantation.
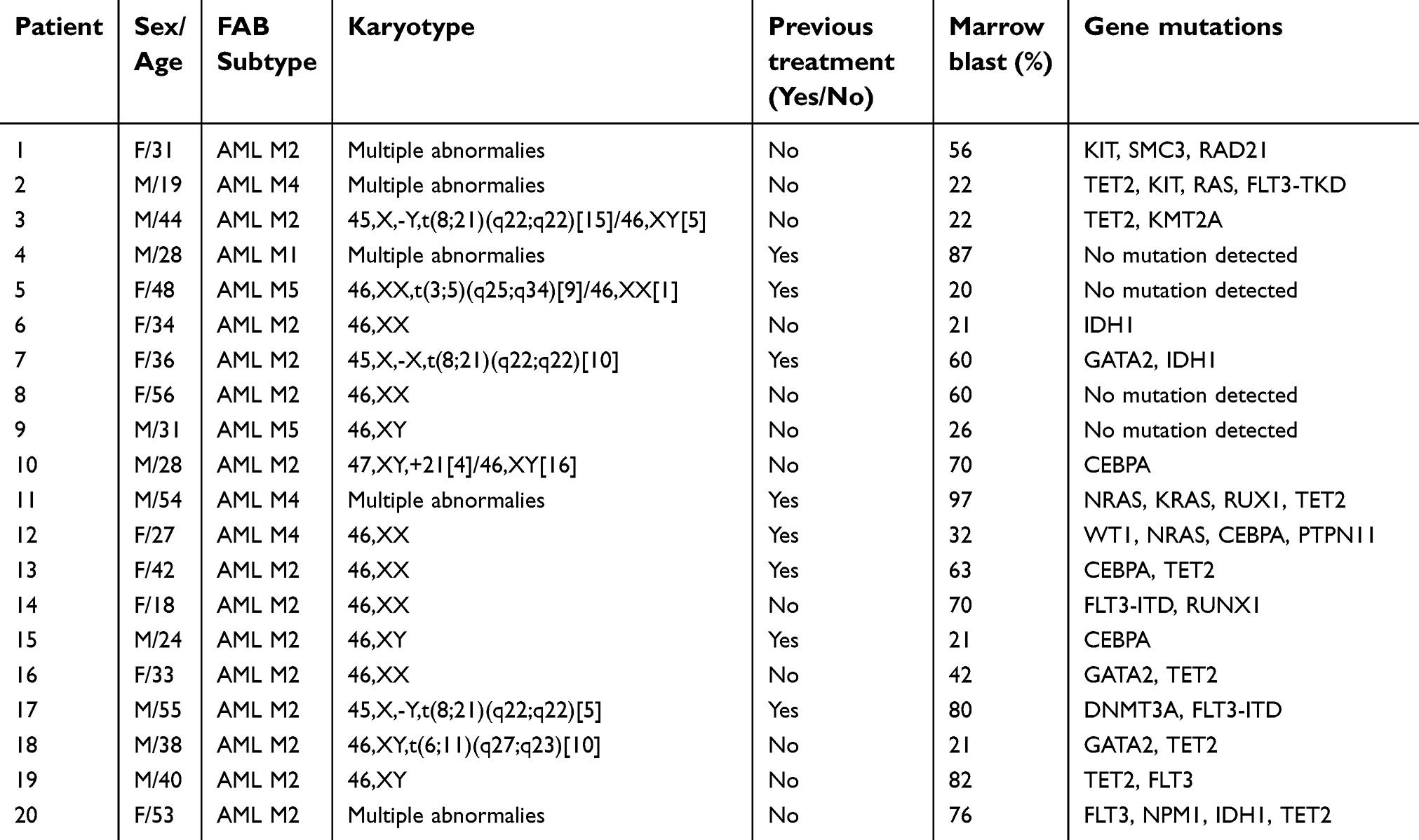 | Table 1 Patient demography |
Presence and frequency of TIL
AML TIL, which were CD3+ and capable to produce IFN-γ, were detected in bone marrow samples of 50% (10/20) of the patient cohort (Figure 1A and B). Positive Elispot responses were identified in Pt. 1, Pt. 2, Pt. 3, Pt. 6, Pt. 7, Pt. 10, Pt. 13, Pt. 14, Pt. 15, and Pt. 16. Negative Elispot responses were identified in Pt. 4, Pt. 5, Pt. 8, Pt. 9, Pt. 11, Pt. 12, Pt. 17, Pt. 18, Pt. 19, and Pt. 20. The spot counts of AML TIL were shown in Table S1. The median spot counts were 152 (range: 70–250).
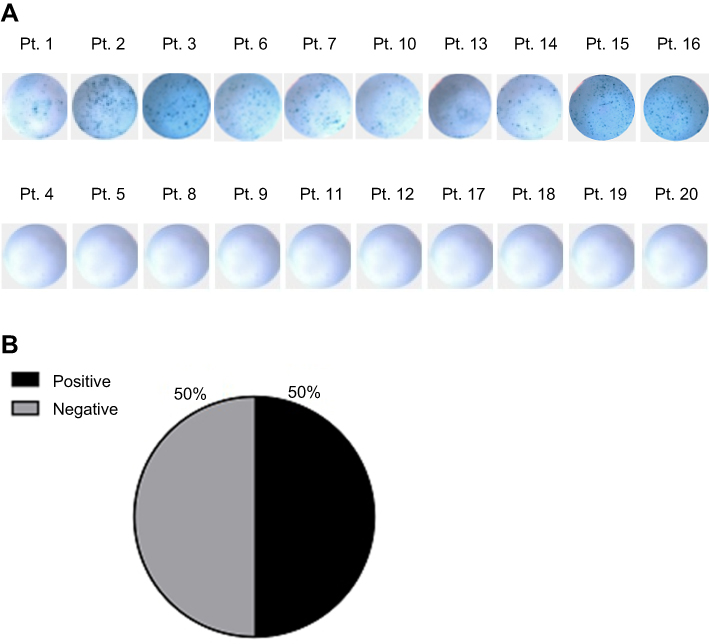 | Figure 1 IFN-γ Elispot. (A) AML TIL for each patient were co-cultured overnight with autologous AML cells. Representative images of duplicate experiments are shown. (B) Distribution chart of AML patients with and without detectable TIL.Abbreviations: AML, acute myeloid leukemia; Pt. patient; TIL, tumor-infiltrating lymphocytes. |
Expansion of TIL
We tested the expandability of AML TIL in 10 bone marrow samples and compared to that of circulating lymphocytes from 10 peripheral blood samples collected from the patient cohort in 14-day co-culture system using irradiated feeder cells of unrelated PBMC supplemented with anti-CD3, IL-2, IL-7, and IL-15. Bone marrow group yielded a mean ± SD of 8.7 x 109±1.0 x 109 cells being similar to 7.1 x 109±0.8 x 109 derived from the peripheral blood cell group (p=0.093). The folds of increase in cell numbers of 875±102 were noted among bone marrow samples being comparable to 713±80 of the circulating lymphocytes in the peripheral blood samples (p=0.098) (Figure 2).
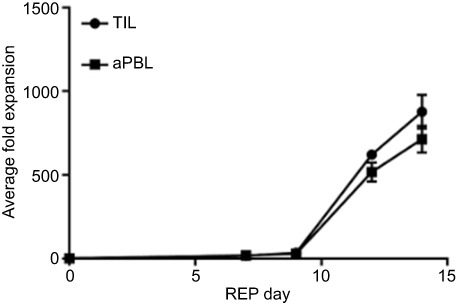 | Figure 2 Growth kinetics of AML TIL (n=10) and AML aPBL (n=10) in triplicate.Abbreviations: aPBL, activated peripheral blood lymphocytes; AML, acute myeloid leukemia; TIL, tumor-infiltrating lymphocytes. |
Phenotypic characterization of expanded TIL
The immunophenotypings of ex vivo expanded TIL and circulating lymphocytes in peripheral blood are shown in Table 2. There were no significant differences of the relative frequencies of the cell subsets, namely, CD3+CD8+ T cells, CD3+CD4+ T cells, and CD3+CD56+ T cells derived from bone marrow and peripheral blood of AML patients, suggesting the culture system could expand AML TIL equally well. However, expressions of CD137 (4-1BB) and CD279 (PD-1) on TIL were significantly up-regulated compared to those expressed on circulating lymphocytes.
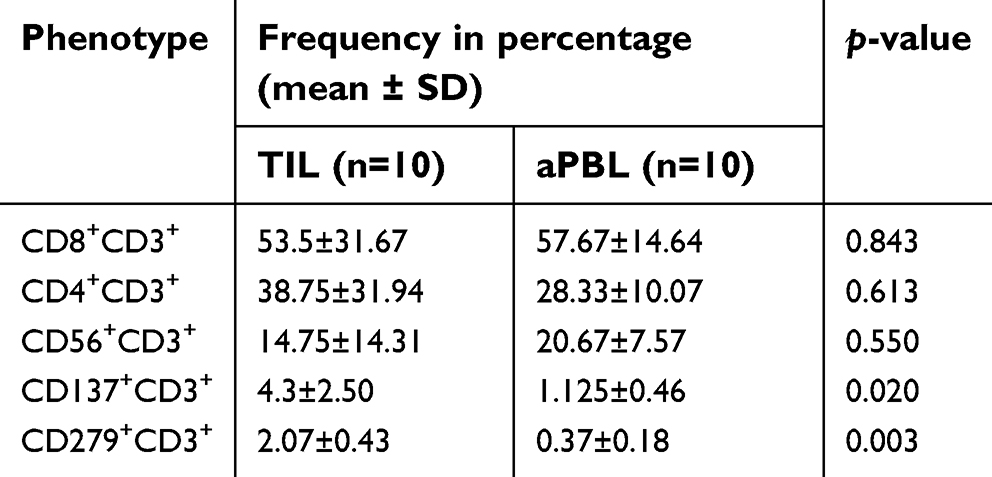 | Table 2 Immunophenotypes of ex vivo expanded TIL and aPBL of AML patients |
Functionality of expanded TIL
Figure 3A shows the quantitation of secretary IFN-γ in the culture supernatants of the 10 bone marrow samples evident of detectable TIL. A mean of 2228.5 pg/mL (range: 670–3900 pg/mL) was found. All readouts were far beyond the cut-off threshold of 200 pg/mL being widely acknowledged in the field.20 CD137 is a costimulatory marker that is induced upon the specific recognition of T cells by their target cell, and reactivity of the TILs was also evaluated by the upregulation of CD137. The baseline CD137 expression in the cultures of AML TILs before and following coincubation with autologous AML cells was determined in the 10 bone marrow samples, supporting the understanding that CD137 serves as a marker of the reactivity of AML (Figure 3B).
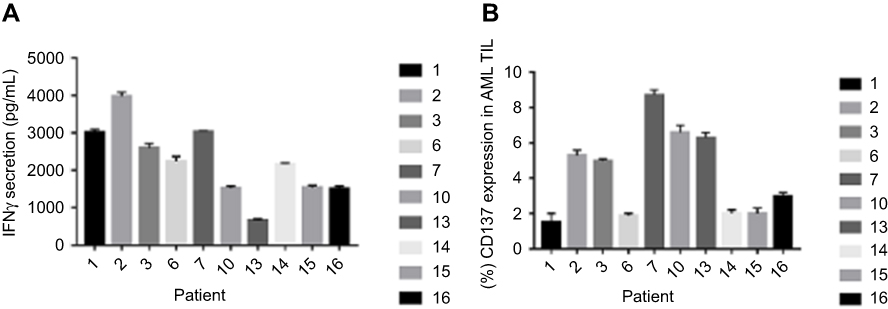 | Figure 3 Overnight co-cultures of AML TIL and autologous AML cells in triplicate. (A) IFN-γ secretion. (B) CD137expression.Abbreviations: AML, acute myeloid leukemia; TIL, tumor-infiltrating lymphocytes. |
In the comparative study of expanded TIL and aPBL of three AML patients, the secretary IFN-γ levels derived from co-cultures of TIL and autologous AML cells were much higher than those from co-cultures of aPBL and autologous AML cells suggesting greater anti-leukemia reactivity (Figure 4A). The anti-leukemia propensity of TIL was further attested in cytotoxicity assays using K562 cells which numbers were normalized to the same numbers of TIL and aPBL in the assays (Figure 4B, Patient 1: TIL-mediated cytotoxicity vs aPBL-mediated; 36.8%±2.2% vs 18.7%±2.0%; p=0.001, Patient 2: 41.5%±8.5% vs 21.0%±4.5%; p=0.021, Patient 3: 38.1%±5.0% vs 21.6%±3.1%; p=0.008). (Ara-C)-mediated cytotoxicity was 14.93%±0.64% in K562 cells.The anti-leukemia propensity of TIL was also attested in cytotoxicity assays using NB4 cells which numbers were normalized to the same numbers of TIL and aPBL in the assays (Figure 4C, Patient 1: TIL-mediated cytotoxicity vs aPBL-mediated; 28.9%±1.5% vs 10.0%±1.1%; p=0.001, Patient 2: 31.6%±2.5% vs 17.0%±2.5%; p=0.002, Patient 3: 30.0%±3.5% vs 14.0%±2.1%; p=0.025). (Ara-C)-mediated cytotoxicity was 7.34%±0.26% in NB4 cells.
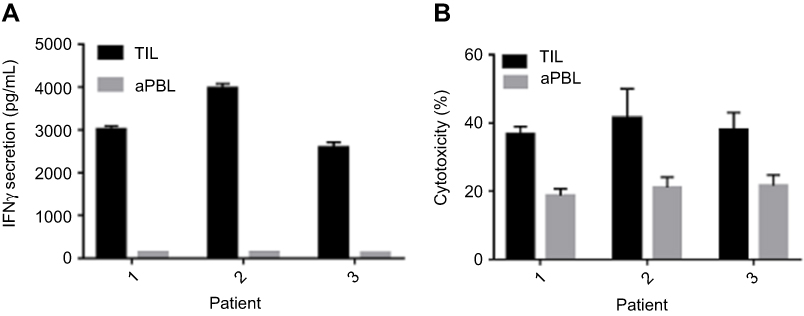 | Figure 4 Comparison of TIL and aPBL on IFN-γ secretion and anti-leukemia reactivity. (A) AML TIL and AML aPBL of patients 1, 2, and 3 were co-cultured with autologous AML cells in triplicate. (B) AML TIL and AML aPBL were co-cultured with K562 cell line in triplicate.Abbreviations: aPBL, activated peripheral blood lymphocytes; AML, acute myeloid leukemia; TIL, tumor-infiltrating lymphocytes. |
Magnetic bead separation of CD137-expressing cells
Immunomagnetic selection of CD137-expressing cells was performed on expanded TIL of Patient 3. The expression of CD137 on TIL before expansion culture was low being 2.66%, but CD137+ cells increased significantly to 6.24% after co-culture with autologous AML cells and was further increased to 42.13% in the CD137+ fraction after the magnetic separation (Figure 5A–C). The immunomagnetic selection enables the enrichment of IFN-γ-producing TIL as evidence of an increase of Elispot counts (Figure S1, pre-selection vs post-selection; 66±3 vs 661±20).
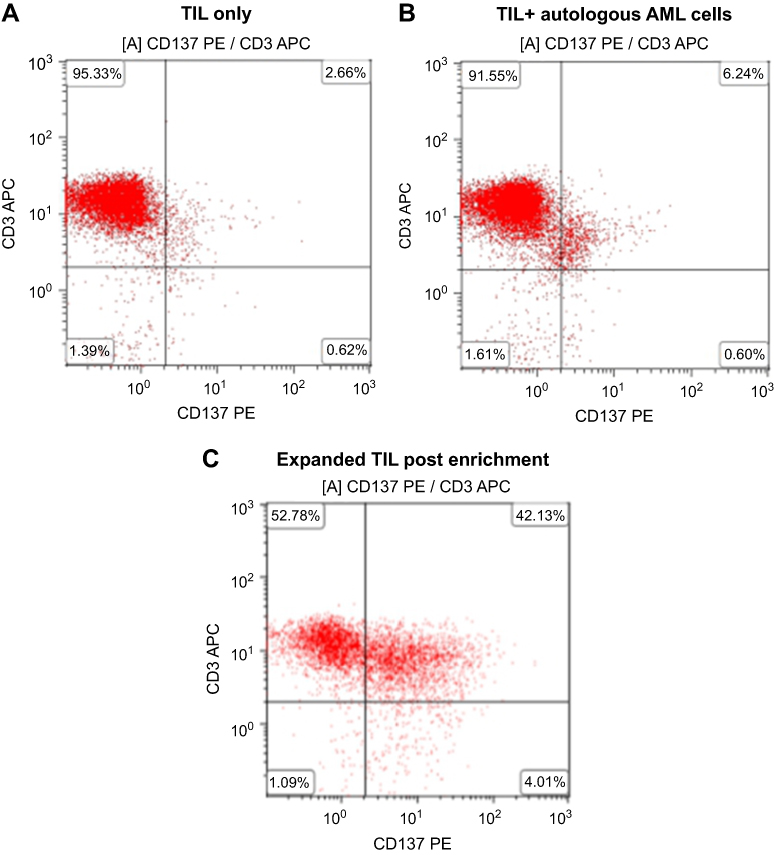 | Figure 5 CD137 separation with magnetic beads enriches TIL from AML Patient 3. (A) TIL. (B) Overnight co-culture of TIL and autologous AML cells. (C) Expanded TIL post-CD137-selection.Abbreviations: AML, acute myeloid leukemia; TIL, tumor-infiltrating lymphocytes. |
 | Figure S1 IFNγ release of CD137 separated TIL compared with unseparated TIL. Abbreviation: TIL, tumor-infiltrating lymphocytes. |
Discussion
The treatment of AML is challenging. Although a majority of AML patients achieve remission after chemotherapy, only a small portion of patients especially those >60 years of age might experience durable responses. Besides, the remission is relatively short. Approximately 20% of AML patients can achieve disease remission at two years. The five-year survival rate of AML patients of <60 years of age is approximately 35%.25 The high rate of relapse after chemotherapy is thought to be related to the intrinsically resistant clonal populations to cytotoxic injury. Allo-HSCT is potentially curative for a subset of patients because of the capacity of allo-reactive lymphocytes to eradicate malignant clonal populations.26 However, the treatment modality is often limited by treatment-related mortality and morbidity.
There is a need for novel therapies for AML including immune-based therapies. To our knowledge, there is little promising immunotherapy available to the treatment of AML. Striking responses have been observed in patients with refractory acute lymphocytic leukemia undergoing immunotherapy with chimeric antigen receptor-T (CAR-T) cell therapy, in which persistent leukemia-specific T cells has led to durable response.27 Unfortunately, identifying a target that is widely expressed on AML cells is challenging. As a result, the application of CAR-T cell therapy in AML patients has proven to be limited. Cellular immunotherapy may be an alternative.
“Neo-epitopes” derived from the tumor are ideal targets. They are unique in tumor cells but not normal tissue. T cells are able to recognize differences between mutated epitopes and non-mutated epitopes because of peptides on the surface of tumor cells presented by MHC molecules. They are “non-self”. They are not subject to central tolerance and may potentially be immunogenic.28 They can elicit T cell response and are often found infiltrating solid tumors and likely play an important role in ACT.
Reports in the literature suggest that ACT using autologous TIL was promising in patients with solid tumors. T cells that are reactive to tumor-specific antigens might mediate tumor regression.29 Twenty percent of metastatic melanoma patients were reported to experience durable complete regression after treatment with autologous TIL in the light of reactivity with autologous tumor cells.30 In the postoperative treatment of non-small cell lung cancer, the three-year survival rate was significantly higher for patients who underwent TIL ACT compared to that of untreated patients.31 In patients with pancreatic adenocarcinomas, infiltration of CD4+ T and CD8+ cells was noted to associate with an improved prognosis and a markedly better five-year survival rate.32 A recent study demonstrated that the infiltration of CD3+ T cells correlated with an improved clinical outcome in patients with epithelial ovarian cancer.33 T cell immune response in patients with solid tumors suggests the potential application of TIL.
Whether AML harbor TIL has been debated for decades, but no direct evidence has so far been reported. In this study, we demonstrate, for the first time, that the presence of AML TIL and they are able to specifically recognize autologous AML cells. The major observation on a small cohort of AML patients is: 1) TIL could be identified in bone marrow of 50% of AML patient cohort as revealed by immunophenotyping and IFN-γ Elispot assay, 2) AML TIL exerted anti-AML reactivity and anti-leukemia cytotoxicity, 3) AML TIL exhibited a higher anti-AML reactivity and anti-leukemia cytotoxicity compared with those of aPBL, 4) AML TIL were expandable, and 5) enrichment of expandable AML-reactive TIL is feasible. More works are needed to explore the potential application of AML TIL in ACT.
The recognition of neo-epitopes is important in the understanding of tumor immunity and host immune defense such as the generation of TIL. For a tumor-specific antigen to be recognized by the human immune system, several conditions must be met: 1) production of mutant peptides by the antigen processing machinery, 2) high affinity of mutant peptides to human leukocyte antigen (HLA), and 3) recognition of mutant peptide-HLA complex by host T cells.34 Studies on solid tumors demonstrated that 0.5–4% of mutations spontaneously trigger T-cell response.18 AML TIL could recognize autologous AML cells as neo-epitopes on AML cells were presented by HLA molecules. Among 10 AML patients whom TIL were not detected, disease-related gene mutations were demonstrated in six patients. Mutant proteins may be very low in quantity or poorly expressed on AML cells. Mutant epitopes may not be properly presented to the HLA molecules. The affinity of the epitopes to the host HLA molecules is very low leading to poor and unstable formation of mutant peptide-HLA complex binding to T cell receptors.
A recent study demonstrated that TIL reactivity represents neo-epitope-driven T-cell responses in solid tumor.35 Three patients with metastatic cervical cancer experienced durable complete regression after treatment with autologous TIL. The reactivity of autologous TIL in terms of IFN-γ release and CD137 upregulation was noted.36 In a study of patient with metastatic triple negative breast cancer, the T cell response against a special mutation was characterized.37 Increases of IFN-γ secretion and CD137 expression on TIL were reported. In this report, we identified anti-leukemia reactivity in AML TIL which could secrete IFN-γ. The expression of CD137 on AML TIL was up-regulated in co-cultures with autologous AML cells. The AML TIL demonstrated a profound anti-leukemia reactivity compared to that of AML aPBL in terms of IFN-γ secretion and cell-mediated cytotoxicity. Our observation attests the findings in solid tumors.
Upon antigen recognition, T-cells evolve significantly with functional and phenotypic changes. The upregulation of co-inhibitory molecule CD279 (PD-1) and the co-stimulatory molecule CD137 (4-1BB) provides the opportunity to use these biomarkers to identify tumor-specific T cells in solid tumors.38 CD137 (4-1BB), a TNFR-family member, is an important regulator of immune responses with specific antigen expression.39 CD279 (PD-1) was used to recognize neo-antigen-reactive T cells, but it was noted that self-antigen-reactive T cells also express CD279 (PD-1).40 A previous study reported that the expression of CD137 (4-1BB), but not CD279 (PD-1), on lymphocytes defines a population of tumor-reactive cells in solid human cancers.30 In this study, the AML TIL have a higher expression of CD137 (4-1BB) and CD279 (PD-1) than those of AML aPBL. The numbers of CD137 (4-BB)-expressing cells were upregulated following co-cultures with autologous AML cells. Data of the study are in accordance with previous reports suggesting that CD137 (4-1BB) may be a potential biomarker for identification and enrichment of AML-reactive TIL. Tumor-reactive TIL numbers are low in solid tumors. The ex-vivo expansion of TIL may be amenable to yield a therapeutic dose of functional TIL for ACT. An enrichment of CD137+ cells further enhances the purity of TIL from IL-7- and IL-15-supplemented expansion cultures. More works are needed to confirm the preliminary findings in this study.
Conclusion
We first demonstrate TIL in 50% of AML patient cohort. AML TIL express CD137 (4-1BB) and secret IFN-γ. They exhibit profound anti-AML reactivity and cytotoxicity. Besides, they are expandable in cultures, which might be translated to ACT for AML in the future.
Ethical approval
The study was conducted subject to the approval of the Institutional Review Board of Harbin Medical University and in accordance with the Declaration of Helsinki. Bone marrow and peripheral blood samples were taken with written informed consent of patients and donors.
Abbreviation list
ACT, Adoptive cell therapy; allo-HSCT, Allogeneic hematopoietic stem cell transplantation; AML, Acute myeloid leukemia; aPBL, Activated peripheral blood lymphocytes; APL, Acute promyelocytic leukemia; BMMC, Bone marrow mononuclear cells; ELISA, Enzyme-linked immunosorbent assay; IL, Interleukin; IFN-γ, Interferon-γ; LDH, Lactate dehydrogenase; PBMC, Peripheral blood mononuclear cells; TIL, Tumor-infiltrating lymphocytes.
Acknowledgments
We thank Xiaoyong Hao and Kent Kam Sze Tsang for critical reading and English editing of the manuscript. This work was supported by Grant JJ2017QN0760 from Natural Science Foundation of Heilongjiang Province.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control. 2008;19(4):379–390. doi:10.1007/s10552-007-9097-2
2. Siegel RL, Miller KD, Jemal A. Cancer statistics 2015. CA Cancer J Clin. 2015;65(1):5–29. doi:10.3322/caac.21294
3. D€Ohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi:10.1056/NEJMra1406184
4. Shah A, Andersson TM, Rachet B, Bjorkholm M, Lambert PC. Survival and cure of acute myeloid leukaemia in England, 1971–2006: a population-based study. Br J Haematol. 2013;162(4):509–516. doi:10.1111/bjh.12425
5. Meyers J, Yu Y, Kaye JA, Davis KL. Medicare fee-for-service enrollees with primary acute myeloid leukemia: an analysis of treatment patterns, survival, and healthcare resource utilization and costs. Appl Health Econ Health Policy. 2013;11(3):275–286. doi:10.1007/s40258-013-0032-2
6. Kantarjian H, O’Brien S. Questions regarding frontline therapy of acute myeloid leukemia. Cancer. 2010;116(21):4896–4901. doi:10.1002/cncr.25281
7. Passweg JR, Baldomero H, Bader P, Bonini C, Cesaro S, Dreger P. Hematopoietic stem cell transplantation in Europe 2014: more than 40 000 transplants annually. Bone Marrow Transplant. 2016;51(6):786–792. doi:10.1038/bmt.2016.20
8. Versluis J, Hazenberg CL, Passweg JR, van Putten WL, Maertens J, Biemond BJ. Post-remission treatment with allogeneic stem cell transplantation in patients aged 60 years and older with acute myeloid leukaemia: a time dependent analysis. Lancet Haematol. 2015;2(10):e427–436. doi:10.1016/S2352-3026(15)00148-9
9. Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: myths,controversies, and unknowns. Blood. 2011;117(8):2307–2318. doi:10.1182/blood-2010-10-265603
10. Duval M, Klein JP, He W, Cahn JY. Cairo: hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol. 2010;28(23):3730–3738. doi:10.1200/JCO.2010.28.8852
11. Lichtenegger FS, Schnorfeil FM, Hiddemann W, Subklewe M. Current strategies in immunotherapy for acute myeloid leukemia. Immunotherapy. 2013;5(1):63–78. doi:10.2217/imt.12.145
12. Lichtenegger FS, Krupka C, Köhnke T, Subklewe M. Immunotherapy for acute myeloid leukemia. Semin Hematol. 2015;52(3):207–214. doi:10.1053/j.seminhematol.2015.03.006
13. Cornelissen JJ, Gratwohl A, Schlenk RF, et al. The European leukemianet AML working party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat Rev Clin Oncol. 2012;9(10):579–590. doi:10.1038/nrclinonc.2012.154
14. Stelljes M, Krug U, Beelen DW, et al. Allogeneic transplantation versus chemotherapy as postremission therapy for acute myeloid leukemia: a prospective matched pairs analysis. J Clin Oncol. 2014;32(4):288–296. doi:10.1200/JCO.2013.54.6911
15. Ramos NR, Mo CC, Karp JE, Hourigan CS. Current approaches in the treatment of relapsed and refractory acute myeloid leukemia. J Clin Med. 2015;4(4):665–695. doi:10.3390/jcm4040665
16. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA
17. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–218. doi:10.1038/nature12213
18. Lu YC, Yao X, Crystal JS, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014;20(13):340–3410. doi:10.1158/1078-0432.CCR-13-3045
19. Robbins PF, Lu YC, El-Gamil M, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–752. doi:10.1038/nm.3161
20. Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi:10.1158/1078-0432.CCR-11-0116
21. Besser MJ, Shapira-Frommer R, Itzhaki O, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. 2013;19(17):4792–4800. doi:10.1158/1078-0432.CCR-13-0380
22. Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi:10.1126/science.1076514
23. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–68. doi:10.1126/science.aaa5139
24. Tran E, Turcotte S, Gros A, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–645. doi:10.1126/science.1251102
25. Kahl C, Krahl R, Becker C, et al. Long-term follow-up of the AML97 study for patients aged 60 years and above with acute myeloid leukaemia: a study of the East German haematology and oncology study group (OSHO). J Cancer Res Clin Oncol. 2016;142(1):305–315. doi:10.1007/s00432-016-2126-3
26. Champlin R. Reduced intensity allogeneic hematopoietic transplantation is an established standard of care for treatment of older patients with acute myeloid leukemia. Best Pract Res Clin Haematol. 2013;26(3):297–300. doi:10.1016/j.beha.2013.10.011
27. Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125(26):4017–4023. doi:10.1182/blood-2014-07-591040
28. Wang QJ, Yu Z, Griffith K, Hanada K, Restifo NP, Yang JC. Identification of T-cell receptors targeting KRAS-mutated human tumors. Cancer Immunol Res. 2016;4(3):204–214. doi:10.1158/2326-6066.CIR-15-0188
29. Cohen CJ, Gartner JJ, Horovitz-Fried M, et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest. 2015;125(10):3981–3991. doi:10.1172/JCI80323
30. Ye Q, Song DG, Poussin M, et al. CD137 accurately identifies and enriches for naturally-occurring tumor-reactive T cells in tumor. Clin Cancer Res. 2014;20(1):44–55. doi:10.1158/1078-0432.CCR-13-3045
31. Ben‑Avi R, Farhi R, Ben-Nun A, et al. Establishment of adoptive cell therapy with tumor infiltrating lymphocytes for non-small cell lung cancer patients. Cancer Immunol Immunother. 2018;67(8):1221–1230. doi:10.1007/s00262-018-2174-4
32. Hall M, Liu H, Malafa M, et al. Expansion of tumor-infiltrating lymphocytes(TIL) from human pancreatic tumors. J Immunother Cancer. 2016;4:61. doi:10.1186/s40425-016-0164-7
33. Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi:10.1056/NEJMoa020177
34. Nielsen JS, Sedgwick CG, Shahid A, et al. Toward personalized lymphoma immunotherapy identification of common driver mutations recognized by patient CD8+ T cells. Clin Cancer Res. 2016;22(9):2226–2236. doi:10.1158/1078-0432.CCR-15-2023
35. Linnemann C, van Buuren MM, Bies L, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015;21(1):81–85. doi:10.1038/nm.3773
36. Stevanović S, Draper LM, Langhan MM, et al. Complete regression of metastatic cervical cancer after treatment with human papillomavirus–targeted tumor-infiltrating T cells. J Clin Oncol. 2015;33(14):1543–1550. doi:10.1200/JCO.2014.58.9093
37. Assadipour Y, Zacharakis N, Crystal JS, et al. Characterization of an immunogenic mutation in a patient with metastatic triple negative breast cancer. Clin Cancer Res. 2017;23(15):4347–4353. doi:10.1158/1078-0432.CCR-16-1423
38. Inozume T, Hanada K, Wang QJ, et al. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J Immunother. 2010;33(9):956–964. doi:10.1097/CJI.0b013e3181fad2b0
39. Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi:10.1146/annurev.immunol.23.021704.115839
40. Gros A, Parkhurst MR, Tran E, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433–438. doi:10.1038/nm.4051
Supplementary materials
Methods
Mutations in the following 127 genes were used in the indicated number of patients: CEBPA,NPM1,FLT3-ITD,FLT3-TKD,TP53,KIT,DNMT3A,IDH1,IDH2,MLL,TET2,RUNX1,KRAS,NRAS,ASXL1,WT1,GATA1,SF3B1,SRSF2,U2AF1,ZRSR2,EZH2,CBL,JAK2,SETBP1,ETV6,MPL,CALR,PTPN11,CSF3R,GATA2,STAG2,JKA3,BRAF,TERC,IKZF1,ABL,SH2B3,NOTCH1,FBXW7,CDKN2A,TPMT,IL7R,JAK1,JAK3,RB1,MYD88,ABCB1,ABCC3,AKT2,AKT3,AMER1,APC,ATM,ATRX,BCL2,BCOR,BCORL1,CACNA1E,CARD11,CBLB,CBLC,CCND1,CD79B,CDA,CREBBP,CRLF2,CSF1R,CTLA4,CUX1,CYP2C19,CYP3A4,DIS3,DNAH9,E2A,EGFR,ERCC1,ERG,FAM46C,GNAS,GSTM1,GSTP1,HRAS,ID3,KDM6A,KMT2C,KMT3A,MAP2K4,MAP3K7,MDM2,MEF2B,MLH1,MTHFR,NF2,NOTCH2,NQO1,NT5C2,NTRK1,NTRK2,PDGFRA,PIGA,PIK3CA,PTEN,RAD21,SMAD4,SMC1A,SMC3,STAT5A,STAT5B,SYK,TRAF3,XRCC1,XO,GATA3,EP300,NF1,ETNK1,ANKRD26,DDX41,DKC1,PRPF8,STAT3,TERT,SRP72,ELA2,HAX1,GFI1,BLM,ATG2B,GSKIP,ATM,BCOR,PIGA.
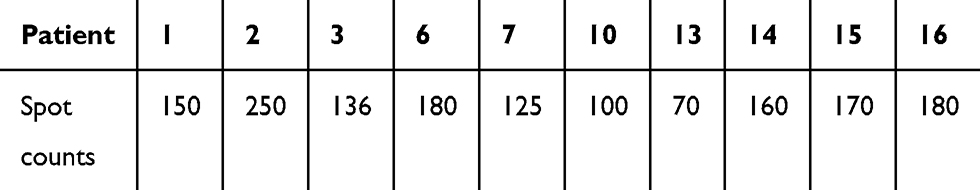 | Table S1 IFN-γ Elispot counts |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.