Back to Journals » Journal of Multidisciplinary Healthcare » Volume 14
Assessing Children with Poor Coordination Can Be Tricky – A Review on Ataxia and Ataxia Mimickers and a Study of Three Children with Severe Epilepsy
Received 15 September 2021
Accepted for publication 16 November 2021
Published 30 November 2021 Volume 2021:14 Pages 3331—3341
DOI https://doi.org/10.2147/JMDH.S251860
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Sydney Martin,1 Michael S Salman2
1Department of Family Medicine, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, Manitoba, Canada; 2Section of Pediatric Neurology, Department of Pediatrics and Child Health, Max Rady College of Medicine, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, Manitoba, Canada
Correspondence: Michael S Salman
Section of Pediatric Neurology, Children’s Hospital, AE 308, 820 Sherbrook Street, Winnipeg, Manitoba, R3A 1R9, Canada
Tel +1204 787 2414
Fax +1204 787 1922
Email [email protected]
Abstract: While ataxia is a relatively common presenting feature in pediatric patients, it represents only one possible cause of uncoordinated movements. Other possible causes of uncoordinated movements include ingestion of toxic substances, musculoskeletal diseases, psychogenic disorders, extrapyramidal movement disorders, peripheral neuropathies, spasticity from any cause, and epilepsy. Therefore, primary health care providers must recognize and exclude other etiologies of uncoordinated movements before attaching the label “ataxia” to any patient presenting with poor coordination. Once the presence of ataxia is confirmed, the cause should be investigated. As ataxia may be vestibular, sensory, or cerebellar in origin, medical practitioners must evaluate the diverse symptoms and signs to effectively differentiate the various types of ataxia. Three case studies are presented to illustrate the complexity associated with the assessment of ataxia. Each case will discuss a pediatric patient who displays cerebellar ataxia as a concurrent feature of a gene-specific developmental and epileptic encephalopathy. These cases will provide an example of how ataxia may be differentiated from other causes of uncoordinated movements related to epilepsy and anti-seizure medications, namely: nonconvulsive seizures, postictal state, and medication side effects or toxicity. The assessment of poor balance can be challenging at times; however, with knowledge of the differential diagnosis of poor balance, medical practitioners will be able to confidently determine the presence of true ataxia from various ataxia mimickers, thereby allowing for timely and accurate diagnosis, and appropriate management.
Keywords: incoordination, ataxia, children, developmental and epileptic encephalopathy
Introduction
Poor coordination is a relatively common presenting symptom and is, therefore, one with which general practitioners should be familiar. Since it carries a broad differential diagnosis, health care providers must differentiate the various possible etiologies to determine management, prognosis, and whether a referral for further investigations is necessary.1
The differential diagnosis for poor coordination includes extrapyramidal movement disorders, ingestion of toxic substances, medication side effects, epilepsy, muscle weakness, spasticity, musculoskeletal disease, psychogenic disorders, and ataxia.1 While these various causes will be outlined in this paper, a specific emphasis will be placed on further understanding and recognizing ataxia. Poor coordination is often thought to be synonymous with ataxia; however, pure ataxia is only one cause of uncoordinated movements. Therefore, mimickers of ataxia must be excluded during the assessment.1 Further, once ataxia is confirmed, the specific system involved must be determined, for example, whether the ataxia is vestibular, sensory, or cerebellar in origin.2 This paper will focus mainly on the presentation of cerebellar ataxia, but both vestibular and sensory ataxia will be briefly discussed. Non-motor manifestations of cerebellar dysfunction will not be addressed in this review.
Developmental and epileptic encephalopathies (DEE) represent a group of neurodevelopmental disorders in which epileptic activity adversely impacts brain function, contributing to cognitive, behavioural, and motor deficits.3–5 Ataxia may be a concurrent feature of DEE.3 There are several other possible causes of uncoordinated movements in patients with DEE, such as nonconvulsive seizures, postictal state, and medication side effects or toxicity.1,6–10 These potential causes must be ruled out first before considering ataxia as a concurrent feature of these disorders.
The purpose of this paper is to illustrate the complexities encountered when assessing ataxia. First, this paper will highlight the differential diagnosis of uncoordinated movements with a specific emphasis on cerebellar ataxia. Then, three case studies will be presented to demonstrate further the intricacies associated with assessing ataxia. Specifically, the case studies will highlight how ataxia may be distinguished from other possible causes of poor coordination related to epilepsy and antiseizure medications. The case studies will discuss three patients who have been diagnosed with various gene-specific DEE. All three patients have ataxia as a concurrent clinical feature.
Differential Diagnosis of Poor Coordination
Extrapyramidal Movement Disorders
Sydenham’s chorea is a neurological disorder most commonly seen as a manifestation of rheumatic fever.11 The disorder is characterized by involuntary choreiform movements, hypotonia, muscle weakness, and gait disturbance. These movements typically worsen with stress and resolve with sleep. In this disorder, antibodies against group A streptococcus cross-react with neurons in the basal ganglia, causing an increased release of dopamine, and as a result, uncontrollable movements.11 These involuntary movements cause poor balance in some patients and may be easily mistaken for ataxia. An important clue in differentiating these movements from ataxia is that they are typically observed when the patient is resting and they become florid during action. Some patients may even become non-ambulatory.
Ingestion of Toxic Substances
The ingestion of alcohol or drugs, exposure to heavy metals, and toxic doses of medications may also cause poor balance.7,8 Several classes of drugs that may cause incoordination include benzodiazepines, antineoplastic, and antiseizure medications. In general, uncoordinated movements tend to occur within days to weeks of introducing a new drug or increasing the dose; however, the development of poor coordination with chronic use has also been reported.8 It is imperative to take a thorough history of any exposure to toxic substances or illicit drugs, as well as a medication history, including any over-the-counter medications. There are several signs that, if present, suggest toxic ingestion including altered mental status and confusion, reduced level or loss of consciousness, seizures, vomiting, and abnormal pupillary size and reactivity.7,8
Epilepsy
Poor coordination may present as a result of nonconvulsive seizure activity, otherwise known as pseudoataxia or epileptic ataxia.7 Acute, fluctuating and more prolonged imbalance may also be a prominent feature in nonconvulsive status epilepticus.11 Such presentation is not true ataxia; instead, decreased level of awareness, myoclonic jerks, and atonic seizures interrupt smooth movements of the limbs, thereby producing pseudoataxia. Often, the patient will appear inattentive or confused.10
In addition to pseudoataxia, uncoordinated movements may occur as a manifestation of the postictal state.6,8 The postictal state occurs immediately after seizure termination and ends with the patient’s return to baseline.8 This state is characterized by sensory, cognitive, and motor deficits, including altered level of awareness, confusion, autonomic dysregulation, and postictal weakness. The associated confusion or weakness seen in the postictal state may be mistaken for ataxia.6,8 Therefore, uncoordinated movements as a manifestation of the postictal state must be considered in the differential diagnosis.
Furthermore, antiepileptic medications may also cause uncoordinated movements, either as a general side effect or as a result of toxic levels.6,7 Therefore, in patients with epilepsy, healthcare providers must also consider the side effects or toxicity associated with antiseizure medications.
Ataxia may also be a concurrent feature in children with DEE and progressive myoclonic epilepsy.3,12 Obtaining a thorough history from a witness is helpful in clarifying why a child with epilepsy may be poorly coordinated.
Weakness, Spasticity, and Clumsiness
Muscle weakness may occur due to lesions in the cerebral cortex, brainstem, spinal cord, peripheral nerves, neuromuscular junction, and muscle.13 Muscle weakness can be easily assessed on clinical exam by looking for decreased resistance to active movement, pronator drift, or asymmetries in arm or leg use.14 Motor weakness may present as poor coordination and can, therefore, be mistaken for ataxia. Spasticity, in which muscles are abnormally stiff with increased resistance to passive stretch from damage to upper motor neurons that leads to the inhibition of fluid movement, may cause poor coordination.1 Looking for additional upper motor signs, such as hyperactive reflexes, Babinski’s sign, and clonus, may provide other diagnostic clues. Finally, unsteady gait may represent clumsiness due to a developmental coordination disorder.
Musculoskeletal Disorders
Specific musculoskeletal disorders, such as irritable hip, may also cause gait disturbance.1 Injury, deformity, or any process causing pain to the lower extremities or back may also cause impaired balance or gait abnormality that can be mistaken for ataxia.14 Typically, a gait disturbance due to pain is termed an antalgic gait.
Psychogenic Disorder
Gait disturbance is a common feature of psychogenic or functional movement disorder.10 With this disorder, the gait disturbance can be differentiated from true ataxia by two main observations. First, the patient will lack any adaptation, such as a wide-based gait, to improve balance. Second, the patient is unlikely to fall, as seen with true ataxia. Furthermore, tremor is the most common presenting feature of psychogenic movement disorder. This tremor may be postural and present both at rest and during action, making it distinguishable from a true cerebellar tremor, which typically occurs during a posture or on action. In addition, the tremor associated with psychogenic disorder will worsen during clinical examination and improve when the patient is distracted.10
Ataxia
Finally, one significant cause of uncoordinated movements is ataxia.1 Ataxia is characterized by impaired coordination of movement and balance associated with a lack of muscle control during voluntary activity.15 In the province of Manitoba in Canada, chronic ataxia was a relatively common presenting symptom in pediatric patients with a median age of ataxia onset of three years.16 The annual crude prevalence rate of chronic ataxia was 2.4 in 10,000, while the annual crude incidence rate was 3.2 in 100,000 in children and adolescents less than 17 years of age during 1991–2008. Many causes of chronic ataxia were evident including Angelman syndrome, ataxia-telangiectasia, posterior fossa stroke, mitochondrial diseases, and DEE.16
Table 1 summarizes the various causes of poor coordination.
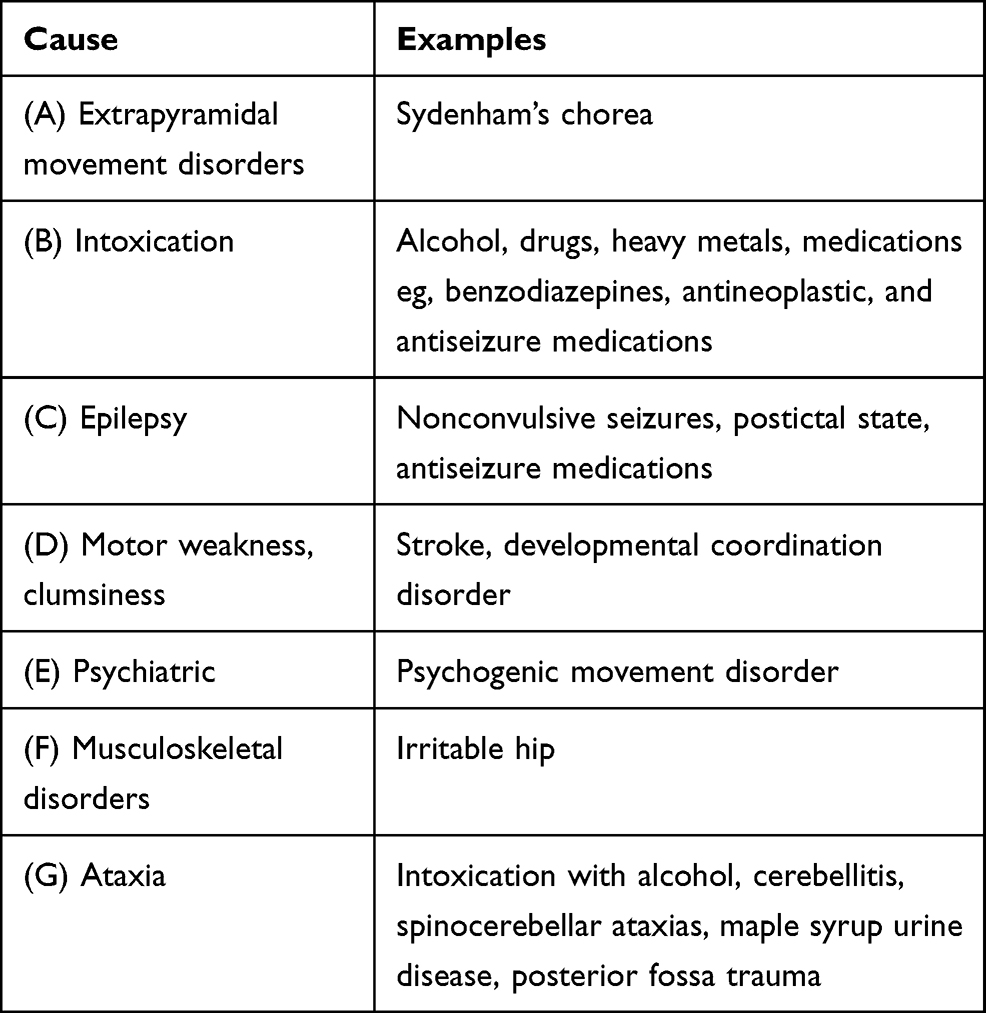 |
Table 1 Causes of Poor Balance and Incoordination |
Causes of Ataxia
Ataxia itself is a presenting symptom, not a disease; therefore, the underlying etiology must be elucidated.2 Ataxia has many causes and a classification scheme. An initial step in evaluating ataxia in pediatric patients is determining the timing of onset.2 Acute onset, typically described as less than 72 hours, suggests toxic, vascular or traumatic etiologies.1 Subacute onset is more likely to be caused by an infectious, inflammatory, or paraneoplastic disorder, whereas chronic ataxia is more likely due to genetic or neurodegenerative disorders.
Acute Causes of Ataxia
When evaluating an acute presentation of ataxia, medical practitioners must rule out three leading causes: stroke, toxicity, and trauma.8,13 Rarely, acute ischemic stroke, particularly involving the posterior cerebral circulation, may result in acute ataxia.10 The ingestion of toxic substances is also a common cause of ataxia, as outlined above.6,7 Traumatic injury may also cause ataxia.14 However, it is unlikely for trauma to cause an isolated presentation. Finally, neoplasms, such as tumours originating in the cerebellum or spinal cord, may also be responsible if they suddenly enlarge or bleed.13
Subacute Causes of Ataxia
Several subacute causes of ataxia should also be considered, including infection, inflammation, and metabolic disorders.14 Some common infectious causes of ataxia include the coxsackie and varicella viruses, which can cause cerebellitis. Rotavirus is another cause of post-infectious cerebellar ataxia.17 Inflammatory conditions, such as multiple sclerosis or acute disseminated encephalomyelitis, may also present with ataxia. Finally, there is a wide range of genetic and metabolic disorders, such as biotinidase deficiency, Hartnup disease, and abetalipoproteinemia, which may also cause ataxia.14
Chronic and Progressive Causes of Ataxia
Typically, chronic ataxia is due to various genetic or neurodegenerative disorders.1 The genetic causes of chronic ataxia are extensive, and numerous etiologies have been reported.16 The most common diagnoses include, but are not limited to, Angelman syndrome, ataxia-telangiectasia, various mitochondrial disorders, some types of DEE, and Rett syndrome.
Types of Ataxia
There are several types of ataxia, including cerebellar, sensory (proprioceptive), and vestibular ataxia (Table 2).2 These types may be distinguished based on the structure from which the dysfunction originates and the associated signs and symptoms. While this paper will focus on cerebellar ataxia, it is important to recognize the other two types.
 |
Table 2 Types of Ataxia |
Vestibular Ataxia
Vestibular ataxia may arise from damage or dysfunction of the vestibular apparatus (semicircular canals, saccules, utricles, and vestibular nerves) or impaired input from these structures to the cerebellum.2,13 Labyrinthitis and vestibular mononeuritis are two examples of vestibular disorders that may result in vestibular ataxia.2,13,14 Vestibular ataxia will often be associated with nausea, vomiting, hearing loss, poor balance, vertigo, and horizontal conjugate nystagmus contralateral to the side affected with the fast phases of the nystagmus directed towards the unaffected side.2,13 A lurching gait, or the tendency of an individual to fall to one side while ambulating, may also be seen.2,14 Typically, the patient will lean towards the ipsilateral side of the lesion.2
Sensory Ataxia
Sensory ataxia, also known as proprioceptive ataxia, results from impaired proprioceptive input from the peripheral nerves and spinocerebellar tracts to the cerebellum.2 The impaired transmission of proprioceptive information leads to sensory loss seen when testing vibration and joint position sense in the toes and fingers. Sensory ataxia may also manifest as the inability to stand with feet together and eyes closed, known as Romberg’s sign and as a high-stepping and feet-slapping gait. Typically, proprioceptive ataxia is worsened when visual cues are removed, as evidenced by a patient’s inability to touch their nose with their eyes closed. Guillain-Barré syndrome is one potential cause of sensory ataxia.2
Cerebellar Ataxia
Cerebellar ataxia results from dysfunction within the cerebellum itself or its afferent and efferent tracts.1,2 The cerebellum has been shown to have several roles including ocular, limbs, speech, cognitive, and behaviour control.1 As such, patients with cerebellar ataxia may present with motor and non-motor symptoms and signs.
Motor Manifestations of Cerebellar Ataxia
The following symptoms and signs help differentiate cerebellar ataxia from vestibular and sensory ataxia, as well as some of the other causes and presentations of uncoordinated movements outlined above.
Ataxic Gait
One of the most common presentations associated with cerebellar dysfunction is an unsteady and wide-based gait.1,2,14 An impaired stance, characterized by an increased width between the feet and the inability to stand with feet together for more than thirty seconds, suggests cerebellar ataxia.2 Truncal instability, which manifests as the oscillation of the body when sitting or standing, may also be present.1,2,14
Poorly Coordinated Voluntary Limb Movements
Several signs have been documented on physical examination in patients with cerebellar dysfunction. Dysmetria refers to when the patient either over-or undershoots their intended target, as seen with the finger-to-nose or heel-to-shin tests.1,2,14 Dysdiadochokinesis, or nonrhythmic and impaired rapid alternating movements, is often seen in cases of cerebellar ataxia. Further, dyssynergia, or the decomposition of movement into sequential tasks, may occur.14 Intention tremor is another common clinical presentation associated with cerebellar dysfunction and is characterized by the increasing amplitude of oscillation at the end of voluntary movement.1,2 Once again, this may be seen when completing the finger-to-nose test. Finally, titubation, which refers to involuntary, rhythmic oscillations of a body part eg, head or trunk, and hypotonia are also possible signs of cerebellar disorders.1,2
Ocular Motor Signs
The cerebellum plays an important role in ocular motor control.1 Several types of nystagmus, including downbeat, upbeat, and gaze-evoked nystagmus, represent signs of cerebellar dysfunction. Saccadic (ie, jerky) smooth ocular pursuit and saccadic dysmetria (ie, inaccurate fast eye movements) may also be present.1
Abnormal Speech
Speech production requires the coordination of numerous muscles located in the mouth and pharyngeal regions.1 Dysarthria is one common sign of cerebellar disease.
Management of Ataxia
The available treatment options for ataxia are limited. However, treatment of the associated symptoms in patients with disorders that involve cerebellar and extracerebellar structures of the central nervous system, is possible.18 Associated symptoms, including spasticity, seizures, hearing loss, sleep disturbance, depression, and anxiety, should be assessed and treated. In some instances, the underlying etiology should be treated. For example, the management of tumors, strokes, and inflammatory disorders, as well as the avoidance of certain medications and toxins, are important in the treatment of ataxia.18
Supportive management of ataxia itself includes physiotherapy, occupational therapy, speech and language therapy, and other rehabilitation specialists.18 Referral to the appropriate and relevant specialists, such as geneticists, endocrinologists, and cardiologists, is also essential in managing multisystem diseases associated with ataxia. Finally, patient education, including discussions regarding diagnosis, prognosis, and counselling, is a vital component in managing ataxia.18
One potential management option for ataxia focuses on motor rehabilitation (see reviews for more details).18,19 While, ultimately, the site and cause of the lesion determine the likelihood of recovery, continuous intensive motor training has been shown to be beneficial. In studies on adults, improvement of ataxia was reported with intensive whole-body coordinative training. However, the lack of motivation to participate in a demanding training regimen is often a barrier in pediatric patients. New technologies have been developed to make the training regimen more entertaining for children. A whole-body controlled video game technology has shown improvement of ataxia in a study on children with progressive spinocerebellar ataxia.18
Transcranial direct current stimulation (tDCS) has also shown promise as a potential management option for patients with degenerative ataxia.18,19 In tDCS, direct current influences cerebellar activity and excitability, which in turn modulates motor learning and impacts cognitive and emotional processes. A double-blinded, randomized, sham-controlled trial showed significant improvement in several performance scores in patients with various causes of ataxia. This study included patients with various degenerative ataxias, such as spinocerebellar ataxia, Friedreich’s ataxia, ataxia with ocular motor apraxia, multiple system atrophy-cerebellar type, fragile-X associated tremor/ataxia syndrome, and sporadic adult-onset ataxia of unknown origin.18–20 tDCS is an appealing option as it is easily delivered, non-invasive, and has no reported side effects.
In two randomized, double-blind, placebo-controlled, Class 1 studies, a decrease in the International Cooperative Ataxia Rating Scale (ICARS) was seen with Riluzole.18,19 More studies are needed to confirm the effectiveness of this medication. Further research may be facilitated by using more quantitative outcome measures and emphasizing specific causes and mechanisms of action.19 Long-term studies will be important in determining treatment options that extend beyond symptomatic treatment.
Developmental and Epileptic Encephalopathies
In DEE, frequent epileptic activity adversely impacts brain function, contributing to cognitive, behavioural, and motor plateauing or regression.4 It represents one group of neurodevelopmental disorders in which ataxia may occur. Of the 184 children with chronic ataxia in Manitoba, Canada, five patients had ataxia as part of a DEE, with a crude prevalence rate of 1.48 per 100,000 children during 1991–2008.16
Over the last few decades, genetic epilepsy panels have expanded and now include several hundred genes, with a proportion of them tailored to investigate genes described in DEE.21 Over 50 genetic variants have been associated with DEE and more are likely to be discovered.5 In addition, the number of forms classified in the Online Mendelian Inheritance in Man (OMIM) continues to increase. For example, between the years 2016–2019, 66 entries were found in OMIM when using the search terms epilepsy, seizure, epileptic syndrome, and epileptic encephalopathy.22 A review that highlights several of the common genes found in DEE is available for the interested reader.5
Clinical Presentation
In general, the symptoms and signs of various diseases that cause DEE tend to be similar. There is no clear genotype-phenotype correlation.3,4 In addition, mutations in a single gene can result in multiple phenotypes, a phenomenon known as phenotypic heterogeneity or pleiotropy.3 Thus, diagnosing the cause of DEE can be challenging.
While several core clinical features are commonly found in DEE, including developmental delay, behavioral difficulties, and intractable epilepsy; ataxia is one less common feature that when present, can narrow the list of diagnostic possibilities in patients with DEE.3 But first the presence of ataxia in patients with DEE needs to be established.
Case Studies
Three case studies are presented to illustrate the potential difficulties encountered in distinguishing the cause of incoordination in patients with DEE. In each case, we discuss a pediatric patient who has a DEE associated with cerebellar ataxia that is caused by a distinct genetic etiology.
Patient A
Background Information
Patient A is a six-year-old male. He presented initially at the age of three years and four months with a seizure after a fall in which he hit his head. The initial seizure consisted of right gaze deviation, nystagmus, and clonic movements in his right upper limb. The duration of the first seizure was unknown. He had two more seizures lasting 20 minutes and 45 seconds, respectively. It was thought that the seizures were due to the fall initially rather than the patient falling as a result of the seizures.
Over the next two years, the patient experienced nine breakthrough seizures and was diagnosed with epilepsy. He was initially started on valproic acid, which required dose optimization to gain better seizure control. Additionally, at four years and two months of age, clobazam was added to improve his seizure control. His final seizure occurred around five and a half years of age. He has since remained seizure-free on two antiseizure medications.
He also has behavioural difficulties and developmental delay, especially with language acquisition. While slow improvement in his development has been noted, at the age of five years and ten months of age, he was still only able to use 50 single words and could not put two words together.
The patient was born to a G1P0 mother at 39 weeks gestation via caesarean section due to failure to progress and followed an uneventful neonatal course. He was born in the Philippines and immigrated to Canada at three years of age. There is no relevant family history of seizure or developmental delay.
Physical Examination
At three years and eight months of age, weight was 15.3 kg (42nd percentile), height 99.1 cm (32nd percentile), and head circumference 53 cm (98th percentile). His cardiovascular, respiratory, and abdominal exam was unremarkable. He had no neurocutaneous stigmata or dysmorphic features. He had full extraocular eye movements, normal pupillary reaction to light, and normal facial movements. He had normal strength and reflexes but was hypotonic. He walked with an ataxic wide-based gait and required assistance to prevent him from falling. At three years and ten months of age, he displayed an intention tremor in his upper limbs bilaterally. Since then, his physical exam has remained unchanged.
Investigations
At the age of three years and ten months, the patient’s electroencephalogram (EEG) showed frequent epileptiform activity over the left temporal-central region. A brain magnetic resonance imaging (MRI) completed at the same age showed left mesial temporal sclerosis but did not show any other abnormalities that could explain his ataxia. The patient also underwent extensive routine, metabolic, and genetic testing (including a microarray CGH), which were all unremarkable. Finally, at six years of age, the patient had whole-exome sequencing, which ultimately provided a diagnosis of CACNA1A-related epileptic encephalopathy following the discovery of a genetic mutation in the calcium channel gene, CACNA1A. Specifically, this patient was heterozygous for a de novo pathogenic variant in the CACNA1A gene designated c.4174G>A, p.Val1392Met. The mode of inheritance is autosomal dominant. The patient’s disease phenotype is consistent with his diagnosis.
Current Status
The patient continues to display intention tremor bilaterally and has an ataxic wide-based gait. The patient’s DEE is associated with chronic ataxia. His ataxia does not wax and wane and is present independent of when his seizures were not controlled or when his antiseizure medication doses were changed. His antiseizure medications’ trough levels are in the therapeutic range.
Patient B
Background Information
Patient B is a seven-year-old male. The patient was referred for investigation of apneic spells associated with stiffness. These spells consisted of the patient becoming stiff, not breathing, rolling back of his eyes, then waking up and vomiting. These episodes occurred daily, beginning at around five months of age. Epilepsy was diagnosed, and as a result, the patient was started on phenobarbital following his initial hospital presentation at ten months of age. After a dosage increase, his seizures were controlled. He remained seizure-free for almost a year at which point the decision was made to switch his medication to levetiracetam as his development was slow (developmental delay may be a side effect of phenobarbital). Treatment with levetiracetam proved to be successful in controlling his seizures. After being seizure-free for two years, he was weaned off the levetiracetam. However, the seizures recurred at four years of age, and as a result, levetiracetam was restarted. Despite optimizing the dose, he continued to have breakthrough seizures, so clobazam was added, and the dose was titrated up several times until seizure control was achieved. The patient’s current treatment regimen has successfully controlled his seizures over the last 20 months.
There were no concerns regarding the patient’s development during his initial visit to the pediatric neurology clinic at 13 months of age. He appeared to be reaching all the appropriate developmental milestones. However, the patient soon began to display developmental delay, particularly with language. He was only able to use one word and two signs to communicate at two years of age. Over the years, the patient gained more words, reaching a documented maximum of 20 words at three years of age. However, his speech regressed and became subsequently incoherent. For the past two years, the patient has been averbal. Further, the patient has hyperactivity, which is currently well controlled on lisdexamphetamine.
The patient was born at 40 weeks’ gestation via caesarean section. There were no complications or exposure to teratogens during pregnancy. He is of Hutterite ethnicity and is the fourth of five children. His parents and siblings are all healthy, except for one older brother with attention deficit hyperactivity disorder (ADHD). A distant family history of seizures is present; however, no further details are available.
Physical Examination
At 13-months of age, the patient weight was 11.7 kg (85–97th percentile), height 76.5 cm (50th percentile), and his head circumference 46.5 cm (50th percentile). His cardiovascular, respiratory, and abdominal exam was unremarkable. He did not display any neurocutaneous lesions, and his Wood’s lamp dermatological exam (used to look for hypopigmented skin patches as seen in patients with tuberous sclerosis) was unremarkable. He did not have any dysmorphic features. His pupils were equal and reactive to light and he had full extraocular eye movements. There was no evidence of nystagmus. Initially, his motor exam, including tone, strength, coordination, and reflexes, was unremarkable. However, at two years and five months of age he developed an ataxic wide-based gait with a tendency to fall, intention tremor bilaterally in his upper limbs, and decreased tone. At the age of four years and two months, he displayed dysmetria in his upper limbs. Over the next couple of months, his gait became steadier but was consistently ataxic and wide-based. There have been no further changes on subsequent physical exams.
Investigations
At one year of age, an EEG showed intermittent interictal epileptiform discharges and intermittent polymorphic slow wave activity over the left fronto-temporal region. A brain MRI at 17 months of age was unremarkable. The patient had extensive routine, metabolic, and genetic testing (including a microarray CGH), which were all unremarkable. At seven years of age, whole-exome sequencing revealed a diagnosis of STXBP1- encephalopathy following the discovery of a genetic mutation in the syntaxin binding protein 1 gene, STXBP1. Specifically, he was heterozygous for a de novo pathogenic variant in the STXBP1 gene, c.568C>T, p.Arg190Trp. This variant follows an autosomal dominant inheritance pattern. His phenotype is consistent with this disease.
Current Status
The patient continues to display significant developmental delay, particularly with expressive language. He has an ataxic wide-based gait. He is hypotonic and has intention tremor with dysmetria in his upper limbs. His seizures are controlled on the two antiseizure medications.
Patient C
Background Information
Patient C is a 16-year-old female with celiac disease. She had two hospital admissions for seizures occurring when she was three years old. Before the initial admission, she had four seizures over a five-hour period. During these episodes, her eyes rolled up, her arms jerked, and her legs flexed. The seizures lasted between 30 seconds and two minutes. A month later, she was admitted to hospital again due to another cluster of seizures, during which she experienced nine seizures, each lasting between five and 30 seconds over a period of 24 hours. She was started on valproic acid following the diagnosis of epilepsy.
Over the next several months, the patient continued to experience frequent clusters of seizures spread over a few hours. During that time, her valproic acid dose was optimized. When she was five years old, the seizure clusters were controlled with the introduction of clobazam. Over the next several years, she continued to experience frequent breakthrough seizures. Eventually, at the age of nine years lamotrigine was added, which improved her seizure control and ultimately allowed for the clobazam to be weaned and discontinued. Despite the optimization of lamotrigine dose, she continued to experience breakthrough seizures.
At the age of 12 years, the patient experienced another seizure exacerbation. Levetiracetam was added and it significantly improved her seizure control; however, she began to display behavioural issues, a recognized side effect of this medication. As a result, the dose was decreased but she continued to experience mild behavioural problems. She was started on vitamin B6, which has been shown anecdotally to reduce levetiracetam-induced behavioural disturbances. The addition of vitamin B6 successfully improved her behaviour. She continued to experience infrequent seizures between 13 and 14 years of age, so both levetiracetam and lamotrigine doses were optimized further. Currently, her seizures are well controlled with valproic acid, levetiracetam, and lamotrigine with rare breakthrough seizures.
Physical Examination
At five years and seven months of age, the patient weight was 25.7 kg (90–95th percentile), height 118 cm (90th percentile), and head circumference 50 cm (25–50th percentile). Her cardiovascular, respiratory, and abdominal exam was unremarkable. She did not display any dysmorphic features. Her pupils were equal and reactive to light, and her extraocular eye movements were full. She had normal smooth ocular pursuit and saccades. She did not have nystagmus. She was hypotonic, but strength and reflexes were normal. She displayed head titubation, intention tremor, and dysmetria in her upper limbs. She also had an ataxic but not wide-based gait. At six years of age, she began to display truncal titubation. At seven years and six months of age, her speech became dysarthric, and her ataxia worsened with the development of a wide-based gait. By the age of 11 years, her gait became more ataxic and wide-based, and both her intention tremor and head titubation worsened. There have been no further changes on subsequent physical exams.
Investigations
The patient has undergone extensive routine, metabolic, and genetic workup (including karyotype and methylation studies for Angelman syndrome), which have all been unremarkable. An EEG at four years of age showed epileptiform activity, mainly located over the left parietal region with spread to the central and temporal regions. Her brain MRI was normal. Around seven years of age, a mutation in the methyl-CpG binding protein 2 (MeCP2) gene was found, and the patient was diagnosed with Rett syndrome. Specifically, she had a de novo mutation in exon 4 of the MeCP2 gene, c.1157_1197del, p.Leu386HisfsX5. Rett syndrome follows an X-linked dominant inheritance pattern. Her clinical phenotype is consistent with this syndrome.
Current Status
The patient’s ataxia remains stable. She continues to display intention tremor and dysmetria in her upper limbs, truncal titubation, and an ataxic wide-based gait. She may have up to two nocturnal brief seizures per month, if any.
Discussion
In our three case studies, it was evident that all patients displayed signs of cerebellar dysfunction. Each of the three patients has intention tremor and an ataxic wide-based gait. Further, both patients B and C have dysmetria. These three features are all examples of cerebellar dysfunction.1,2,14 The lack of signs associated with vestibular ataxia such as direction-dependent nystagmus or sensory ataxia with worsening of symptoms on closing the eyelids, further reinforces that the patients display cerebellar ataxia.2,12
One area in which differentiating ataxia from other causes of uncoordinated movements is challenging is in individuals with DEE or intractable seizures. There are several potential causes of uncoordinated movements occurring in individuals with epilepsy that can be mistaken for ataxia.1,6–10 Incoordination may result from nonconvulsive status epilepticus, which is often associated with decreased level of awareness, confusion, myoclonic jerks, and interrupted smooth movements of the limbs, leading to pseudoataxia.6,10 In addition, motor deficits, confusion, sleepiness, and lethargy occurring in the postictal phase of an epileptic seizure may also be mistaken for ataxia, especially if the seizure was not witnessed.8
Further, antiseizure medications may also cause poorly coordinated movements through several mechanisms.6,7 These include poor coordination as a general side effect, due to the sedating effect of the medication, or due to toxic levels of antiseizure medications. Therefore, in patients with epilepsy, the above three factors, ie, nonconvulsive seizures, postictal state, and medication effects, must be ruled out first before determining that a patient displays ataxia, especially when their “ataxia” is fluctuating.1,6–10
In each of the three patients discussed above, the three causes of poorly coordinated movements that may be seen in the setting of DEE and antiseizure medications have been ruled out. The trough levels of their medications have been monitored regularly since starting treatment to ensure that toxic levels are not present. Further, the ataxia has been present constantly in all three patients despite several medication and dose changes, suggesting that the ataxia is not the result of the medications’ side effect. Their ataxia did not fluctuate. Therefore, it is unlikely to be a manifestation of a postictal state or nonconvulsive seizures, especially during the time periods when their seizures were fully controlled. Their ataxia is a clinical feature of their disease phenotype.
While our understanding of DEE has expanded significantly over the past twenty years, they remain a complex group of neurodevelopmental disorders with variable presentations.4 Currently, numerous genetic etiologies have been discovered in children with DEE.3,5 The above case studies highlight three of these genes: CACNA1A, STXBP1, and MeCP2. Mutations in both CACNA1A and STXBP1 genes have been associated with infantile ataxias and DEE, thereby suggesting a common end result and phenotype in both disorders.23 In addition, the use of whole-exome sequencing has facilitated the diagnosis of the first two cases underscoring the importance of genetic advances in the discovery of new disease-causing genetic mutations. Epilepsy gene panels provide an alternative way to investigate such patients, if their CGH microarray results are unremarkable.
Currently, how these genetic mutations lead to the development of ataxia is not well understood. Thus far, several potential mechanisms have been suggested. For example, CACNA1A encodes calcium channels found in Purkinje cells in the cerebellum, and thus any alteration in their structure or function, caused by a genetic mutation, may cause ataxia, while mutations in STXBP1 gene have been found to impact the release of neurotransmitters from synaptic vesicles, which may alter the balance of excitation and inhibition.23 In contrast, it has been suggested that MeCP2 plays a role in transcription repression and mediating spontaneous neurotransmission and short-term synaptic plasticity.24 Thus, mutations in this ubiquitous protein have a more widespread effect on several neural processes. Further research is needed to increase our understanding of the pathological mechanisms of ataxia to facilitate improved and targeted treatment and management strategies.
Conclusion
Ataxia is a relatively common presenting symptom in childhood but represents only one possible cause of uncoordinated movements or poor balance. Therefore, health care providers must recognize and rule out several other causes and mimickers of ataxia to accurately determine the presence of ataxia and allow for timely management. Furthermore, awareness of the various types of ataxia (cerebellar, vestibular, and sensory) and their clinical presentations is important.
The case studies discussed highlight the complexity in recognizing ataxia as a concurrent feature of DEE and provide an example of the approach healthcare providers need to take when presented with a patient showing uncoordinated movements or poor balance. While these case studies highlight specific examples of DEE associated with ataxia, the underlying principles to differentiate the various causes of poor coordination can be applied to similar clinical settings with an appropriate clinical approach.
Consent
All parents gave verbal and written consent for the cases to be published.
Acknowledgment
We thank the children described in this manuscript and their parents for giving us permission to publish their cases. This manuscript was based on a capstone project submitted to the Faculty of Graduate Studies at the University of Manitoba in partial fulfillment of the requirements for the degree of Master of Physician Assistant Studies.
Funding
No specific funding was received for this work.
Disclosure
The authors declare that there are no conflicts of interest relevant to this work.
References
1. Salman MS. Clinical features, assessment, and management of patients with developmental and other cerebellar disorders. In: Marzban H, editor. Development of the Cerebellum from Molecular Aspects to Diseases. Switzerland: Springer International Publishing AG; 2017:407–422.
2. Ashizawa T, Xia G. Ataxia. Contin Lifelong Learn Neurol. 2016;22(4):1208–1226. doi:10.1212/CON.0000000000000362
3. McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016;15(3):304–316. doi:10.1016/S1474-4422(15)00250-1
4. Kalser J, Cross JH. The epileptic encephalopathy jungle – from Dr West to the concepts of aetiology-related and developmental encephalopathies. Curr Opin Neurol. 2018;31(2):216–222. doi:10.1097/WCO.0000000000000535
5. Specchio N, Curatolo P. Developmental and epileptic encephalopathies: what we do and do not know. Brain. 2021;144(1):32–43. doi:10.1093/brain/awaa371
6. Marcián V, Filip P, Bareš M, Brázdil M. Cerebellar dysfunction and ataxia in patients with epilepsy: coincidence, consequence, or cause? Tremor Hyperkinetic Mov. 2016;6:376. doi:10.5334/tohm.331
7. van Gaalen J, Kerstens FG, Maas RPPWM, Härmark L, van de Warrenburg BPC. Drug-induced cerebellar ataxia: a systematic review. CNS Drugs. 2014;28(12):1139–1153. doi:10.1007/s40263-014-0200-4
8. Pottkämper JCM, Hofmeijer J, van Waarde JA, van Putten MJAM. The postictal state — what do we know? Epilepsia. 2020;61(6):1045–1061. doi:10.1111/epi.16519
9. Javadzadeh M, Hassanvand Amouzadeh M, Sadat Esmail Nejad S, Abasi E, Alipour A, Mollamohammadi M. Ataxia in childhood: epidemiological, clinical and neuroradiologic features, and the risk of recurrence. Iran J Child Neurol. 2017;11(3):1–6.
10. Poretti A, Benson J, Huisman T, Boltshauser E. Acute ataxia in children: approach to clinical presentation and role of additional investigations. Neuropediatrics. 2012;44(03):127–141. doi:10.1055/s-0032-1329909
11. Risavi BL, Iszkula E, Yost B. Sydenham’s chorea. J Emerg Med. 2019;56(6):119–121. doi:10.1016/j.jemermed.2019.02.012
12. Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol. 2015;15(3):164–171. doi:10.1136/practneurol-2014-000994
13. Caffarelli M, Kimia AA, Torres AR. Acute ataxia in children: a review of the differential diagnosis and evaluation in the emergency department. Pediatr Neurol. 2016;65:14–30. doi:10.1016/j.pediatrneurol.2016.08.025
14. Overby P, Kapklein M, Jacobson RI. Acute ataxia in children. Pediatr Rev. 2019;40(7):332–343. doi:10.1542/pir.2017-0223
15. Pavone P, Praticò AD, Pavone V, et al. Ataxia in children: early recognition and clinical evaluation. Ital J Pediatr. 2017;43(1):6. doi:10.1186/s13052-016-0325-9
16. Salman MS, Lee EJ, Tjahjadi A, Chodirker BN. The epidemiology of intermittent and chronic ataxia in children in Manitoba, Canada. Dev Med Child Neurol. 2013;55(4):341–347. doi:10.1111/dmcn.12081
17. Kirik S, Aslan M, Özgör B, Güngör S, Aslan N. Acute ataxia in childhood: clinical presentation, etiology, and prognosis of single-center experience. Pediatr Emerg Care. 2021;37(3):e97. doi:10.1097/PEC.0000000000002104
18. Salman MS. Epidemiology of cerebellar diseases and therapeutic approaches. Cerebellum. 2018;17(1):4–11. doi:10.1007/s12311-017-0885-2
19. Zesiewicz TA, Wilmot G, Kuo S-H, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology. Neurology. 2018;90(10):464–471. doi:10.1212/WNL.0000000000005055
20. Benussi A, Dell’Era V, Cotelli MS, et al. Long term clinical and neurophysiological effects of cerebellar transcranial direct current stimulation in patients with neurodegenerative ataxia. Brain Stimul. 2017;10(2):242–250. doi:10.1016/j.brs.2016.11.001
21. Test ID: ESPAN - Clinical: epilepsy/seizure genetic panels by next-generation sequencing (NGS), varies. Available from: https://www.mayocliniclabs.com/test-catalog/Clinical+and+Interpretive/603346.
22. Hebbar M, Mefford HC. Recent advances in epilepsy genomics and genetic testing. F1000Res. 2020;9:F1000 Faculty Rev–185. doi:10.12688/f1000research.21366.1
23. Valence S, Cochet E, Rougeot C, et al. Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med. 2019;21(3):553–563. doi:10.1038/s41436-018-0089-2
24. Na ES, Monteggia LM. The role of MeCP2 in CNS development and function. Horm Behav. 2011;59(3):364–368. doi:10.1016/j.yhbeh.2010.05.014
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.