")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Assessing the potential impact of non-proprietary drug copies on quality of medicine and treatment in patients with relapsing multiple sclerosis: the experience with fingolimod
Authors Correale J , Chiquete E, Milojevic S, Frider N, Bajusz I
Received 17 April 2014
Accepted for publication 5 June 2014
Published 25 June 2014 Volume 2014:8 Pages 859—867
DOI https://doi.org/10.2147/DDDT.S66398
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jorge Correale,1 Erwin Chiquete,2 Snezana Milojevic,3 Nadina Frider,3 Imre Bajusz3
1Raúl Carrea Institute for Neurological Research, Foundation for the Fight against Infant Neurological Illnesses, Buenos Aires, Argentina; 2Department of Neurology and Psychiatry, Salvador Zubirán National Institute of Medical Science and Nutrition, Mexico City, Mexico; 3Novartis Pharma AG, Basel, Switzerland
Background: Fingolimod is a once-daily oral treatment for relapsing multiple sclerosis, the proprietary production processes of which are tightly controlled, owing to its susceptibility to contamination by impurities, including genotoxic impurities. Many markets produce nonproprietary medicines; assessing their efficacy and safety is difficult as regulators may approve nonproprietary drugs without bioequivalence data, genotoxic evaluation, or risk management plans (RMPs). This assessment is especially important for fingolimod given its solubility/bioavailability profile, genotoxicity risk, and low-dose final product (0.5 mg). This paper presents an evaluation of the quality of proprietary and nonproprietary fingolimod variants.
Methods: Proprietary fingolimod was used as a reference substance against which eleven nonproprietary fingolimod copies were assessed. The microparticle size distribution of each compound was assessed by laser light diffraction, and inorganic impurity content by sulfated ash testing. Heavy metals content was quantified using inductively coupled plasma optical emission spectrometry, and levels of unspecified impurities by high-performance liquid chromatography. Solubility was assessed in a range of solvents at different pH values. Key information from the fingolimod RMP is also presented.
Results: Nonproprietary fingolimod variants exhibited properties out of proprietary or internationally accepted specifications, including differences in particle size distribution and levels of impurities such as heavy metals. For microparticle size and heavy metals, all tested fingolimod copies were out-of-specification by several-fold magnitudes. Proprietary fingolimod has a well-defined RMP, highlighting known and potential mid- to long-term safety risks, and risk-minimization and pharmacovigilance procedures.
Conclusion: Nonproprietary fingolimod copies produced by processes less well controlled than or altered from proprietary production processes may reduce product reproducibility and quality, potentially presenting risks to patients. Safety data and risk-minimization strategies for proprietary fingolimod may not apply to the nonproprietary fingolimod copies evaluated here. Market authorization of nonproprietary fingolimod copies should require an appropriate RMP to minimize risks to patients.
Keywords: fingolimod, multiple sclerosis, risk management plan, bioequivalence, nonproprietary medicine
Introduction
Multiple sclerosis (MS) is a chronic, inflammatory neurodegenerative disorder that takes either progressive or relapsing forms. It is associated with significant long-term physical and cognitive disability. There are several treatment options that can delay the progress of this loss of function and modify the course of the disease. Access to high-quality medicines is essential for all patients; substandard-quality medicines have the potential to cause unexpected side effects or to fail to slow disease progression, which are particularly negative effects in chronic diseases such as MS. The relapsing form of MS is treated with different disease-modifying therapies, including beta-interferons, glatiramer acetate, natalizumab, fingolimod, and teriflunomide. Fingolimod, a once-daily oral sphingosine-1-phosphate receptor modulator, was initially approved in 2010 for the treatment of relapsing MS, and its efficacy has been established across all four measures of disease activity (relapse rate, magnetic resonance imaging outcomes, brain volume loss, and disability progression).1 Fingolimod has also shown superior efficacy to an established injectable disease-modifying therapy, intramuscular interferon beta-1a.2
Fingolimod is a small molecule that binds with high affinity to four of the five known sphingosine-1-phosphate receptors. It acts on the peripheral immune system by redirecting lymphocytes to lymphoid tissues, thereby keeping pathogenic lymphocytes out of the central nervous system; it also exerts effects directly within the central nervous system.3 Owing to its specific mechanisms of action, precise controls are required in fingolimod manufacture in order to ensure consistent and predictable treatment outcomes.
The production of fingolimod employs tightly controlled processes and good manufacturing practices to ensure batch-to-batch reproducibility, content uniformity, and product stability, which contribute to a manageable benefit/risk profile. This is important because fingolimod is incompatible with many excipients and the dose in the final product is low (0.5 mg). The potential for the formation of impurities is of particular concern because a chemical used in the initial phase of the manufacturing process has genotoxic properties. The proprietary manufacturer has designed the production process, controls, and quality testing to ensure that genotoxic impurity levels in fingolimod are substantially below the threshold of toxicological concern established by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).4 For relapsing MS, consistency of the final product is especially important, owing to the unpredictability of relapse occurrence, lack of validated biomarkers to determine fingolimod dose insufficiency, and the irreversible brain volume loss and disability progression associated with the disease.
In some localities, such as China, South Asia, Eastern Europe, Africa, and Latin America, nonproprietary versions of drugs are widely manufactured; in addition, regulatory requirements, such as the need for bioequivalence testing or product quality evaluation, including impurity and genotoxicity studies, are often less stringent than in regions such as the USA or European Union. In certain countries, generic versions of proprietary drugs and copies are considered to be in the same category, and the latter are approved by some health authorities. The crucial difference, however, is that the term “generic drug” refers to variants of proprietary drugs that undergo bioequivalence and quality testing, which is not necessarily the case for those termed as “copies”. Although copies contain the same active ingredient, concentration, pharmaceutical form, and dosage as the proprietary and generic drugs, they do not necessarily meet quality specifications set by the relevant regulatory bodies, which may potentially impact their effectiveness and safety profile. In addition, some medications (including fingolimod) are sensitive to varying levels of temperature and humidity, and so need very specific storage and transportation conditions. Therefore, a nonproprietary drug may not be bioequivalent to the proprietary version, or of equal quality. As mentioned, the availability of high-quality medicines is essential for patients. Although nonproprietary therapies can appear attractive owing to their low acquisition price, this may be offset by the potential risks, such as lack of treatment efficacy or unexpected safety issues from impurities or altered bioavailability, arising from poor quality control in excipient selection, manufacturing processes, and packaging.
This study presents a quality evaluation of proprietary fingolimod and several nonproprietary fingolimod copies for particle size distributions, and the content of inorganic materials and heavy metals. In addition, the need for bioequivalence testing as a consequence of the Biopharmaceuticals Classification System (BCS) class 2 behavior of fingolimod and its mid- to long-term safety-monitoring program are addressed in the context of the potential implications for nonproprietary fingolimod.
Methods
Drugs tested
Fingolimod manufactured under the patent holder’s proprietary method was used as the reference substance for each test. Eleven nonproprietary fingolimod drug variants were obtained from different manufacturers in the People’s Republic of China via the public trade process. All tests were performed by Novartis International Pharmaceutical Branch Ireland.
Microparticle size testing
Several drops of a dispersing liquid (1% Span 80 [Sigma-Aldrich Co, St Louis, MO, USA] in n-hexadecane [EMD Millipore, Billerica, MA, USA]) were added to 0.1 g of each test substance in a cuvette, and then mixed with a vortex to form a smooth and homogeneous paste. Further dispersion liquid was added to dilute the paste to a final volume of 3–6 mL before mixing again. Following the preparation of the test dispersion, the cumulative volume distribution was determined using a Sympatec HELOS laser diffraction sensor (Sympatec GmBH, Clausthal-Zellerfeld, Germany) with focal length of 50 mm, optical concentration of at least 5%, and measurement duration of 20 seconds. The particle size for each test substance was determined at the undersize values of 10%, 50%, and 90% (X10, X50, and X90, respectively) from the cumulative volume distribution. Owing to the limited availability of some nonproprietary fingolimod variants, only ten were tested.
Sulfated ash testing
Sulfated ash testing was used to measure the amount of inorganic impurities present in proprietary fingolimod and each nonproprietary test substance. Approximately 1 g ± 1 mg of each test substance was weighed into a platinum crucible that had been ignited to a constant weight. The test substance was then saturated with concentrated sulfuric acid, the crucible carefully heated until the test substance carbonized, then slow heating continued until production of most of the sulfurous fumes ceased. The crucible was then ignited, allowed to cool, and the residue weighed. The procedure was repeated until the residue attained constant mass or until the mass was lower than the threshold for out-of-specification levels of inorganic impurities.
Heavy metals testing
Heavy metals testing was conducted to assess the presence of nickel (Ni), lead (Pb), palladium (Pd), iron (Fe), copper (Cu), and zinc (Zn) using inductively coupled plasma optical emission spectrometry (Varian Vista-PRO; Agilent Technologies Inc., Santa Clara, CA, USA). The cut-off threshold was not more than 10 parts per million (ppm) for the total of all six heavy metals. Of the eleven nonproprietary fingolimod drugs, only three were tested for the presence of heavy metals owing to limited availability.
The test substance (0.2 g) was weighed in a digestion tube, and 4 mL of nitric acid plus 0.2 mL of perchloric acid were added. Digestion was performed using closed-vessel decomposition at 250°C–260°C for at least 45 minutes in a high-pressure autoclave (UltraCLAVE; Milestone Inc., Shelton, CT, USA). Once complete, the digestion apparatus was allowed to cool, the digestion tubes were opened, and the contents were made up to 10.0 mL with water after the addition of 0.1 mL of the stock internal standard solution (the concentration of each element in the standard stock solution was 0.1 μg/mL). Readings were then taken using a Varian Vista-PRO spectrometer (Agilent Technologies Inc.). The presence of individual metals in the test samples was determined from a peak at the relevant wavelength for each metal; the concentration of each metal analyte (ppm) was calculated by multiplying the analyte concentration in the test solution (μg/mL), the volume of test solution (mL), and the dilution factor, then dividing that value by the test sample mass (g). If the total for all metals assessed in a test substance exceeded 10 ppm, the test substance was considered to be out-of-specification based on proprietary specifications.
Unspecified impurities
Testing for the presence of nonspecific impurities was conducted using high-performance liquid chromatography with ultraviolet light detection. Two reference solutions were prepared: for the first, 30 mg±0.01 mg fingolimod hydrochloride (HCl) was weighed into a 50 mL volumetric flask, then dissolved in and diluted to volume with solvent (mobile phase A [water with 0.1% phosphoric acid] and mobile phase B [acetonitrile] 1:1 v/v). For the second reference solution, 5.0 mL of the first reference solution was diluted to 50 mL with solvent, then 2.5 mL of the resulting solution was diluted to 50.0 mL with solvent (corresponding to a 0.5% solution). Peak areas for specified impurities as well as for any unspecified impurity in the chromatogram of the test solution and the peak area of fingolimod HCl in the second reference solution were then determined, and the percentage content for each of the defined peaks and any unspecified impurity was calculated.
Solubility
The solubility of fingolimod was quantified using a range of solvents (Table 1).
 | Table 1 Solvents used for fingolimod solubility testing |
Risk monitoring and minimization
Fingolimod has a well-defined risk management plan (RMP). In addition to background information on MS pathophysiology, epidemiology, and treatment, the program provides a database of key safety findings from preclinical and clinical studies as well as from real-world experience with fingolimod. The RMP describes potential and identified safety risks associated with fingolimod that result from adverse effects or interaction with other pharmaceutical agents; key patient populations for which data are lacking are also identified. The information is updated regularly in order to incorporate new safety information and risk-minimization strategies as they become available.
One particular area of importance in this context is the initial pharmacodynamic effect of fingolimod on heart rate. Fingolimod induces a transient dose-dependent reduction in heart rate. With the proprietary fingolimod 0.5 mg formulation, this effect is well described and mostly benign, with less than 1% of patients reporting symptomatic bradycardia. However, nonproprietary formulations could potentially show different results and present a risk to the patient.
Results
Microparticle size
All of the nonproprietary fingolimod copies tested (ten samples) were found to be out-of-specification relative to fingolimod, with out-of-specification magnitudes observed as high as five-fold in excess of proprietary fingolimod (Table 2).
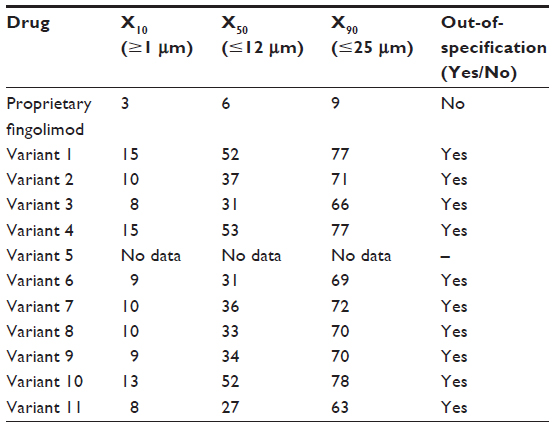 | Table 2 Microparticle size distributions for fingolimod and nonproprietary fingolimod copies |
Sulfated ash, heavy metals, and unspecified impurities
Testing for impurities revealed that nonproprietary fingolimod copies contained levels of impurities that ranged from three-fold to 20-fold higher than those present in fingolimod produced using the proprietary manufacturing procedures. Depending on the test used, 46%–100% of fingolimod copies exceeded the acceptable impurity level threshold. The findings are summarized in Table 3, and presented in greater detail in Figures 1–3.
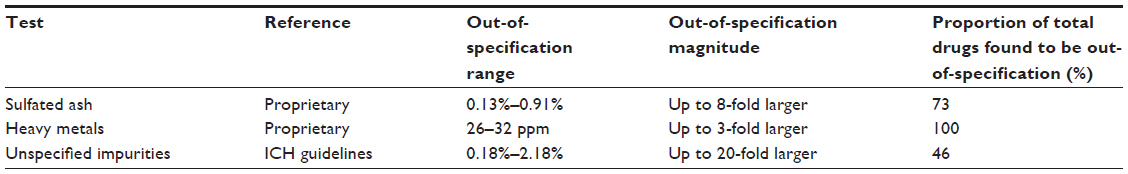 | Table 3 Summary of impurity testing results for nonproprietary fingolimod variants |
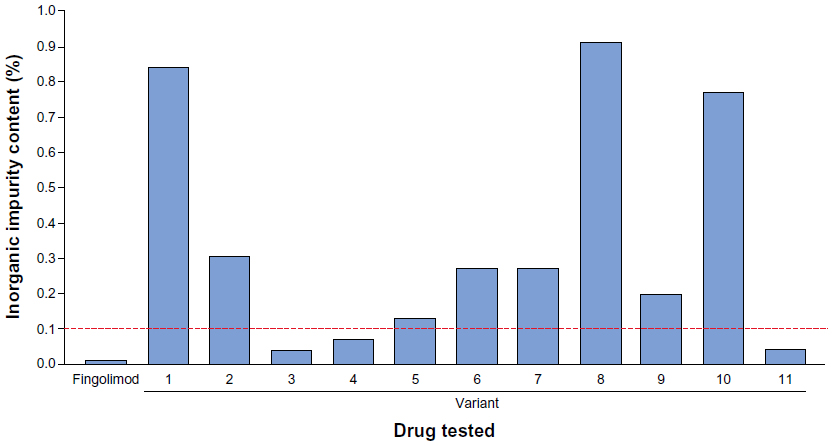 | Figure 1 Sulfated ash test results for fingolimod and eleven nonproprietary fingolimod variants. |
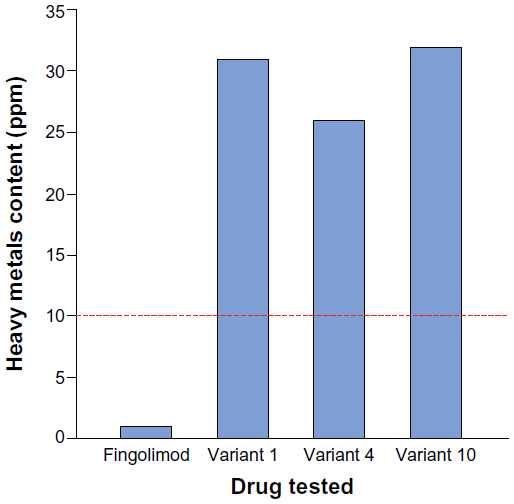 | Figure 2 Heavy metals test results for fingolimod and three nonproprietary fingolimod copies. |
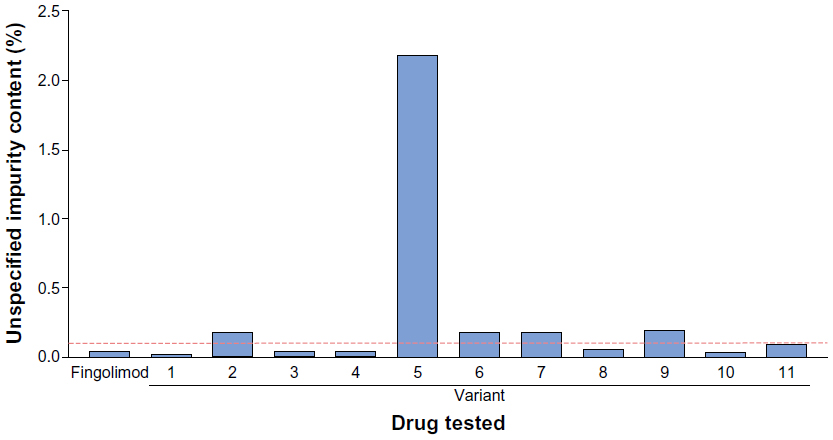 | Figure 3 Unspecified impurities in fingolimod and nonproprietary fingolimod as detected by high-performance liquid chromatography. |
Sulfated ash
Sulfated ash testing showed eight of eleven nonproprietary fingolimod variants to be out-of-specification, with the variant that was furthest out-of-specification yielding eight-fold higher levels of inorganic impurities than the out-of-specification threshold (Figure 1).
Heavy metals
Testing for heavy metals was carried out on only four drugs (proprietary fingolimod and three nonproprietary fingolimod variants) owing to limited availability of nonproprietary copies. All three fingolimod variants tested were found to have a heavy metals content over 10 ppm, meaning that they were out of specification. The largest magnitude of difference between a nonproprietary fingolimod variant and the threshold was a greater than three-fold higher heavy metals content (for variants 1 and 10) (Figure 2).
Unspecified impurities
High-performance liquid chromatography testing for unspecified impurities revealed that five of eleven fingolimod copies exceeded the out-of-specification threshold. The furthest out-of-specification copy was variant 5, which contained impurity levels approximately 20-fold higher than the acceptable threshold (Figure 3).
Solubility
Proprietary fingolimod was found to have an extremely low solubility in neutral pH media (<0.01 mg/mL in pH 6.8 phosphate buffer [at both 25°C and 37°C]), although it was freely soluble in highly acidic conditions (pH 1.0 0.1 N HCl) and in deionized water.
Risk monitoring of fingolimod
The RMP examines identified and potential safety issues and their management, and is updated regularly based on new safety data derived from postmarketing and ongoing studies. Gaps in current knowledge are identified, such as patient subgroups that are less well studied. Risks to health from potential or identified drug-related adverse effects and the prevalence and severity of such effects are addressed in detail. Potential metabolic pathways and medicinal products that fingolimod may interact with are also discussed. Information on risk-minimization procedures, the general pharmacovigilance strategy, and ongoing and proposed safety studies is presented. The fingolimod RMP provides significant benefit to patients and regulators by providing clear, detailed information on various aspects of the use of fingolimod. The RMP has also enabled incorporation of post-approval safety data; these data are collected from a larger number of patients than are included in clinical trials and also have the advantage of relating to the real-world setting. Furthermore, the RMPs are a requirement of regulatory authorities in certain localities, and they can prove useful in supporting changes to the marketing authorization that may be needed in the future.
Unfortunately, in many countries, regulatory agencies do not have a comprehensive system of pharmacovigilance, and the task of monitoring the safety and efficacy of products is carried out by the holders of market authorization.
Discussion
The technical development of proprietary fingolimod required a precise analytical approach during research and development, as well as adherence to good manufacturing practices to ensure batch-to-batch consistency, blend and content uniformity, and quality and stability of the final product. Storage and transportation in the post-manufacture phase are critical to maintaining these characteristics, owing to the sensitivity of fingolimod to changes in temperature and humidity. Since the dose strength of fingolimod is low (0.5 mg), qualifying as a specialized pharmaceutical dose form according to some health authority definitions,5 blend and content uniformity of the product are critical parameters for achieving the controlled efficacy and safety profile. These considerations are particularly important in the case of therapies for diseases that are classed as a high sanitary risk, such as MS. Sanitary risk is defined as the appearance or possibility of disease complications that may be life-threatening or that endanger the physical or mental integrity of the patient, and/or produce serious adverse reactions.
This study has evaluated nonproprietary fingolimod copies manufactured by eleven different producers in the People’s Republic of China compared with proprietary fingolimod for parameters anticipated to be of critical importance for product quality. The key findings are that the nonproprietary fingolimod copies were all out-of-specification, often by a substantial degree, for at least one quality criterion: microparticle size, content of heavy metals, unspecified impurities, or inorganic materials. These substandard drugs could potentially impact on treatment efficacy and safety via alterations in bioavailability or because of unexpected toxicities.
Differences between proprietary and nonproprietary drugs can arise from a variety of sources, such as manufacturing procedures, changes in the composition of the active pharmaceutical ingredient (API), packaging, degradation of the product (eg, due to temperature changes), inadequate quality controls, and deviation from good manufacturing practices. Particle size changes could impact on product solubility (smaller particles have a higher surface-area-to-volume ratio, and dissolve more quickly) and absorption, hence affecting drug bioavailability.6–9 This potentially affects the efficacy and safety profile, which may lead to treatment below the expected therapeutic level (in cases of reduced bioavailability) and/or increased risk of adverse effects for the patient (in cases of increased bioavailability). The latter may include new signals not reported for proprietary fingolimod, or an increased incidence of known adverse effects that have a low incidence with the 0.5 mg proprietary formulation but that are known to have a dose-dependent effect. The larger particles found in variants may also be more likely to be affected by diet. It is established that the pharmacodynamics of proprietary fingolimod are unaffected by food intake,10,11 which is thought to be in part a function of its small particle size.11 Therefore, the variant forms of fingolimod tested in this study may display different properties from the proprietary form in the presence or absence of food, a phenomenon that has been documented for other drugs.12–14
This study also assessed the presence of heavy metals, inorganic materials, and unspecified impurities. Some metal ions, such as zinc and copper, are essential for normal physiological functioning;15,16 however, excess intake or the ingestion of nonessential metals such as cadmium, lead, and mercury may pose risks to health. Elevated copper, zinc, and iron levels are associated with neurological disorders such as Alzheimer’s and Parkinson’s diseases,17–19 while lead exposure has been implicated in reproductive problems,20 impaired neurological functioning and development,21 and hypertension.22 Manufacturing vessels, packaging, and storage or production conditions such as ultraviolet light or heat can all cause or exacerbate the leaching of metal ions into the pharmaceutical product.23,24 In this study, all of the nonproprietary fingolimod variants tested were above the proprietary threshold of 10 ppm for heavy metals, with two variants showing values more than three times this value.
As with heavy metals, other inorganic materials such as solvents, reagents, ligands, catalysts, metals, and salts can enter pharmaceutical products via manufacturing processes, vessels, or packaging. Eight of eleven (73%) nonproprietary fingolimod copies tested exceeded the out-of-specification threshold for inorganic materials, with the highest out-of-specification value found to be eight times higher than the accepted proprietary threshold. Similarly, five of eleven (45%) fingolimod copies exceeded the ICH threshold for unspecified impurities. The presence of impurities that are out-of-specification could potentially result in unexpected side effects, either from the impurity itself or via an impact on the API, which could affect therapeutic efficacy or introduce unexpected toxicities.
In addition to the impurities tested in this study, proprietary fingolimod genotoxic impurity levels are substantially below the threshold of toxicological concern established by the ICH.4 To that effect, it is important to perform a comprehensive risk assessment, using the most precise and up-to-date analytical techniques, to minimize the risk of product contamination by impurities, in particular potentially genotoxic impurities, that may arise from starting materials, process intermediates, related impurities, and reagents.
Another essential attribute of fingolimod is stability. A comprehensive range of standard excipient-testing studies was employed in the development process of fingolimod; several excipients tested led to the appearance of degradation products. Degradation products potentially decrease the amount of drug available in each formulation, which may have an impact on treatment efficacy. Appropriate excipients are also critical for manufacturing robustness and reproducibility, and the product’s performance. It should therefore be a standard part of the regulatory approval process to undertake 2 years of real-time stability studies of nonproprietary fingolimod to determine product shelf-life.
Profiling of fingolimod in the context of the BCS has shown that fingolimod has atypical biopharmaceutical behavior. It has an extremely low solubility in neutral pH media (less than 0.01 mg/mL at pH 6.8 at 25°C and 37°C), although fingolimod permeability could not be measured owing to its intrinsic characteristics; however, its bioavailability is relatively high (>90%).25 This could suggest BCS class 2 behavior (ie, low solubility, high permeability).26 This atypical profile re-emphasizes the necessity for health authorities to require bioequivalence testing before approval of nonproprietary fingolimod variants. Pharmacokinetic/pharmacodynamic studies have also demonstrated that proprietary fingolimod displays a slow absorption rate, with a broad absorption phase and a maximum concentration generally reached at about 16 hours post-administration.10,25 Orally administered fingolimod phosphate results in a sharp increase in concentration, with maximum concentration reached after approximately 6–8 hours; terminal half-lives for both forms of fingolimod are about 6–9 days.11 These characteristics again support the necessity for bioequivalence testing in order to obtain approval for any nonproprietary variants of fingolimod. According to the World Health Organization, in vivo bioequivalence testing is essential for drugs whose API is highly sensitive (eg, because of agents used in manufacturing or impurities), or that are at high risk of differences in therapeutic efficacy resulting from variation in bioavailability from the originator drug.27 Such testing is required in the USA and European Union for approval of generic medicines; however, nonproprietary copy medicines are made available in other markets without conducting such studies.
In addition, it is unclear whether an RMP is available for any nonproprietary fingolimod copies. In some regions, RMPs are submitted as part of the application for marketing authorization of a drug or when significant changes are made to the marketing authorization of an approved drug.28,29 RMPs aim to provide a proactive means of dealing with drug-related safety issues, as opposed to merely reactively collecting data.29 As is standard for drugs produced by the proprietor, fingolimod has an extensive and well-developed RMP that is updated regularly to incorporate new data. As discussed, the significant microparticle size distribution differences between fingolimod and nonproprietary fingolimod copies, and the high levels and wide range of impurities present in the latter, suggest that strategies and information outlined in the fingolimod RMP may not be directly applicable to medicines containing nonproprietary fingolimod. The overall effect of this is that treatment disruptions or discontinuations become more likely with the use of nonproprietary fingolimod copies, resulting in the loss of expected treatment effects; this could potentially result in failure to slow disability, or cause additional or more extreme side effects.
In summary, substandard drug copies are an important public health issue, with both clinical and economic implications. Patients may be placed at risk if they take nonproprietary fingolimod variants that fail to meet proprietary and internationally accepted specifications for quality parameters, such as impurities or particle-size distributions, or that may potentially employ poorly controlled or altered manufacturing processes. In addition, the solubility, bioavailability, and pharmacokinetic/pharmacodynamic profile of the proprietary fingolimod API make it important that producers of nonproprietary fingolimod establish bioequivalence and efficacy of their APIs. This, and implementation of RMPs, should be essential to obtain approval from health authorities across the globe, because it cannot be assumed that data for the originator drug will be applicable to nonproprietary versions.
Acknowledgments
Novartis Pharmaceuticals Corporation funded the study and the editorial support for the preparation of this manuscript, which was provided by Robert M Gillies, PhD, from Oxford PharmaGenesis™ Ltd. The authors would like to thank Guido Jordine and Alfons Roth for their valuable contributions.
Disclosure
Dr Jorge Correale is a board member of Merck Serono Argentina, Merck Serono LATAM, and Novartis Argentina. He has received reimbursement for developing educational presentations for Merck Serono Argentina, Merck Serono LATAM, Biogen Idec Argentina, Teva Tuteur Argentina, and Novartis LATAM, as well as professional travel/accommodation grants.
Dr Erwin Chiquete has received research grants from Sanofi and Ferrer Grupo, has served as research adviser for Sanofi, Novartis, and Genzyme, and has received speaker honoraria from Novartis Mexico, Genzyme, and Ferrer Grupo.
Dr Snezana Milojevic, Dr Nadina Frider, and Dr Imre Bajusz are employees and stockholders of Novartis Pharmaceuticals Corporation.
The authors report no other conflicts of interest in this work.
References
Kappos L, Radue EW, O’Connor P, et al; FREEDOMS Study Group. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. | |
Cohen JA, Barkhof F, Comi G, et al; TRANSFORMS Study Group. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. | |
Groves A, Kihara Y, Chun J. Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J Neurol Sci. 2013;328(1–2):9–18. | |
European Medicines Agency. ICH Guideline M7 on Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk. London: European Medicines Agency; 2013. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/02/WC500139217.pdf. Accessed June 6, 2014. | |
European Medicines Agency. Annex II to note for guidance on process validation CHMP/QWP/848/99 and EMEA/CVMP/598/99: Non-standard processes. London: European Medicines Agency; 2004. Available from: https://www.tga.gov.au/pdf/euguide/qwp205403en.pdf. Accessed June 6, 2014. | |
Jounela AJ, Pentikäinen PJ, Sothmann A. Effect of particle size on the bioavailability of digoxin. Eur J Clin Pharmacol. 1975;8(5):365–370. | |
Desai MP, Labhasetwar V, Amidon GL, Levy RJ. Gastrointestinal uptake of biodegradable microparticles: effect of particle size. Pharm Res. 1996;13(12):1838–1845. | |
Chaumeil JC. Micronization: a method of improving the bioavailability of poorly soluble drugs. Methods Find Exp Clin Pharmacol. 1998;20(3):211–215. | |
Johnson KC, Swindell AC. Guidance in the setting of drug particle size specifications to minimize variability in absorption. Pharm Res. 1996;13(12):1795–1798. | |
Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Single-dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57(5):586–591. | |
David OJ, Kovarik JM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clin Pharmacokinet. 2012;51(1):15–28. | |
Ouellet D, Grossmann KF, Limentani G, et al. Effects of particle size, food, and capsule shell composition on the oral bioavailability of dabrafenib, a BRAF inhibitor, in patients with BRAF mutation-positive tumors. J Pharm Sci. 2013;102(9):3100–3109. | |
Aungst BJ, Nguyen NH, Taylor NJ, Bindra DS. Formulation and food effects on the oral absorption of a poorly water soluble, highly permeable antiretroviral agent. J Pharm Sci. 2002;91(6):1390–1395. | |
Wu Y, Loper A, Landis E, et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: a Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int J Pharm. 2004;285(1–2):135–146. | |
Uauy R, Olivares M, Gonzalez M. Essentiality of copper in humans. Am J Clin Nutr. 1998;67(Suppl 5):952S–959S. | |
Shankar AH, Prasad AS. Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr. 1998; 68(Suppl 2):447S–463S. | |
Desai V, Kaler SG. Role of copper in human neurological disorders. Am J Clin Nutr. 2008;88(3):855S–858S. | |
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci. 1998;158(1):47–52. | |
Youdim MB, Ben-Shachar D, Riederer P. Iron in brain function and dysfunction with emphasis on Parkinson’s disease. Eur Neurol. 1991; 31 Suppl 1:34–40. | |
Apostoli P, Kiss P, Porru S, Bonde JP, Vanhoorne M. Male reproductive toxicity of lead in animals and humans. ASCLEPIOS Study Group. Occup Environ Med. 1998;55(6):364–374. | |
Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN. The long-term effects of exposure to low doses of lead in childhood. An 11-year follow-up report. N Engl J Med. 1990;322(2):83–88. | |
Navas-Acien A, Guallar E, Silbergeld EK, Rothenberg SJ. Lead exposure and cardiovascular disease – a systematic review. Environ Health Perspect. 2007;115(3):472–482. | |
Bohrer D, do Nascimento PC, Binotto R, Becker E. Influence of the glass packing on the contamination of pharmaceutical products by aluminium. Part III: Interaction container-chemicals during the heating for sterilisation. J Trace Elem Med Biol. 2003;17(2):107–115. | |
Fliszar KA, Walker D, Allain L. Profiling of metal ions leached from pharmaceutical packaging materials. PDA J Pharm Sci Technol. 2006;60(6):337–342. | |
Kovarik JM, Hartmann S, Bartlett M, et al. Oral-intravenous crossover study of fingolimod pharmacokinetics, lymphocyte responses and cardiac effects. Biopharm Drug Dispos. 2007;28(2):97–104. | |
Food and Drug Administration. Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System. Silver Spring: Food and Drug Administration; 2000. Available from: http://www.gmp-compliance.org/guidemgr/files/3618FNL.PDF. Accessed June 6, 2014. | |
World Health Organization. Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. WHO Technical Report Series. Geneva; World Health Organization; 2006:347–390. Available from: http://apps.who.int/medicinedocs/documents/s19639en/s19639en.pdf. Accessed June 6, 2014. | |
European Medicines Agency. Guideline on Good Pharmacovigilance Practices (GVP): Module V – Risk Management Systems. London: European Medicines Agency; 2012. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500129134.pdf. Accessed June 6, 2014. | |
Lis Y, Roberts MH, Kamble S, J Guo J, Raisch DW. Comparisons of Food and Drug Administration and European Medicines Agency risk management implementation for recent pharmaceutical approvals: report of the International Society for Pharmacoeconomics and outcomes research risk benefit management working group. Value Health. 2012;15(8):1108–1118. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.