")
Back to Journals » Drug Design, Development and Therapy » Volume 12
Asiatic acid ameliorates CC l4-induced liver fibrosis in rats: involvement of Nrf2/ARE, NF-κB/IκBα, and JAK1/STAT3 signaling pathways
Authors Fan J, Chen Q, Wei L, Zhou X, Wang R, Zhang H
Received 11 July 2018
Accepted for publication 29 August 2018
Published 26 October 2018 Volume 2018:12 Pages 3595—3605
DOI https://doi.org/10.2147/DDDT.S179876
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Jie Fan,1,* Qingshan Chen,2,* Liwen Wei,1 Xiaoming Zhou,3 Rong Wang,4 Hai Zhang1
1Department of Pharmacy, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, China; 2Department of Pharmacy, Shanghai Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai 200438, China; 3Department of Endocrinology and Metabolism, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, China; 4Department of Pharmacy, Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 201999, China
*These authors contributed equally to this work
Purpose: Currently, there are no effective therapies for liver fibrosis; hence, the development of anti-liver fibrosis agents is urgently needed. Here, we attempted to investigate the therapeutic effect and mechanism of asiatic acid (AA) on liver fibrosis, mainly focusing on the impact of AA on nuclear erythroid 2-related factor 2/antioxidant response element (Nrf2/ARE), nuclear factor-kappa B (NF-κB)/IκBα, and JAK1/signal transducer and activator of transcription 3 (STAT3) signaling pathways.
Methods: Rats were induced liver fibrosis by carbon tetrachloride (CCl4) for 6 weeks and concomitantly treated with AA (5 and 15 mg/kg) or vehicle by daily gavage. After AA treatment, the morphology of liver tissue was analyzed by H&E and Masson’s trichrome staining, and serum biochemical indicators were also assayed. Thereafter, the protein levels of Nrf2, HO-1, NQO-1, GCLC, NF-κB, IκBα, JAK1, p-JAK1, STAT3, and p-STAT3 were determined by Western blotting.
Results: Our results showed that AA treatment dramatically ameliorated CCl4-induced oxidative stress, inflammation, and fibrosis in rats. The expression of nuclear Nrf2 was increased after AA treatment, whereas cytoplasm Nrf2 levels were decreased. The protein expression of Nrf2 target proteins including HO-1, NQO-1, and GCLC was significantly increased by AA treatment. Furthermore, AA treatment decreased the levels of nuclear NF-κB to inhibit NF-κB/IκBα signaling pathway. In addition, we also found that AA treatment regulated JAK1/STAT3 signaling by decreasing the phosphorylation levels of JAK1 and STAT3.
Conclusion: These results demonstrate that AA ameliorates CCl4-induced liver fibrosis in rats by regulating Nrf2/ARE, NF-κB/IκBα, and JAK1/STAT3 signaling pathways, which suggests that AA might be a new antifibrosis agent that improves liver fibrosis.
Keywords: asiatic acid, liver fibrosis, Nrf2-ARE, NF-κB/IκBα, JAK1/STAT3
Introduction
Liver fibrosis, excessive deposition of collagen and extracellular matrix (ECM) in the liver, is the result of the wound-healing response to chronic liver damage triggered by a variety of causes, including hepatitis virus infection, alcohol consumption, or nonalcoholic steatohepatitis.1,2 If not treated promptly, liver fibrosis can progress to liver cirrhosis, ultimately leading to liver failure or even death. Some studies have reported that liver fibrosis process is reversible, and early intervention can prevent its progression.3,4 Therefore, it would be of great value to develop new agents for prevention and therapeutic intervention strategies to treat liver fibrosis.
During liver fibrosis progression, hepatocellular damage and inflammation trigger complex cellular events that result in hepatic stellate cells (HSCs) activation and ECM deposition.5–7 The mechanism of liver fibrosis has been widely investigated. Oxidative stress is likely an important phenomenon that may result in hepatocellular damage and inflammatory responses.8,9 Previous studies have found that targeting nuclear erythroid 2-related factor 2/antioxidant response element (Nrf2/ARE) pathway to inhibit oxidative stress-mediated hepatocellular damage could attenuate liver fibrosis.10,11 Furthermore, nuclear factor-kappa B (NF-κB) is a key transcription factor that can modulate several steps in the inflammatory cascade by inducing the expression of inflammatory genes. Several investigations have revealed that inhibition of NF-κB/IκBα signaling pathway ameliorated the severity of liver fibrosis.12,13 In addition, recent studies suggest that signal transducer and activator of transcription 3 (STAT3) is an important transcription factor associated with liver inflammation, fibrosis, and cancer.14,15 Several studies have shown that STAT3 promotes HSCs survival, proliferation, and activation, thus contributing to liver fibrosis.16–18
Asiatic acid (AA), a bioactive compound extracted from the Centella asiatica, has multiple pharmacologic effects, including antioxidative, anti-inflammatory, and hepatoprotective activities.19–22 Recently, AA was found to have protective effects against CCl4-induced liver fibrosis by blocking TGF-β/Smad signaling pathways in vivo and in vitro.23 In addition, our previous studies have reported that AA attenuated CCl4-induced liver fibrosis in rats by regulating the PI3K/AKT/mTOR and Bcl-2/Bax signaling pathways.24 However, further molecular mechanisms underlying the protective effect of AA against CCl4-induced liver fibrosis remain largely unclear, and whether this protective effect is associated with inhibition of JAK1/STAT3 signaling pathway needs to be further clarified. Therefore, the aim of this study was to evaluate the therapeutic effect of AA against CCl4-induced liver fibrosis in rats, and to investigate the impact of AA on Nrf2/ARE, NF-κB/IκBα, and JAK1/STAT3 signaling pathways to elucidate its possible action mechanism.
Materials and methods
Chemicals and reagents
AA was purchased from Shanghai Nature Standard R&D and Biotech Co., Ltd. (purity 98.0%; molecular weight 488.70; Shanghai, China). Carbon tetrachloride (CCl4) was purchased from Shenzhen Xunye Chemical (Shenzhen, China). The biochemical kits of malondialdehyde (MDA), superoxide dismutase (SOD), and glutathione peroxidase (GSH-Px) were purchased from Jiancheng Institute of Biotechnology (NanJing, China). The trizol reagent, primescript RT reagent, and real time-PCR kit were purchased from TaKaRa (Dalian, China). Monoclonal anti-Nrf2, anti-HO-1, anti-NQO-1, anti-GCLC, histone H3, and β-actin antibodies were obtained from Abcam (Cambridge, MA, USA). Monoclonal antibodies against NF-κB, IκBα, JAK1, phospho-JAK1, STAT3, and phospho-STAT3 were purchased from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies used in Western blotting were goat anti-rabbit IgG (H+L) (Bioworld Technology, Shanghai, China). Secondary antibodies used in immunohistologic staining were purchased from Dako (Glostrup, Denmark). Secondary antibodies used in immunofluorescence staining were purchased from Abcam.
Animals and experimental design
A total of 48 male Sprague Dawley rats aged 10 weeks (200±20 g) were purchased from the B&K Universal Group Ltd. (Shanghai, China). They were maintained in a controlled environment individually with 12:12 hours light/dark cycle at a temperature of 21°C ± 2°C and a relative humidity of 55%. Food and water were provided. Animal welfare and experimental procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and were approved by the Animal Ethics Committee of Shanghai 9th People’s Hospital, Shanghai Jiao Tong University School of Medicine (Approval ID: 2017874).
Rats were divided randomly into four groups (n=12 for each group) as follows: the sham control group; the model group; the 5 mg/kg AA group; and the 15 mg/kg AA group. Liver fibrosis was induced by CCl4 orally (1 mL/kg, diluted in 50% peanut oil) for 6 weeks (twice a week).25,26 Meanwhile, the rats in the 5 mg/kg AA or 15 mg/kg AA groups were orally given AA (5 or 15 mg/kg) suspended in a 0.5% carboxymethyl cellulose sodium (CMC–Na) mixture once a day for 6 consecutive weeks. The rats in the sham control group simultaneously received the same volumes of normal CMC–Na mixture. The rats were weighed and anesthetized with pentobarbital sodium at the end of 6 weeks. Serum samples were obtained for analyses of liver functions, and a portion of the liver tissue was immediately fixed in 10% formalin for pathologic examination.
Histologic analysis
The liver samples were subjected to H&E staining for histologic examination to determine the liver fibrosis degree. Microscopic fields in all liver sections were randomly selected for examination by a light microscope (Nikon, Tokyo, Japan). The scoring of liver fibrosis degree was evaluated following the criteria: 0, no obvious fibrosis; 1, fibrosis present: collagen fibers that extend from the portal triad or central vein to peripheral regions; 2, mild fibrosis: few collagen fibers extending without formation of compartments; 3, moderate fibrosis: collagen fibers with formation of “pseudo leaves”; 4, severe fibrosis: many collagen fibers with thickening of partial compartments and formation of “pseudo lobes”. Meanwhile, liver sections were stained by Masson’s trichrome to estimate collagen deposition, which stains collagen fibers blue. Five different fields were randomly observed in each slice (×100), and three slices were selected in each group. The quantitative assays of collagen deposition were determined via the image software Image-Pro Plus 6.0 (Media Cybernetics Inc., Silver Spring, MA, USA) according to the procedure. All histologic examinations were undertaken by a very experienced pathologist blinded to the study protocol.
Biochemical analysis
For liver function examination, serum alanine transaminase (ALT) and aspartate transaminase (AST) were determined by using commercial reagent kit (Roche Diagnostic, Mannheim, Germany) according to the manufacturer’s protocol. Liver fibrosis serum markers hyaluronic acid (HA), laminin (LN), collagen type IV (IV-C), and procollagen III N-terminal peptide (PIIINP) were measured by radioimmunoassay kits (Beifang Biotechnology, Beijing, China). In addition, liver MDA, SOD, and GSH-Px activities were determined with the corresponding biochemical kits (JianCheng Biotechnology, Nanjing, China). All the procedures were performed based on the manufacturers’ protocols.
Western blotting analysis
To detect the protein expression of Nrf2, HO-1, NQO-1, GCLC, NF-κB, IκBα, JAK1, phospho-JAK1, STAT3, and phospho-STAT3, Western blotting was performed. The extraction and isolation of nuclear and cytoplasmic protein were performed according to the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Jiangsu, China). First, the tissue was homogenized in the cytoplasmic protein extraction agent supplemented with phenylmethanesulfonyl fluoride (PMSF) on the ice. After placing on ice for 15 minutes, the tissue was centrifuged for 5 minutes at 1,500 g at 4°C, and the supernatant was partially cytoplasmic protein. Next, the pellet was vortexed for 5 seconds with cytoplasmic protein extraction agent A, incubated for 15 minutes on ice, the cytoplasmic protein extraction agent B was added, vortexed for 5 seconds, and incubated on ice for 1 minute. Then the samples were centrifuged for 5 minutes at 14,000 g at 4°C, and the supernatant was cytoplasmic protein. Finally, the pellet was resuspended in nuclear protein extraction agent supplemented with PMSF, the pellet was vortexed 20 times for 30 minutes, centrifuged for 10 minutes at 14,000 g, and the supernatant was the nuclear proteins. Then, equal amounts of protein were subjected to 10% SDS-PAGE followed by immunoblotting using the following antibodies: rabbit anti-Nrf2, HO-1, NQO-1, GCLC, NF-κB, IκBα, JAK1, phospho-JAK1, STAT3, phospho-STAT3, histone H3, and β-actin at 4°C. On the next day, the membranes were washed and then incubated with secondary antibody at room temperature. The Western blot bands were visualized using an enhanced chemiluminescence system (Fusion FX7 Spectra, Vilber Lourmat, Eberhardzell, Germany) and analyzed by Quantity One (Bio-Rad) according to the standard method.
Quantitative real-time PCR analysis
PCR method is used to detect the expression of related mRNA in liver tissue. Total RNA was extracted using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and evaluated for concentration and purity through Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific, Shanghai, China). Then, the total RNA was reverse transcribed by PrimeScript RT reagent kit (Takara, Shiga, Japan). The primer sequences were designed and synthesized by Sangon Biotechnology (Shanghai, China) and are shown in Table S1. The target mRNA expression was quantified with SYBR Green kits (Takara, Shiga, Japan) in Step One Real-Time PCR System (Applied Biosystems, Warrington, UK). The thermal cycling conditions were as follows: 1. Holding Stage: 95°C, 30 seconds; 2. Cycling Stage: 95°C, 5 seconds; 60°C, 34 seconds; 40 cycles; 3. Melt Curve Stage: 95°C, 15 seconds; 60°C, 1 minute; 95°C, 15 seconds. Glyceraldehyde phosphate dehydrogenase (GAPDH) was used as the reference gene. The expression levels were measured in terms of the cycle threshold (Ct) and then normalized to GAPDH expression using the 2−ΔΔCt method.
Immunohistologic staining analysis
Immunohistologic examinations were carried out to detect the expression of p-JAK1 and p-STAT3. Briefly, liver sections were deparaffinized and treated with 3% H2O2 to block endogenous peroxidase activity. Antigen retrieval was performed in citrate buffer. After cooling, the sections were treated with 5% BSA to block nonspecific protein binding. The sections were incubated with anti-p-JAK1 (1:200) and anti-p-STAT3 (1:200) overnight at 4°C. Meanwhile, sections incubated with PBS alone were set as negative controls. Finally, the sections were washed with PBS, incubated with a biotinylated secondary antibody (1:1) and then with an avidin–biotin–peroxidase complex, and stained with DAB. All sections were imaged by microscope.
Immunofluorescence staining
The expressions of p-JAK1 and p-STAT3 were also detected by immunofluorescence staining. After being deparaffinized and blocked with 1% BSA, the liver tissue sections were incubated with primary antibodies anti-p-JAK1 (1:100) and anti-p-STAT3 (1:200), overnight at 4°C. Meanwhile, sections incubated with PBS alone were set as negative controls. Subsequently, the sections were incubated with the fluorescent secondary conjugated Alexa Fluor-488 or Alexa Fluor-555 at room temperature for 2 hours after PBS washes. The cell nuclei were counterstained with DAPI, and all stained sections were observed and photographed by fluorescence microscope (Nikon Fluorescence Microscope, Tokyo, Japan).
Statistical analysis
The data are presented as the means ± SD. Comparisons were performed using one-way ANOVA in GraphPad Prism 5. A value of P<0.05 was considered statistically significant.
Results
AA protects against CCl4-induced liver injury in rats
During the experiment, no deaths occurred in control group, but three rats died in model group, two deaths occurred in 5 mg/kg AA group, and two deaths occurred in 15 mg/kg AA group. As shown in Figure 1A–C, the rats administered CCl4 resulted in increasing of liver weight and liver index (liver/body weight ratio) in the model group, while treatment with AA (5 and 15 mg/kg) significantly decreased liver weight and liver index. Furthermore, H&E staining of liver sections showed large area of hepatocyte degeneration, necrosis, and leukocyte infiltration induced by CCl4 in the model group. However, AA treatment (especially 15 mg/kg group) effectively ameliorated CCl4-induced pathologic lesions, as indicated by only a small, localized injury and necrosis (Figure 1D and E). In addition, serum ALT and AST are the important biochemical indexes of liver injury. As expected, the ALT and AST levels were markedly elevated in the model group, while AA treatment (especially 15 mg/kg group) significantly attenuated the upregulation of ALT and AST levels by CCl4 (Figure 1F and G). These results confirmed that AA had a protective effect against CCl4-induced liver injury in a dose-dependent manner.
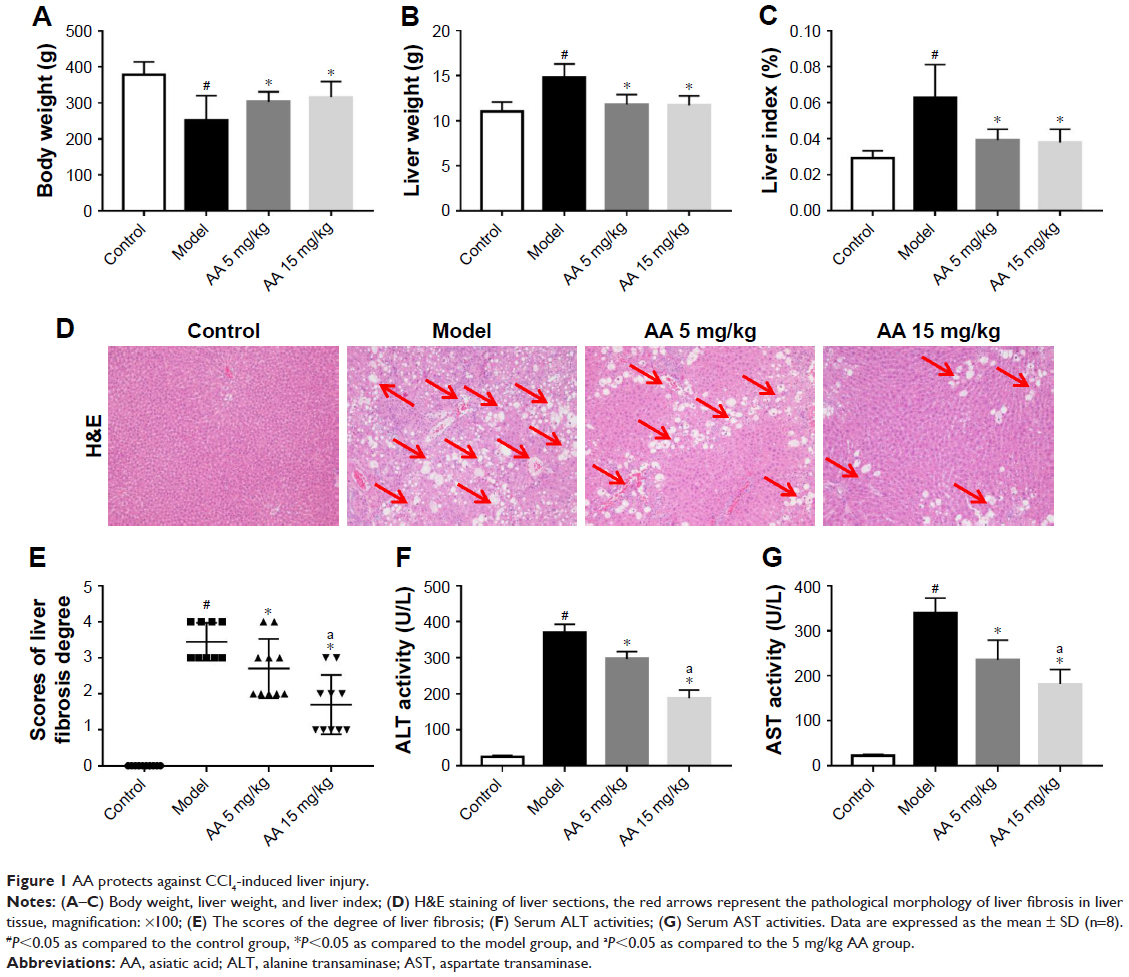 | Figure 1 AA protects against CCl4-induced liver injury. |
AA ameliorates CCl4-induced liver fibrosis in rats
To determine the effect of AA on CCl4-induced liver fibrosis, Masson’s trichrome staining of liver sections was performed to evaluate the effect of AA on collagen deposition. The results showed that collagen deposition in the model group was increased by 40-fold compared to the control rats. However, AA treatment markedly attenuated liver fibrosis, with a 46% decrease in the 5 mg/kg group and a 68% decrease in the 15 mg/kg group (Figure 2A and B). Meanwhile, the serum biomarkers of liver fibrosis further demonstrated that liver fibrosis was attenuated by AA treatment in rats. As shown in Figure 2C, these serum biomarkers (HA, LN, IV-C, and PIIINP) were significantly increased in CCl4-treated rats, but treatment with AA (especially 15 mg/kg group) decreased their content (P<0.05). These results suggested that AA could ameliorate liver fibrosis induced by CCl4 in vivo.
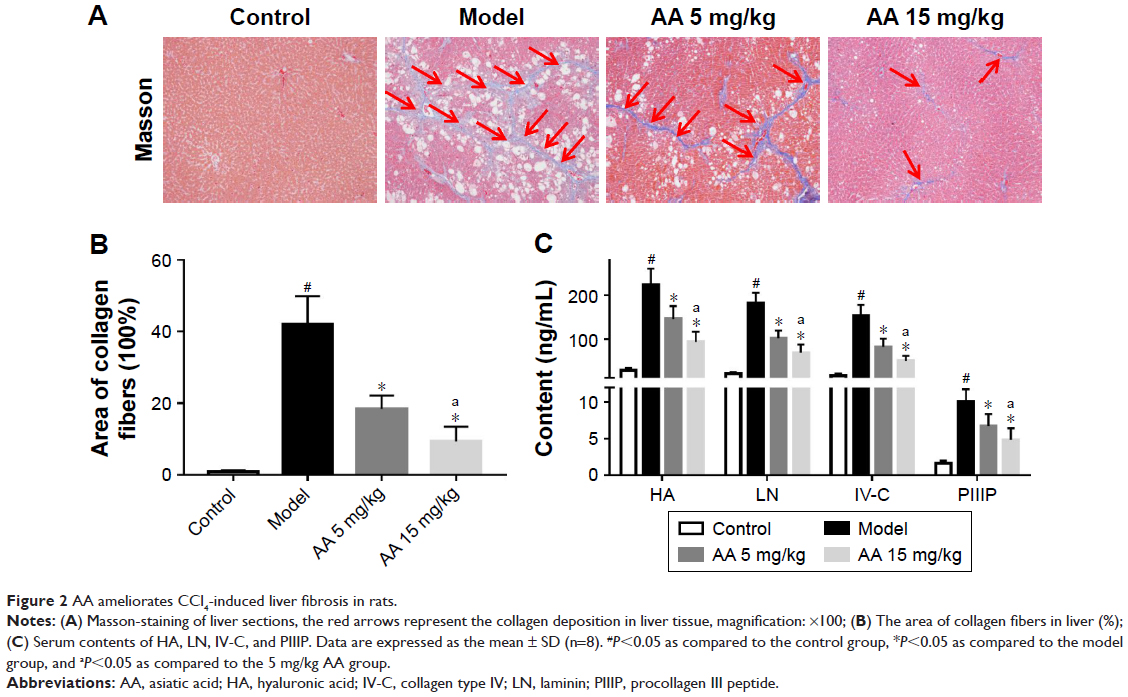 | Figure 2 AA ameliorates CCl4-induced liver fibrosis in rats. |
AA regulates CCl4-induced oxidative stress and Nrf2/ARE signaling pathway
To determine the effect of AA on CCl4-induced oxidative stress in rats, the activities of MDA, SOD, GSH-Px, and Nrf2/ARE pathway were determined. As shown in Table 1, compared to control rats, the content of MDA was significantly elevated in the model group, while the activities of SOD and GSH-Px were markedly decreased. However, these aberrant changes were partly reversed by 5 and 15 mg/kg AA treatment in a dose-dependent manner, suggesting that AA could block CCl4-induced oxidative stress. Next, the expression levels of proteins involved in Nrf2/ARE pathway were measured to explore whether AA activated this way to counteract oxidative damage-induced CCl4. The results showed that AA treatment significantly increased the nuclear accumulation of Nrf2 and decreased Nrf2 in the cytoplasm compared to that in control group (Figure 3A and B). Furthermore, the expression levels of Nrf2 target proteins HO-1, NQO-1, and GCLC were obviously increased after AA treatment at 15 mg/kg (Figure 3C). These results revealed that activation of Nrf2/ARE signaling pathway by AA treatment might suppress CCl4-induced oxidative stress.
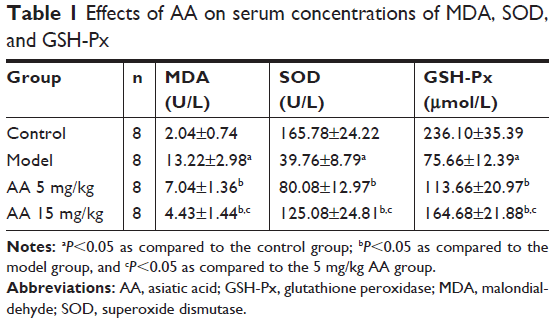 | Table 1 Effects of AA on serum concentrations of MDA, SOD, and GSH-Px |
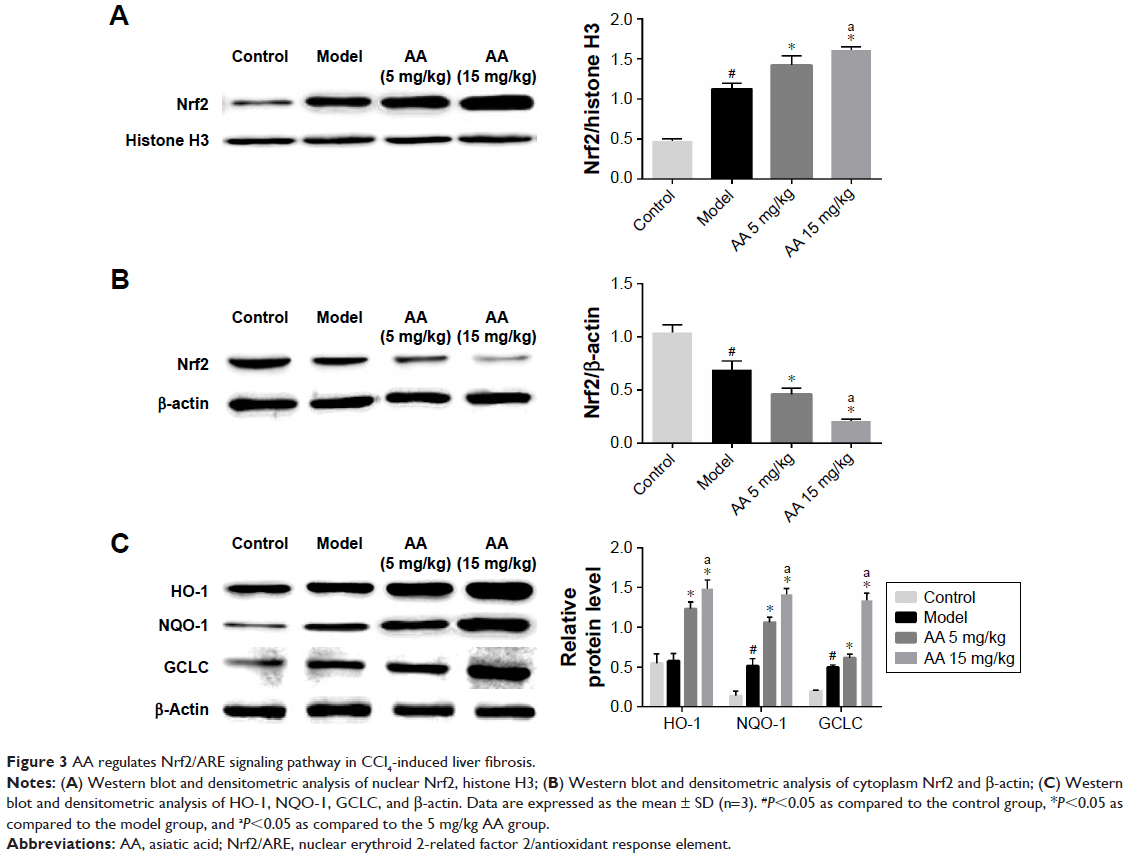 | Figure 3 AA regulates Nrf2/ARE signaling pathway in CCl4-induced liver fibrosis. |
AA suppresses CCl4-induced inflammation and NF-κB/IκBα signaling pathway
To determine the effect of AA on CCl4-induced inflammation in rats, the mRNA expressions of inflammatory cytokines tumor necrosis factor alpha (TNF-α), Cox-2, IL-6, IL-1β, TGF-β, and NF-κB/IκBα pathway were determined. As shown in Figure 4A, we found that the expressions of TNF-α, Cox-2, IL-6, IL-1β, and TGF-β were significantly increased in the model group, while their levels were all reduced by 5 and 15 mg/kg AA treatment in a dose-dependent manner. Furthermore, the expression levels of proteins involved in NF-κB/IκBα pathway were measured to explore whether AA activated this way to suppress inflammation-induced CCl4. The data indicated that nuclear NF-κB expression was inhibited after AA treatment compared to that in the control rats. Meanwhile, CCl4 treatment resulted in decreased protein levels of cytoplasm NF-κB and IκBα, which were significantly reversed by AA treatment in a dose-dependent manner (Figure 4B and C). All these results indicated that inhibition of NF-κB/IκBα signaling pathway by AA treatment might suppress CCl4-induced inflammatory response.
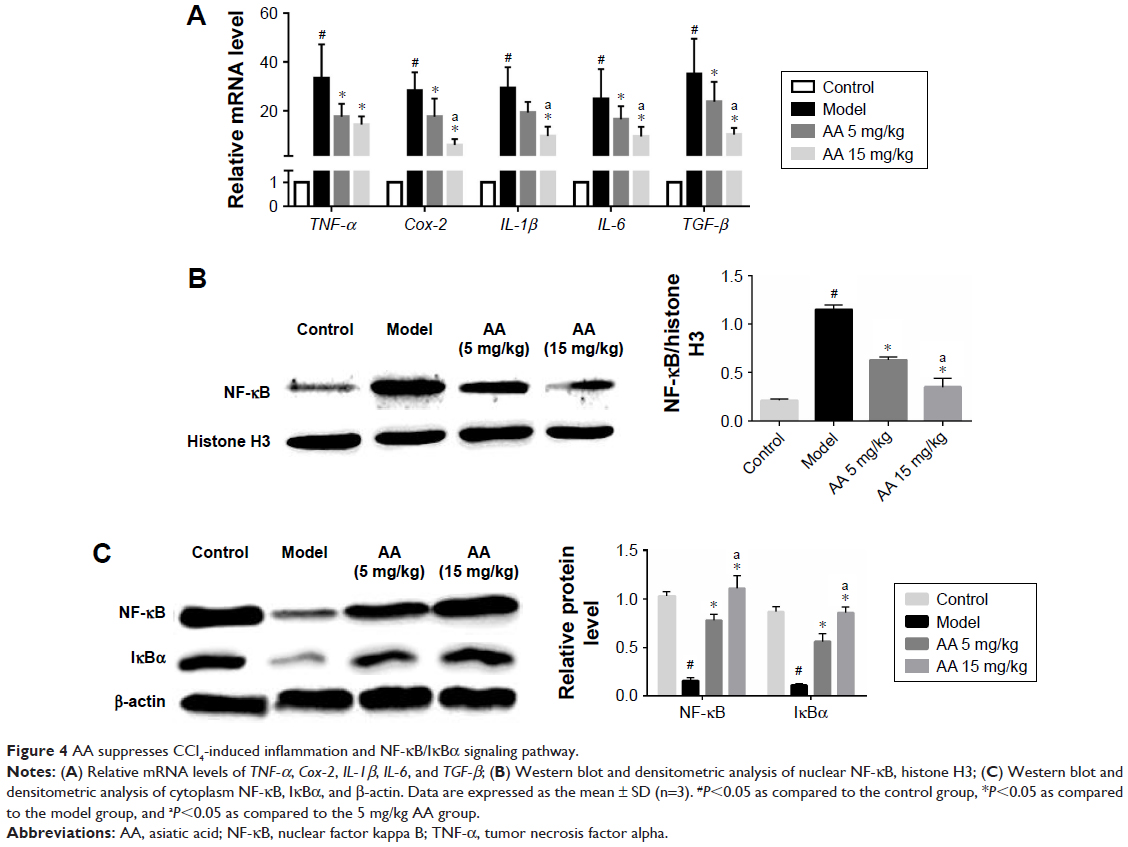 | Figure 4 AA suppresses CCl4-induced inflammation and NF-κB/IκBα signaling pathway. |
AA inhibits JAK1/STAT3 signaling pathway in CCl4-induced liver fibrosis
JAK1/STAT3 signaling is a well-known signaling pathway that plays a critical role in inflammation and HSC activation during liver fibrosis. Therefore, we examined whether the JAK1/STAT3 signaling pathway was involved in the AA-mediated anti-liver fibrosis effects. Compared to the control group, we clearly found that the phosphorylation of JAK1 and STAT3 was significantly increased in the model group (P<0.05). However, AA treatment suppressed CCl4-induced activation of the JAK1/STAT3 signaling pathway and reversed the elevated levels of p-JAK1 and p-STAT3 in a dose-dependent manner. Meanwhile, the total expression of JAK1 and STAT3 remains unchanged (Figure 5A). Furthermore, immunohistochemistry and immunofluorescence staining clearly showed that AA treatment decreased the expression of p-JAK1 and p-STAT3 in a dose-dependent manner (Figure 5B and C). These results suggest that AA protected the liver against liver fibrosis by inhibiting the JAK1/STAT3 signaling pathway.
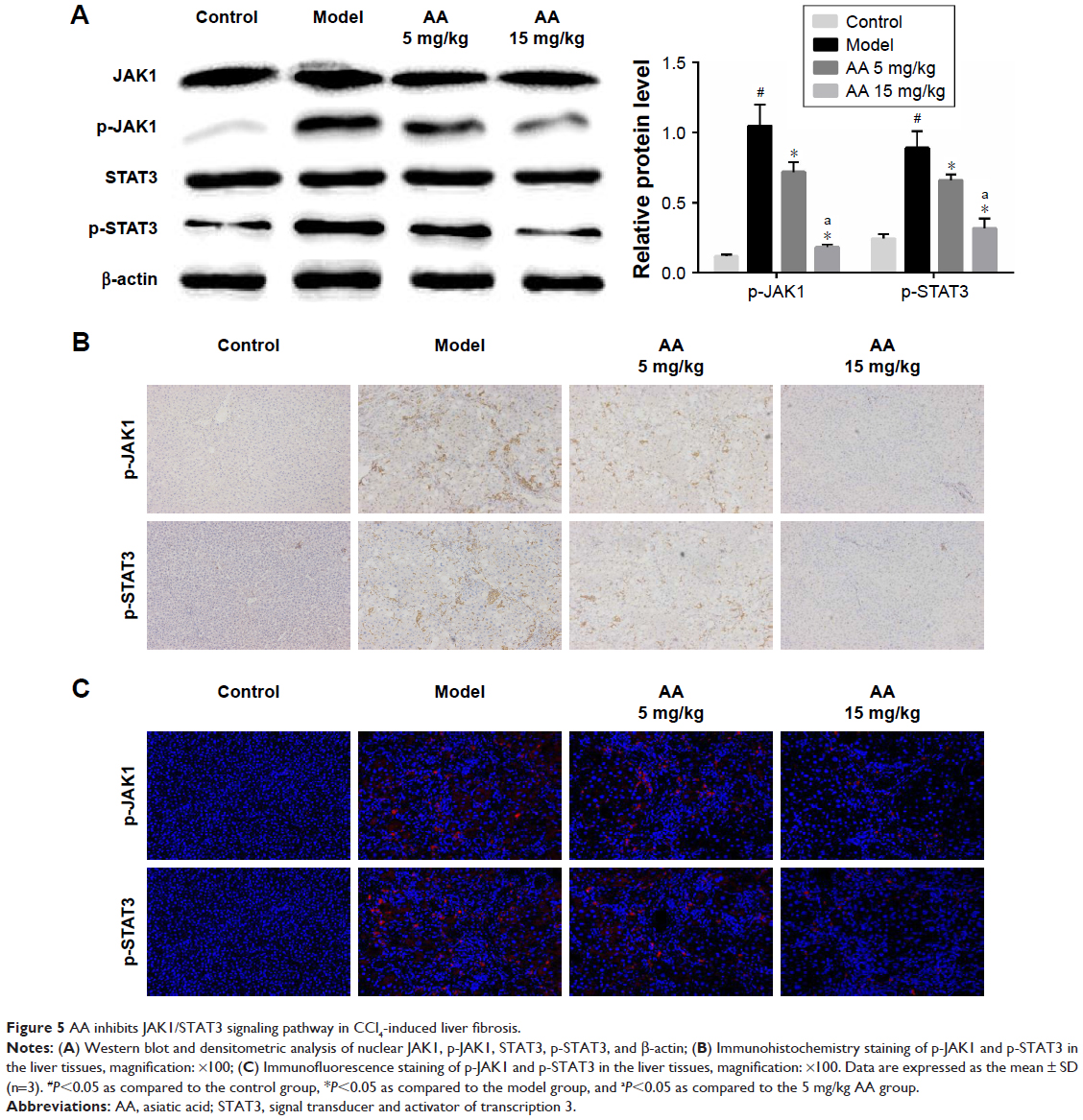 | Figure 5 AA inhibits JAK1/STAT3 signaling pathway in CCl4-induced liver fibrosis. |
Discussion
Liver fibrosis is excessive accumulation of ECM proteins that results from various chronic liver diseases, while there are no effective therapies for liver fibrosis at present.27 As we have known, the development of mild liver injury and liver fibrosis is accompanied by oxidative stress, inflammatory response, activation of HSCs, production of ECM, and hepatocyte necrosis. AA has been known as a natural compound extracted from the Terminalia catappa L., which has antioxidative and anti-inflammatory properties.21 Our previous studies have suggested that AA protects the liver against hepatocyte apoptosis, and inhibits HSC activation and ECM synthesis.23,24 In this study, the results demonstrated that AA alleviated oxidative stress-mediated hepatocyte damage, inflammation, and HSCs activation in rats, which suggested that AA has therapeutic potential against mild liver injury and liver fibrosis.
In this study, serum transaminase (ALT and AST) and H&E staining reflected the severity of CCl4-induced liver fibrosis in rats. When AA was administered after CCl4 treatment, rats were protected against CCl4-induced liver fibrosis as evidenced by lower serum ALT/AST levels and improved liver morphology and histology. In addition, liver fibrosis serum markers (HA, LN, IV-C, and PIIIP) further confirmed that AA could attenuate liver fibrosis. These findings showed that AA could ameliorate chronic liver injury and liver fibrosis induced by CCl4 in rats, which indicate that AA is a potential compound for treating liver fibrosis.
It is well known that oxidative stress is the crucial initiating step of liver fibrosis by inducing hepatocyte damage, inflammation, and HSCs activation in the liver.28 Previous studies have shown that AA exerts its beneficial effects through attenuating oxidative stress.24,29 In the present study, our data also showed that AA effectively inhibited CCl4-induced oxidative stress, as shown by decreased MDA levels and increased levels of SOD and GSH-Px in the rats treated with AA. To further explore the molecular mechanism of AA-inhibited oxidative stress, the Nrf2/ARE signaling pathway was next investigated. Nrf2 is a well-known transcription factor that regulates the expression of various antioxidant genes that are responsible for GSH synthesis and antioxidant defense system through ARE in the cell against oxidative stress.30,31 Recent studies have found that Nrf2 activators dramatically inhibited liver fibrosis and Nrf2-null mice were more susceptible to liver fibrosis compared to wild-type mice, suggesting that Nrf2 is a potential target to treat liver fibrosis.10,32,33 In addition, several studies have reported that AA could activate Nrf2 pathways to exhibit antioxidant and anti-inflammatory activities.34,35 In the current study, AA markedly increased the expression level of nuclear Nrf2 as compared to the model group. Furthermore, the expressions of Nrf2-target proteins HO-1, NQO-1, and GCLC were increased in the AA-treated group. These results indicated that AA protection against liver fibrosis may be through activating the Nrf2-ARE pathway to inhibit oxidative stress-mediated hepatocyte damage in the liver fibrosis rats.
Inflammation is the microenvironment of liver fibrosis and promotes the development of liver fibrosis. Hepatocyte damage promotes secretion of inflammatory cytokines and directly induces HSCs activation.36 In this study, the mRNA levels of the inflammatory cytokines and biomarkers TGF-β1, Cox-2, TNF-α, IL-6, and IL-1β were significantly increased in the model group, but they were significantly attenuated by AA treatment. NF-κB is a transcription factor that acts as a key regulator of inflammation and cell death, thus exerting a major role in liver fibrosis.37 In the absence of activating signals, NF-κB is normally sequestered in the cytoplasm by inhibitor of IκBα. Activation of NF-κB induces the translocation of NF-κB from the cytosol to the nucleus and facilitates the transcription of target genes. Previous studies have reported that AA exerts its anti-inflammatory effect by modulating NF-κB/IκBα signaling pathway.38,39 Whether AA induces NF-κB/IκBα signaling to inhibit CCl4-induced liver fibrosis has not been determined. In this study, CCl4 treatment activated NF-κB and increased nuclear NF-κB expression, whereas AA treatment significantly decreased the expression level of nuclear NF-κB. In addition, AA treatment also increased the protein levels of cytoplasm NF-κB and IκBα. Taken together, our findings demonstrated that AA could attenuate the release of inflammatory cytokines and inhibit the activation of NF-κB in the CCl4-induced liver fibrosis.
Another notable finding was that the JAK1/STAT3 pathway was involved in the protective effect of AA against CCl4-induced liver fibrosis in rats. JAK1/STAT3 signaling pathway can be activated by many cytokines, growth factors, and hormones, which plays a critical role in hepatic fibrogenesis.40,41 STAT3 activation has been detected in several liver diseases, including liver injury, liver steatosis, fibrosis, and hepatocellular carcinoma.15 Previous studies have reported that hepatocyte-specific STAT3 knockout mice display a higher degree of liver fibrosis compared to the wild-type mice in various models of liver fibrosis.42 A recent study suggests that activation of STAT3 in kupffer cells promotes HSCs survival and proliferation.43 In addition, a recent study found that sorafenib and its derivative SC-1 can ameliorate liver fibrosis through STAT3 inhibition in HSCs.44 In this study, the phosphorylation levels of JAK1 and STAT3 were apparently increased in the model group compared to the control group, indicating JAK1/STAT3 pathway being activated in liver fibrosis. After AA treatment, the phospho-JAK1 and phospho-STAT3 levels were significantly attenuated, indicating that the anti-liver fibrosis effect of AA is dependent on the suppression of the JAK1/STAT3 signaling pathway.
In conclusion, this study clearly demonstrated that AA attenuated the development of liver fibrosis through multiple mechanisms. First, AA activates Nrf2-ARE pathway to inhibit oxidative stress-mediated hepatocyte damage. Furthermore, it is reasonable that AA suppressed NF-κB/IκBα and JAK1/STAT3 signaling pathway to inhibit inflammation and HSCs activation. These results suggest that AA has therapeutic potential against liver fibrosis and may provide a novel mechanism for inducing antifibrotic effects. However, the effects of AA on liver fibrosis induced by a different etiology need to be examined in the future.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (81773797, 81803815), the Fund of Shanghai Jiao Tong University School of Medicine (JDYX2017QN008), Shanghai municipal medical and discipline construction projects (2017ZZ02015), and the Fundamental Research Funds for the Central Universities (1515219051).
Disclosure
The authors report no conflicts of interest in this work.
References
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. | ||
Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–841. | ||
Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gastroenterol. 2015;39 Suppl 1:S60–S63. | ||
Ellis EL, Mann DA. Clinical evidence for the regression of liver fibrosis. J Hepatol. 2012;56(5):1171–1180. | ||
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. | ||
Iwaisako K, Jiang C, Zhang M, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111(32):E3297–E3305. | ||
Ray K. Liver: hepatic stellate cells hold the key to liver fibrosis. Nat Rev Gastroenterol Hepatol. 2014;11(2):74. | ||
Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35(2):297–306. | ||
Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21(3):49–98. | ||
Xu W, Hellerbrand C, Köhler UA, et al. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab Invest. 2008;88(10):1068–1078. | ||
Yang JJ, Tao H, Huang C, Li J. Nuclear erythroid 2-related factor 2: a novel potential therapeutic target for liver fibrosis. Food Chem Toxicol. 2013;59:421–427. | ||
Son G, Iimuro Y, Seki E, Hirano T, Kaneda Y, Fujimoto J. Selective inactivation of NF-kappaB in the liver using NF-kappaB decoy suppresses CCl4-induced liver injury and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293(3):G631–G639. | ||
Oakley F, Mann J, Nailard S, et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166(3):695–708. | ||
Wang H, Lafdil F, Kong X, Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci. 2011;7(5):536–550. | ||
Gao B, Wang H, Lafdil F, Feng D. STAT proteins – key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J Hepatol. 2012;57(2):430–441. | ||
Deng YR, Ma HD, Tsuneyama K, et al. STAT3-mediated attenuation of CCl4-induced mouse liver fibrosis by the protein kinase inhibitor sorafenib. J Autoimmun. 2013;46:25–34. | ||
Mair M, Blaas L, Österreicher CH, Casanova E, Eferl R. JAK-STAT signaling in hepatic fibrosis. Front Biosci. 2011;16:2794–2811. | ||
Mair M, Zollner G, Schneller D, et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology. 2010;138(7):2499–2508. | ||
Zhou X, Tang L, Xu Y, Zhou G, Wang Z. Towards a better understanding of medicinal uses of Carthamus tinctorius L. in traditional Chinese medicine: a phytochemical and pharmacological review. J Ethnopharmacol. 2014;151(1):27–43. | ||
Xu Y, Yao J, Zou C, et al. Asiatic acid protects against hepatic ischemia/reperfusion injury by inactivation of Kupffer cells via PPARγ/NLRP3 inflammasome signaling pathway. Oncotarget. 2017;8(49):86339–86355. | ||
Lv H, Qi Z, Wang S, Feng H, Deng X, Ci X. Asiatic acid exhibits anti-inflammatory and antioxidant activities against lipopolysaccharide and d-galactosamine-induced fulminant hepatic failure. Front Immunol. 2017;8:785. | ||
Dong SH, Liu YW, Wei F, Tan HZ, Han ZD. Asiatic acid ameliorates pulmonary fibrosis induced by bleomycin (BLM) via suppressing pro-fibrotic and inflammatory signaling pathways. Biomed Pharmacother. 2017;89:1297–1309. | ||
Tang LX, He RH, Yang G, et al. Asiatic acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One. 2012;7(2):e31350. | ||
Wei L, Chen Q, Guo A, Fan J, Wang R, Zhang H. Asiatic acid attenuates CCl4-induced liver fibrosis in rats by regulating the PI3K/AKT/mTOR and Bcl-2/Bax signaling pathways. Int Immunopharmacol. 2018;60:1–8. | ||
Wang R, Zhang H, Wang Y, Song F, Yuan Y. Inhibitory effects of quercetin on the progression of liver fibrosis through the regulation of NF-κB/IκBα, p38 MAPK, and Bcl-2/Bax signaling. Int Immunopharmacol. 2017;47:126–133. | ||
Chen Q, Zhang H, Cao Y, et al. Schisandrin B attenuates CCl4-induced liver fibrosis in rats by regulation of Nrf2-ARE and TGF-β/Smad signaling pathways. Drug Des Devel Ther. 2017;11:2179–2191. | ||
Popov Y, Schuppan D. Targeting liver fibrosis: strategies for development and validation of antifibrotic therapies. Hepatology. 2009;50(4):1294–1306. | ||
Sánchez-Valle V, Chávez-Tapia NC, Uribe M, Méndez-Sánchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012;19(28):4850–4860. | ||
Lu Y, Kan H, Wang Y, et al. Asiatic acid ameliorates hepatic ischemia/reperfusion injury in rats via mitochondria-targeted protective mechanism. Toxicol Appl Pharmacol. 2018;338:214–223. | ||
Dayoub R, Vogel A, Schuett J, et al. Nrf2 activates augmenter of liver regeneration (ALR) via antioxidant response element and links oxidative stress to liver regeneration. Mol Med. 2013;19:237–244. | ||
Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284(20):13291–13295. | ||
Ni HM, Woolbright BL, Williams J, et al. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J Hepatol. 2014;61(3):617–625. | ||
Köhler UA, Kurinna S, Schwitter D, et al. Activated Nrf2 impairs liver regeneration in mice by activation of genes involved in cell-cycle control and apoptosis. Hepatology. 2014;60(2):670–678. | ||
Kamble SM, Patil CR. Asiatic acid ameliorates doxorubicin-induced cardiac and hepato-renal toxicities with Nrf2 transcriptional factor activation in rats. Cardiovasc Toxicol. 2018;18(2):131–141. | ||
Qi Z, Ci X, Huang J, et al. Asiatic acid enhances Nrf2 signaling to protect HepG2 cells from oxidative damage through Akt and ERK activation. Biomed Pharmacother. 2017;88:252–259. | ||
Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127(1):55–64. | ||
Luedde T, Schwabe RF. NF-κB in the liver – linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–118. | ||
Park JH, Seo YH, Jang JH, Jeong CH, Lee S, Park B. Asiatic acid attenuates methamphetamine-induced neuroinflammation and neurotoxicity through blocking of NF-kB/STAT3/ERK and mitochondria-mediated apoptosis pathway. J Neuroinflammation. 2017;14(1):240. | ||
Yun KJ, Kim JY, Kim JB, et al. Inhibition of LPS-induced NO and PGE2 production by asiatic acid via NF-kappa B inactivation in RAW 264.7 macrophages: possible involvement of the IKK and MAPK pathways. Int Immunopharmacol. 2008;8(3):431–441. | ||
Kong X, Horiguchi N, Mori M, Gao B. Cytokines and STATs in liver fibrosis. Front Physiol. 2012;3:69. | ||
Gao B. Cytokines, STATs and liver disease. Cell Mol Immunol. 2005;2(2):92–100. | ||
Horiguchi N, Lafdil F, Miller AM, et al. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology. 2010;51(5):1724–1734. | ||
Nieto N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology. 2006;44(6):1487–1501. | ||
Su TH, Shiau CW, Jao P, et al. Sorafenib and its derivative SC-1 exhibit antifibrotic effects through signal transducer and activator of transcription 3 inhibition. Proc Natl Acad Sci U S A. 2015;112(23):7243–7248. |
Supplementary material
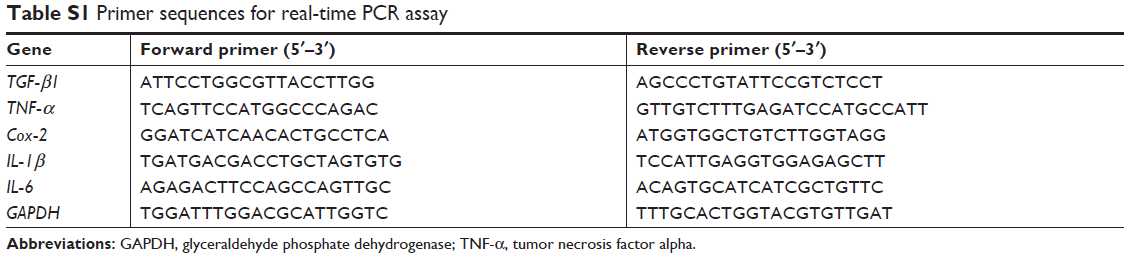 | Table S1 Primer sequences for real-time PCR assay |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.