Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Asenapine for bipolar disorder
Authors Scheidemantel T, Korobkova I, Rej S, Sajatovic M
Received 1 September 2015
Accepted for publication 9 November 2015
Published 4 December 2015 Volume 2015:11 Pages 3007—3017
DOI https://doi.org/10.2147/NDT.S78043
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Thomas Scheidemantel,1 Irina Korobkova,2 Soham Rej,3,4 Martha Sajatovic1,2
1University Hospitals Case Medical Center, 2Case Western Reserve University School of Medicine, Cleveland, OH, USA; 3Department of Psychiatry, University of Toronto, Toronto, ON, 4Geri PARTy Research Group, Jewish General Hospital, Montreal, QC, Canada
Abstract: Asenapine (Saphris®) is an atypical antipsychotic drug which has been approved by the US Food and Drug Administration for the treatment of schizophrenia in adults, as well as the treatment of acute manic or mixed episodes of bipolar I in both adult and pediatric populations. Asenapine is a tetracyclic drug with antidopaminergic and antiserotonergic activity with a unique sublingual route of administration. In this review, we examine and summarize the available literature on the safety, efficacy, and tolerability of asenapine in the treatment of bipolar disorder (BD). Data from randomized, double-blind trials comparing asenapine to placebo or olanzapine in the treatment of acute manic or mixed episodes showed asenapine to be an effective monotherapy treatment in clinical settings; asenapine outperformed placebo and showed noninferior performance to olanzapine based on improvement in the Young Mania Rating Scale scores. There are limited data available on the use of asenapine in the treatment of depressive symptoms of BD, or in the maintenance phase of BD. The available data are inconclusive, suggesting the need for more robust data from prospective trials in these clinical domains. The most commonly reported adverse effect associated with use of asenapine is somnolence. However, the somnolence associated with asenapine use did not cause significant rates of discontinuation. While asenapine was associated with weight gain when compared to placebo, it appeared to be modest when compared to other atypical antipsychotics, and its propensity to cause increases in hemoglobin A1c or serum lipid levels appeared to be similarly modest. Asenapine does not appear to cause any clinically significant QTc prolongation. The most commonly reported extra-pyramidal symptom associated with asenapine was akathisia. Overall, asenapine appears to be a relatively well-tolerated atypical antipsychotic, effective in the treatment of acute manic and mixed episodes of BD.
Keywords: asenapine, bipolar, manic episode, mixed episode, depressive features, safety, tolerability
Introduction
Bipolar disorder (BD) is a complex, multifaceted, multisystem illness, affecting up to 1% of the world’s population.1 Characterized primarily by manic, hypomanic, and depressive mood episodes, it remains one of the most disabling illnesses worldwide.2,3
The management of BD includes a comprehensive evaluation. History-taking includes screening for manic, hypomanic, and depressive episodes, as well as past or current psychotic symptoms and suicidality, as well as previous course of mood episodes (bipolar I or II, continuous cycling). The majority of patients will experience psychiatric comorbidity (eg, anxiety disorders, learning disorders, personality disorders) and/or alcohol or substance abuse/dependence during their lifetime.1 Physical comorbidity occurs commonly, with patients having an average of 2.5–3.5 medical conditions, while cognitive dysfunction occurs in up to 30% of patients, particularly in old age.1,4 In fact, along with the depressive phase of illness,5 the most disabling aspects of BD are physical comorbidity and cognitive dysfunction.6 These important clinical issues require an interdisciplinary integrated collaborative care approach with primary care doctors, specialist physicians, psychologists, nurses, social workers, occupational therapists, and colleagues.
Mood stabilizing agents such as lithium and valproate have been effective treatments in many patients,7 as have some of the novel antipsychotic medications.8 In addition to pharmacological treatment, there are psychotherapies, including psychoeducation,9 cognitive behavioral therapy,10 and interpersonal and social rhythm therapy.11 However, many patients do not achieve remission on existing treatments.12 There remains a need for agents that are effective for patients who often do not respond adequately to other therapies (eg, patients experiencing mixed episodes). One such agent may be the second-generation antipsychotic drug, asenapine (Saphris®; Actavis, Parsippany, NJ, USA). In this review, we explore the pharmacology, efficacy, safety, tolerability, and patient-focused perspectives related to asenapine in the treatment of patients with BD.
Pharmacology
Asenapine is a chemically distinct, atypical antipsychotic medication of the dibenzooxepino pyrrole class, and has a tetracyclic structure. Chemically, asenapine possesses two chiral centers; however, it is produced as the racemate.13
Asenapine is unique among atypical antipsychotics in its sublingual mode of administration. When administered sublingually, asenapine has a bioavailability of approximately 35%, while its oral bioavailability, if swallowed, is approximately only 2%.13,14 This is distinct even from the orally disintegrating formulations that are available for risperidone, olanzapine, and aripiprazole, all of which ultimately must be swallowed in order to achieve absorption of the drug. For this reason, the manufacturer’s insert recommends that patients not eat or drink anything within 10 minutes following administration of the drug. However, Gerrits et al15 has shown that consuming water at 5 minutes following a 5 mg dose of asenapine decreased the plasma levels by only 10%, and that consuming water 2 minutes following a similar dose lowered drug exposure by approximately 20%.13 It is not likely that these differences in drug exposure are clinically meaningful, as the reductions in exposure are actually less than the mean interindividual exposure variability which is as high as 26% when administered with perfect adherence to US Food and Drug Administration (FDA) guidelines.14 Additionally, Gerrits et al15 studied the bioavailability of asenapine in an open-label, randomized, 3-way crossover study of healthy male volunteers when the drug was administered in either the sublingual, supralingual, or buccal location. Interestingly, buccal administration resulted in only slightly higher total drug exposure, while supralingual administration was determined to be essentially bioequivalent to the sublingual route.15
Asenapine is produced as both an unflavored dissolving tablet and a black cherry flavored tablet, and is available in either 5 mg or 10 mg tablets. There is no dose titration required for initiation of treatment, according to the manufacturer’s insert, and daily dosing regimens are between 5 mg twice daily to 10 mg twice daily. The product label recommends specific starting doses based on the clinical scenario. In relation to BD, the recommended starting dose for a patient in a manic or mixed episode is 10 mg twice daily, and 5 mg twice daily if used in combination with either lithium or valproate.13 The time to reach steady-state plasma drug levels is around 3 days when dosed twice daily. The observed half-life of asenapine is approximately 24 hours,13,16 which would suggest that once-daily dosing is at least theoretically feasible, and a recently completed study (NCT01549041) investigated this issue; however, as of yet, no published data are available.
The mean time to achieve maximum drug plasma concentrations (Tmax) after a single 5 mg dose is 1 hour with a range of 0.5–1.5 hours. A single 5 mg dose of asenapine will achieve 79% D2 receptor occupancy approximately 36 hours after administration.16 Plasma asenapine is highly (95%) protein bound, primarily to albumin and alpha1acid glycoprotein. Asenapine shows excellent penetration across the blood–brain barrier, and the available evidence indicates that it is not a substrate of P-glycoprotein transporters.16
Asenapine is metabolized to 38 different metabolites, none of which have any significant functional activity at receptors. The primary mechanism of metabolism is via glucuronidation through UDP glucuronosyltransferase 1A4 (UGT1A4), producing asenapine-N-glucuronide. The other major metabolite of asenapine is produced through demethylation, resulting in N-desmethylasenapine, primarily via CYP1A2, with only minor contributions from CYP3A4 and CYP2D6. Clinically relevant increases in total asenapine exposure have only been observed in severe hepatic impairment, and doses should be decreased accordingly. However, no dose adjustments are needed for patients with mild or moderate hepatic impairment or renal impairment. Asenapine and its metabolites are excreted in essentially equal proportions in urine and feces.16
Comparable to nearly all other atypical antipsychotics, asenapine has affinity at a wide variety of serotonergic, dopaminergic, noradrenergic, and histaminic receptors. One of the most consistent features of the atypical antipsychotics as a class of drugs is a high ratio of 5HT2A to D2 binding affinity. Asenapine has a 19-fold higher binding affinity for 5HT2A receptors compared to D2 receptors, which is very similar to ratios for the atypical antipsychotics risperidone, olanzapine, clozapine, and ziprasidone. Asenapine acts as an antagonist at multiple serotonergic receptors. Asenapine’s high affinity for 5HT2C and H1 receptors is very similar to that of both clozapaine and olanzapine, yet the weight gain liability of asenapine is substantially less than the latter two drugs.17 Similarly, asenapine has a high affinity for multiple dopamine receptors, with highest affinity for the D3 receptor (Ki =0.42 nM). Comparable to other atypical antipsychotics, asenapine possesses antagonist activity at H1 receptors (Ki =1.0 nM) and is the only known atypical antipsychotic to possess H2 antagonism (Ki =6.2 nM). Interestingly, asenapine has virtually no affinity for muscarinic acetylcholine receptors.16 Finally, asenapine itself has no binding affinity for either metabotropic glutamate receptors or the ionotropic NMDA, AMPA, and kainate glutamate receptors.17
Efficacy studies in bipolar mania
In 2009, asenapine was approved by the FDA for the treatment of acute mania. Since then, evidence of asenapine’s efficacy/effectiveness in acute mania has accumulated, courtesy of various randomized controlled trials (RCTs), open-label, and observational studies (Table 1).18–23
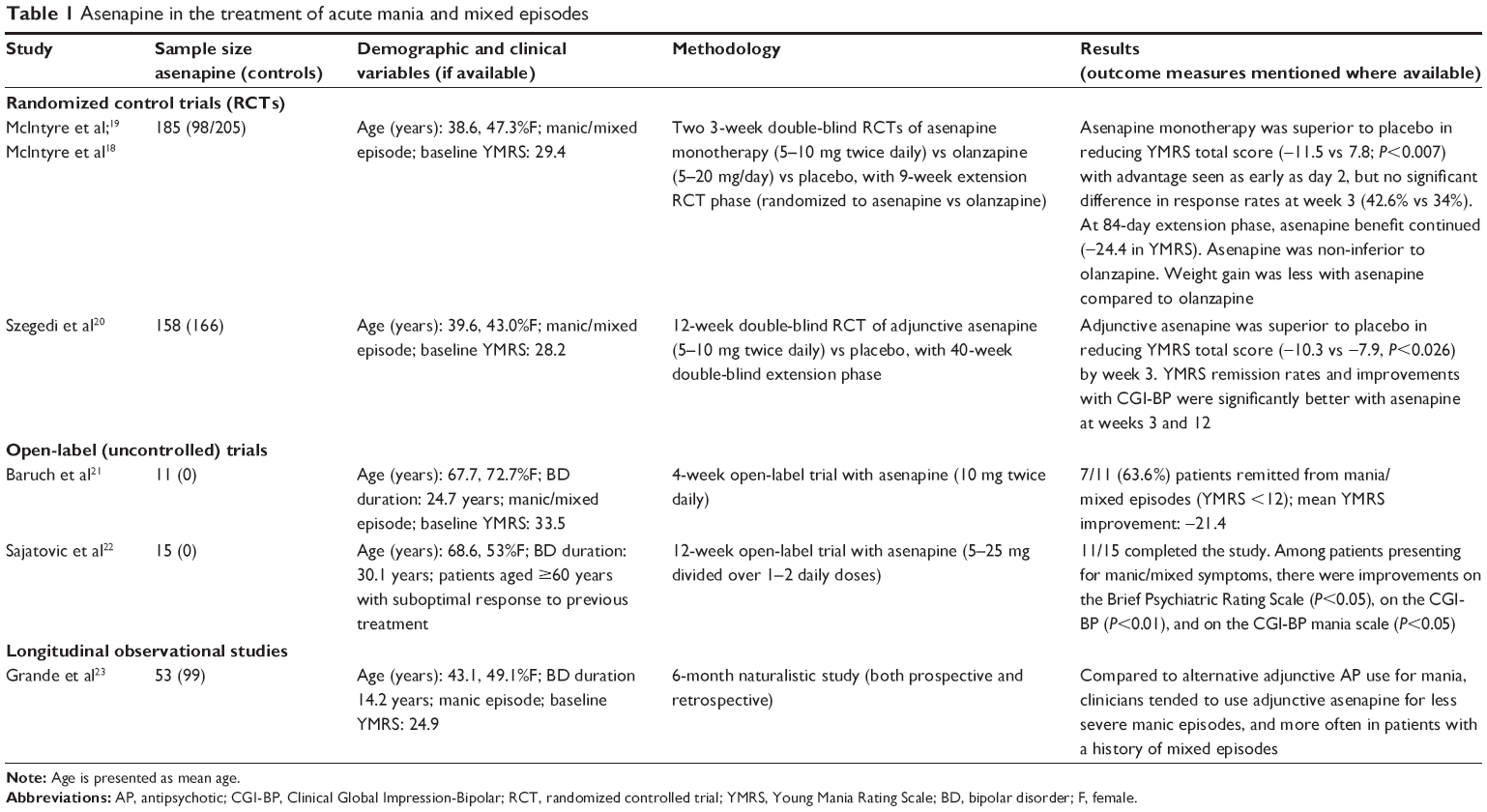 | Table 1 Asenapine in the treatment of acute mania and mixed episodes |
Two 3-week double-blind placebo- and olanzapine- controlled RCTs of asenapine monotherapy,18,19 and one 12-week double-blind of adjunctive asenapine20 demonstrated superiority over placebo in reducing Young Mania Rating Scale (YMRS) scores. Compared to placebo, remission rates, as well as Clinical Global Impression-Bipolar (CGI-BP) scores, were improved at the 3-week and 12-week follow-ups.20 Asenapine and olanzapine-treated groups did not differ significantly with regards to decrease in YMRS over an initial 3-week RCT (asenapine 5–10 mg twice daily vs olanzapine 5–20 mg/day vs placebo) and 9-week RCT extension phases (asenapine vs olanzapine).18,19
Similar findings were obtained in other recent investigations. In two uncontrolled open-label studies21,22 of older adults aged ≥60 (n=11 and n=15, respectively), asenapine dosed at 5–25 mg/day was effective for acute manic/mixed episodes. Improvements were observed in the YMRS, Brief Psychiatric Rating Scale, and the CGI-BP scores at the 4- and 12-week follow-ups.21,22
In a 6-month naturalistic study of adjunctive asenapine (n=53) and other adjunctive antipsychotics (n=99) in the treatment of acute mania, asenapine tended to be used by clinicians for less severe manic episodes and in patients with a history of mixed episodes.23
Early improvements with asenapine may help clinicians identify whether a patient may eventually benefit from this medication during acute mania. In a pooled analysis of two 3-week acute mania trials, asenapine use was associated with broad improvement across all eleven YMRS items, with asenapine separating from placebo as early as 2 days postinitiation.24 Both 2-day and 7-day improvements in YMRS scores appeared to predict the subsequent 3-week response to mania; an absence of YMRS improvement at the 7-day follow-up was particularly predictive of subsequent non-response.25
Based on the available evidence, asenapine is efficacious in the treatment of acute mania, including in the treatment of mixed episodes. Treatment gains become apparent at day 2, but are more prominent at week 3 and thereafter. Compared to younger adults, patients aged ≥60 with mania/mixed episodes seem to have similar treatment benefits with asenapine.21,22 McIntyre et al has found asenapine to be non-inferior to olanzapine.18,19 Further studies will be required to confirm this and compare asenapine with other bipolar pharmacotherapies in the treatment of acute mania.
Analyses in bipolar depression and maintenance treatment
Bipolar depression
There have not yet been any published studies specifically assessing the efficacy of asenapine in bipolar depression. A registered clinical trial (NCT01807741) is actively recruiting participants to evaluate the role of asenapine in the treatment of bipolar depression. However, there have been several published studies of post hoc analyses of original study data focusing specifically on patients presenting with bipolar mixed states, and the efficacy of asenapine in the management of depressive symptoms (Table 2). All of the following studies utilized study data reported by McIntyre et al (NCT00159744 and NCT00159796), which compared asenapine to either olanzapine or placebo in the treatment of bipolar patients in acute manic or mixed states.18,19
 | Table 2 Asenapine in the treatment of depressive symptoms of bipolar disorder |
Azorin et al26 performed a post hoc analysis on patients meeting Diagnostic and Statistical Manual of Mental Disorders IV Text Revision (DSM-IV-TR) diagnostic criteria for mixed episodes and reported efficacy data of improvement of Montgomery–Åsberg Depression Rating Scale (MADRS) scores from baseline at study weeks 3 and 12.26 They identified 295 bipolar patients meeting criteria for a mixed episode at the initiation of the study (asenapine n=107, olanzapine n=122, and placebo n=66). Of those patients, 102 entered the 9-week extension phase (asenapine n=46 vs olanzapine n=56). At study week 3, asenapine was found to be statistically significantly superior to placebo with changes in MADRS scores (Least squares [LS] mean ± standard error [SE]) of −8.2±0.9 for asenapine vs −4.5±1.2 for placebo (P=0.009).26 Interestingly, the difference between olanzapine (−6.5±0.8) and placebo was not statistically significant at week 3 (P=0.181). However, by week 12, MADRS score improvement for asenapine (−11.9±2.6) vs olanzapine (−7.9±1.8) was not found to be statistically significant (P=0.138). Asenapine also showed significantly greater improvement in the MADRS subscale items of inner tension, reduced appetite, and inability to feel, compared to placebo (P<0.05). However, only on the subscale item of inner tension did asenapine perform better than olanzapine (P<0.05).26
Berk et al27 examined the same primary data set, focusing on patients presenting in mixed mood episodes with moderate-to-severe depression. A total of 98 patients (asenapine n=33, olanzapine n=39, and placebo n=26) meeting inclusion criteria (DSM-IV-TR criteria for moderate-to-severe depression and MADRS ≥20) were identified for this analysis. Similar to the findings reported by Azorin et al,26 decreases in MADRS scores in the asenapine group were statistically significantly greater than placebo at day 7, day 21, and at endpoint (last observation carried forward [LOCF]). Decreases in MADRS scores in the asenapine group were statistically significantly greater compared to olanzapine at day 7, but were not significantly different at day 21 or endpoint.27 Again, asenapine showed significant improvement in MADRS scores for subscale items of reported sadness, inner tension, and inability to feel, compared to placebo at day 21, but outperformed olanzapine only on the inability to feel item. Treatment with olanzapine showed no significant improvement on any MADRS subscale item compared to placebo at day 21.27
McIntyre et al28 took a slightly different approach in their analysis of the data, and chose to examine the effect of particular depressive symptoms on remission of depression (defined as MADRS score ≤12) at study day 21 and endpoint (LOCF).28 Six core depressive features were chosen for evaluation, along with what McIntyre et al identified as the corresponding MADRS or Positive and Negative Syndrome Scale (PANSS) item or items. The six features and their linked items are as follows: 1) depressed mood (MADRS item 1 or 2), 2) fatigue and loss of energy (MADRS item 7), 3) diminished interest/pleasure (MADRS item 8), 4) psychomotor retardation (PANSS item G7), 5) worthlessness and guilt (MADRS item 9), and 6) suicidal thinking (MADRS item 10). The key finding from this study indicated that, while remission rates of depression decreased with increasing numbers of depressive symptoms (≥2 or ≥3 of the above-listed core features) in the placebo and olanzapine treatment groups, remission rates remained quite stable in patients treated with asenapine, regardless of the number of depressive symptoms. The remission rate in the asenapine group was significantly greater compared to placebo (P≤0.05) at endpoint (LOCF).28
Finally, Szegedi et al29 examined three distinct subsets of patients from the initial efficacy studies by McIntyre et al18,19: 1) patients with a baseline MADRS score ≥20; 2) patients with a baseline CGI-BP score ≥4; and 3) all patients presenting with mixed episodes at study outset. Szegedi et al reported on the following primary endpoints: overall change in MADRS scores, CGI-BP scores, and PANSS Marder Anxiety/Depression Factor scores from baseline to study days 7 and 21 in each of the three patient groups. In addition, they reported on the overall remission rates (defined as a MADRS score ≤12) in the three subpopulations.29 In all three subpopulations, the decrease in MADRS scores from baseline in the asenapine-treated group was statistically significantly greater than placebo at both trial day 7 and trial day 21. Only in the patient group with a baseline MADRS score ≥20 did asenapine show statistically significant improvement over olanzapine, and this result was only observed at study day 7 (change in MADRS scores for asenapine [LS mean ± SE, −11.3±1.5] and for olanzapine [LS mean ± SE, −4.5±1.6], P=0.02). In none of the three subpopulations did olanzapine show significant improvement in MADRS scores at day 7 or day 21, compared to placebo. Similarly, rates of remission of depressive symptoms in all three patient populations were significantly greater in the asenapine-treated group when compared to placebo at both days 7 and 21. Again, remission rates in all three patient populations were significantly greater in the asenapine group when compared to the olanzapine group, but this was only observed at day 7.29 In the MADRS ≥20 patient group, remission rates at day 7 for asenapine vs olanzapine were 57% and 25% (P=0.006), respectively. In the CGI-BP ≥4 population, remission rates at day 7 for asenapine vs olanzapine were 68% and 45%, respectively (P=0.031).In the mixed-episode patient population, remission rates at day 7 for asenapine vs olanzapine were 76% and 55% (P=0.007), respectively. While remission rates in the asenapine group were higher than the olanzapine-treated group at day 2, this did not reach statistical significance. Lastly, consistent with above data, improvement in CGI-BP and PANSS Marder Anxiety/Depression Factor scores from baseline were significantly greater in all three populations when asenapine was compared to placebo, but only trended towards statistical significance when comparing asenapine to olanzapine.29
Bipolar maintenance
There have been no clinical trials specifically designed to evaluate the efficacy of asenapine in the maintenance phase of the treatment of BD. However, two trials evaluating the efficacy of asenapine in the treatment of acute manic or mixed episodes have included extension phases beyond the initial evaluation phase, measuring response and remission rates out to 52 weeks.30,20
Szegedi et al20 assessed the efficacy of asenapine as an adjunctive treatment for acute mania in adults with BD. Asenapine was added in a flexible-dosing protocol to open-label doses of either lithium or valproate. The primary endpoint was the change from baseline YMRS scores, and was assessed at weeks 3 and 12. Patients completing the 12 weeks of the initial core study were eligible to participate in the 40-week extension study, regardless of YMRS score at week 12. Patients were not re-randomized prior to entering the extension phase, and were continued on the dose of asenapine they were on at the completion of the core study. Mean (standard deviation [SD]) YMRS scores of patients entering the blinded extension phase were 27.6 (6.3) in the asenapine arm and 28.0 (6.1) in the placebo arm. Patients taking adjunctive asenapine, regardless of baseline mood stabilizer (lithium vs valproate), had statistically significantly greater improvement in YMRS scores at weeks 2, 3, and 12 of the core study compared to placebo. However, at week 52 there was no statistically significant difference between asenapine and placebo based on LOCF analysis. Similarly, at week 52, neither response rates (asenapine 68.4%, placebo 78.8%) nor remission rates (asenapine 65.8%, placebo 78.8%) were statistically significantly different between the two groups, with response defined as ≥50% decrease in YMRS score from baseline, and remission defined as a total YMRS score ≤12.20 McIntyre et al30 also reported on the long-term treatment of acutely manic or mixed bipolar patients with asenapine compared to olanzapine. Patients completing the initial 12-week core study were eligible to continue in the 40-week extension phase of the study, with flexible dosing of asenapine compared to active treatment with flexible dosing of olanzapine. The primary endpoints of McIntyre et al’s study were safety and tolerability; however, efficacy assessments of response rates and remission rates were reported as secondary endpoints. Of the 308 patients who completed the initial 12-week core study, 218 were enrolled in the extension phase and received at least one dose of medication. Response rates and remission rates at week 52 (both 97.8% for asenapine and both 98.4% for olanzapine) were not found to be statistically significant using observed cases data. Likewise, response and remission rates (both 93.4% for asenapine and both 95.2% for olanzapine) were not statistically significant using endpoint data with LOCF analysis.30 It should be noted that neither of these studies were designed or sufficiently powered to assess the time-to-intervention for relapse or recurrence of mood episodes (a more clinically meaningful measure of an agent’s utility in the maintenance phase of bipolar treatment).
Use in pediatric and adolescent patients with bipolar manic or mixed episodes
In February 2015, two phase III clinical trials (NCT01244815 and NCT01349907) assessing the efficacy and safety of asenapine in pediatric patients with BD were completed.31,32 The results of these studies prompted the FDA to approve asenapine for use in pediatric patients with bipolar I with acute manic or mixed episodes in March of 2015.33 At the time of this writing, these data have not been published.
The first trial (NCT01244815) was a 3-week core trial of the safety and efficacy of asenapine in pediatric patients with BD (presenting with either a mixed or manic mood state) in a hospital or ambulatory setting. A total of 403 patients aged 10–17 years were enrolled in the study. Patients were randomized in a 1:1:1:1 fashion to either placebo, fixed-dose asenapine 2.5 mg BID, fixed-dose asenapine 5 mg BID, or fixed-dose asenapine 10 mg BID. The primary outcome measure of this study was the change in YMRS scores at study day 21 compared to baseline (Table 3). Secondary outcome measures included changes at study day 21 from baseline on the following rating instruments: CGI-BP; CGI-BP mania; CGI-Depression; Children’s Depression Rating Scale, Revised total score; Children’s Global Assessment Scale total score current; and Pediatric Quality of Life Enjoyment and Satisfaction Questionnaire total score. Regarding the primary outcome measure of change in YMRS score, patients receiving asenapine showed statistically significant improvement in symptoms compared to placebo, with more pronounced improvement with doses at or above 5 mg BID. Similarly, for all doses of asenapine, response rate (decrease in YMRS scores >50%) at study day 21 was significantly greater than placebo. Further review of secondary measure outcomes is beyond the scope of this review.
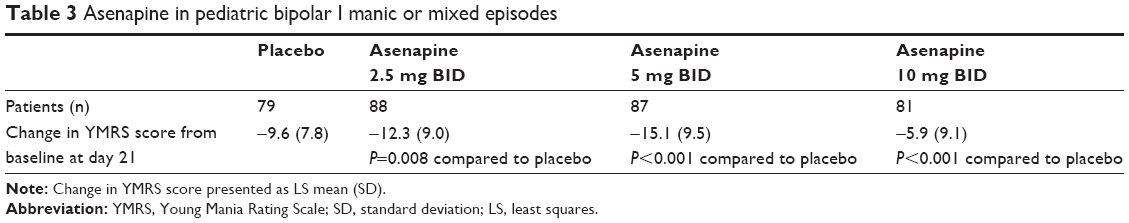 | Table 3 Asenapine in pediatric bipolar I manic or mixed episodes |
The second study (NCT01349907) was a 40-week extension study involving the same study population. The patient doses in the active treatment arms of the core study were all titrated to a dose of asenapine 10 mg BID with flexible dosing thereafter. A total of 322 patients were enrolled in the extension study (asenapine, n=241 and placebo, n=81). The primary outcome measure was the occurrence of any clinical or laboratory adverse event through study day 350. Secondary outcome measures included overall change in YMRS score from baseline to study day 350, remission rate (defined by YMRS ≤12) at study endpoint, time to response failure, as well as change from baseline for all of the aforementioned rating instruments at study day 350. At the time of this writing, no statistical data have been provided for either primary or secondary outcome measures in the extension study.
Safety and tolerability
Second-generation antipsychotics can be associated with significant tolerability issues such as sedation, sexual dysfunction, long-term complications such as metabolic side effects (dyslipidemia, insulin resistance, weight gain), and raised prolactin levels.34
Somnolence
Sedation/somnolence occurring with antipsychotic treatments can negatively affect a patient’s quality of life and cause noncompliance with medications. In BD, however, sedation can be beneficial in managing insomnia and agitation. In a post hoc analysis35 of ten clinical trials in a bipolar monotherapy cohort, the incidence of somnolence with asenapine, olanzapine, and placebo was 23.8%, 26.4%, and 6.4%, respectively. Somnolence occurred early in the course of treatment and was of limited duration. Median time to onset and duration of somnolence with asenapine, olanzapine, and placebo were 1, 2, and 2 days, respectively, and 7, 8.5, and 5 days, respectively. Treatment-emergent somnolence with asenapine was not associated with an increased risk of discontinuation of treatment.35 Overall, somnolence was the single most common adverse event with asenapine treatment. It was described as usually transient with the highest incidence during the 1st week of treatment.36
Metabolic parameters
BD and metabolic syndrome share features of hormonal, immunologic, and autonomic nervous system dysregulation which can make patients more vulnerable to increased risk of metabolic syndrome, diabetes, and vascular disease.37 A meta-analysis of 37 studies (N=6,983) concluded that 37.3% of patients with BD had metabolic syndrome, almost twice the rate in the general population.38 In two 3-week, randomized, double-blind, placebo-controlled, parallel-group studies of asenapine in acute manic or mixed episodes associated with BD, changes from baseline in metabolic parameters, laboratory values, and vital signs were not considered clinically significant.39 In a post hoc analysis of 17 asenapine trials (13 schizophrenia and four bipolar mania), mean changes in total cholesterol, LDL cholesterol, and HDL cholesterol did not differ significantly between asenapine and placebo.40 Triglyceride levels did not change substantially with asenapine, but decreased with placebo (1.8 vs –12.2 mg/dL). In the placebo-controlled population pool, change in fasting glucose was significantly different for asenapine compared with placebo (1.9 vs −1.6 mg/dL). There was no difference in the incidence of clinically relevant changes in hemoglobin A1c. In the olanzapine-controlled population pool, changes in fasting glucose levels were not significantly different between asenapine and olanzapine (2.0 vs 3.3 mg/dL). The incidence of clinically relevant hemoglobin A1c changes was higher with olanzapine. Fasting triglyceride levels ≥200 mg/dL occurred in 22.0% and 10.4% of olanzapine- and asenapine-treated patients, respectively. Overall, asenapine produced minimal changes in serum lipids and glucose (compared with placebo), and is associated with decreases in triglycerides and total cholesterol when compared with olanzapine.40
Body weight and BMI
In a double-blind, placebo-controlled study (N=488), the incidence of clinically significant weight gain (≥7%) with asenapine, placebo, and olanzapine was 7.2%, 1.2%, and 19.0%, respectively, and mean weight change was 0.9, 0.1, and 2.6 kg, respectively.19 In a 40-week extension study, the number needed to harm for clinically significant weight gain with asenapine was 7.30
In a 12-week randomized, placebo-controlled trial of adjunctive asenapine, the incidence of clinically significant weight gain (≥7% from baseline) was 19.5% with asenapine and 5.2% with placebo in the core study, and 36.6% with asenapine and 19.4% with placebo in the extension. BMI shift to the next highest BMI category was 18.8% in the asenapine group and 7.5% in the placebo group in the core study, and 22.0% of the asenapine group and 11.1% of the placebo group in the extension.20 In a post hoc analysis of 17 asenapine trials, a body weight change resulting in an increase of ≥5 BMI units occurred in 5.3% of olanzapine-treated patients vs 1.2% in asenapine-treated patients. Asenapine-treated patients gained consistently more weight than placebo-treated patients (1.2 vs 0.14 kg), but significantly less than olanzapine-treated patients (0.9 vs 3.1 kg).40 Overall, the magnitude of weight gain with asenapine or olanzapine was inversely proportional to baseline BMI (with patients of underweight or normal BMI experiencing more weight gain than overweight or obese patients).40
Extrapyramidal symptoms
In a double-blind placebo-controlled study (N=488), the incidence of extrapyramidal symptoms (EPS) was 10.3%, 3.1%, and 6.8% with asenapine, placebo, and olanzapine, respectively.19 In a 9-week double-blind extension trial (N=504), the incidence of EPS was 10% with placebo, 15% with asenapine, and 13% with olanzapine.41 In a 40-week extension study, the most commonly reported EPS was akathisia, occurring in 11.4% of asenapine and 10.3% of olanzapine patients.30 In a 12-week randomized, placebo-controlled study the incidence of EPS related adverse events was 9.5% with asenapine and 12.0% with placebo during the core study; 22.0% with asenapine and 16.7% with placebo during the extension.20
Cardiovascular adverse effects
Asenapine did not seem to adversely affect cardiac function.20 Effects of asenapine on QTc were assessed in a randomized, placebo-controlled, double-blind, escalating-dose study with doses up to supratherapeutic levels of 20 mg BID. Asenapine appeared to have small effects on the QT interval, less than quetiapine, and below the threshold considered to be clinically significant. Upper bounds of the 95% confidence intervals were 7.5 milliseconds, and no patient experienced a QTc ≥500 milliseconds.42 Even though asenapine has high affinity for the α1 adrenergic receptor, rates of dizziness and syncope were low.35
Hormonal adverse effect
In both the 12-week study and the 40-week extension, changes from baseline in vital signs and most laboratory parameters, including prolactin, were minimal.20 Asenapine appears to have a low propensity to cause prolactin elevation. The prolactin profile of asenapine appears to be similar to that of clozapine.43 This is advantageous over the long term, since hyperprolactinemia can increase the risk of sexual dysfunction, osteoporosis, hip fractures, galactorrhea, gynecomastia, breast cancer, and pituitary tumors.33
Other side effects
Asenapine has no appreciable affinity for muscarinic receptors and induces few anticholinergic side effects.44 Side effects occurring twice as frequently with asenapine as placebo (and in >10% of subjects) included depression, dizziness, nausea, parkinsonism, tremor, and constipation.30 Adverse effects reported by 5% or more of patients and at twice the rate of placebo were sedation, somnolence, depression, constipation, oral hypoesthesia, irritability, and dyskinesia.20
Patient-focused perspectives
Asenapine may be cost-effective and be associated with improved quality of life compared to some other antipsychotics, particularly in the treatment of mixed episodes. In Italian and British pharmacoeconomic analyses, asenapine was associated with fewer costs than olanzapine in the following mixed episodes. Asenapine use was also associated with the prevention of psychiatric hospitalizations, generating a better quality of life, and more quality-adjusted life-years.31,45,46 However, cost benefits were not found in one Spanish study which researched a 6-month naturalistic treatment with adjunctive asenapine vs other adjunctive antipsychotics.23 In a post-hoc analysis of two acute trials in BD-I patients with mixed episodes, asenapine users found improved health-related quality of life compared to olanzapine and placebo-treated patients.47
Most patients treated with asenapine, especially those with manic/mixed symptoms, report satisfaction with their treatment.22
Conclusion
Asenapine is a second-generation antipsychotic medication which has demonstrated efficacy in adults and children with bipolar manic and mixed episodes. While secondary analyses suggest utility in bipolar depression and bipolar maintenance, prospective trials in these therapeutic domains still need to be conducted. Based on clinical trial findings, somnolence appears to be the most common side effect associated with asenapine. Weight gain propensity appears relatively modest, as do effects on serum glucose and lipids. Symptom improvement, particularly in individuals with mania, appears associated with better quality of life.
Overall, asenapine is a useful addition to the treatment armamentarium for patients with BD. However, additional research is needed to better understand its effects in all phases of bipolarity, with long-term use and across the life-span.
Disclosure
Author SR has received funding from the Canadian Institutes of Health Research (CIHR) Fellowship Award. Author MS has received research grants from Pfizer, Merck, Ortho-McNeil Janssen, Janssen, Reuter Foundation, Woodruff Foundation, Reinberger Foundation, National Institute of Health (NIH), and Centers for Disease Control and Prevention (CDC). MS has acted as a consultant to Bracket, Prophase, Otsuka, Pfizer, Amgen, and Sunovion, and has received royalties from Springer Press, Johns Hopkins University Press, Oxford Press, UpToDate, and Lexicomp. MS has also participated in continuing medical education (CME) activities run by the American Physician’s Institute, MCM Education, and CMEology. The other authors report no conflicts of interest.
References
Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1–44. | ||
Whiteford HA, Degenhardt L, Rehm J, et al. Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. Lancet. 2013;382(9904):1575–1586. | ||
Ratnasingham S, Cairney J, Manson H, Rehm J, Lin E, Kurdyak P. The burden of mental illness and addiction in Ontario. Can J Psychiatry. 2013;58(9):529–537. | ||
Lala SV, Sajatovic M. Medical and psychiatric comorbidities among elderly individuals with bipolar disorder: a literature review. J Geriatr Psychiatry Neurol. 2012;25(1):20–25. | ||
Forte A, Baldessarini RJ, Tondo L, Vazquez GH, Pompili M, Girardi P. Long-term morbidity in bipolar-I, bipolar-II, and unipolar major depressive disorders. J Affect Disord. 2015;178:71–78. | ||
Gildengers A, Tatsuoka C, Bialko C, et al. Correlates of disability in depressed older adults with bipolar disorder. Cut Edge Psychiatry Pract. 2013;2013(1):332–338. | ||
Geddes JR, Goodwin GM, Rendell J, et al. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet. 2010;375(9712):385–395. | ||
Loebel A, Cucchiaro J, Silva R, et al. Lurasidone monotherapy in the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):160–168. | ||
Parikh SV, Zaretsky A, Beaulieu S, et al. A randomized controlled trial of psychoeducation or cognitive-behavioral therapy in bipolar disorder: a Canadian Network for Mood and Anxiety treatments (CANMAT) study [CME]. J Clin Psychiatry. 2012;73(6):803–810. | ||
Jones SH, Smith G, Mulligan LD, et al. Recovery-focused cognitive-behavioural therapy for recent-onset bipolar disorder: randomised controlled pilot trial. Br J Psychiatry. 2015;206(1):58–66. | ||
Frank E, Kupfer DJ, Thase ME, et al. Two-year outcomes for interpersonal and social rhythm therapy in individuals with bipolar I disorder. Arch Gen Psychiatry. 2005;62(9):996–1004. | ||
Ghaemi SN, Hsu DJ, Thase ME, et al. Pharmacological Treatment Patterns at Study Entry for the First 500 STEP-BD Participants. Psychiatr Serv. 2006;57(5):660–665. | ||
Forest Pharmaceuticals I. Highlights of Prescribing Information, Saphris (asenapine) sublingual tablets; 2009. Available from: http://pi.actavis.com/data_stream.asp?product_group=1908&p=pi&language=E. Accessed November 12, 2015. | ||
Drug Approval Package, Saphris (Asenapine) Tablet [webpage on the Internet]. MD, USA: US Food and Drug Administration; 2009. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022117s000TOC.cfm. Accessed June 7, 2015. | ||
Gerrits M, de Greef R, Peeters P. Effect of absorption site on the pharmacokinetics of sublingual asenapine in healthy male subjects. Biopharm Drug Dispos. 2010;31(5–6):351–357. | ||
Citrome L. Asenapine for schizophrenia and bipolar disorder: a review of the efficacy and safety profile for this newly approved sublingually absorbed second-generation antipsychotic. Int J Clin Pract. 2009;63(12):1762–1784. | ||
Jardemark K, Marcus MM, Shahid M, Svensson TH. Effects of asenapine on prefrontal N-methyl-D-aspartate receptor-mediated transmission: involvement of dopamine D1 receptors. Synapse. 2010;64(11):870–874. | ||
McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673–686. | ||
McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine in the treatment of acute mania in bipolar I disorder: a randomized, double-blind, placebo-controlled trial. J Affect Disord. 2010;122(1–2):27–38. | ||
Szegedi A, Calabrese JR, Stet L, Mackle M, Zhao J, Panagides J. Asenapine as adjunctive treatment for acute mania associated with bipolar disorder: results of a 12-week core study and 40-week extension. J Clin Psychopharmacol. 2012;32(1):46–55. | ||
Baruch Y, Tadger S, Plopski I, Barak Y. Asenapine for elderly bipolar manic patients. J Affect Disord. 2013;145(1):130–132. | ||
Sajatovic M, Dines P, Fuentes-Casiano E, et al. Asenapine in the treatment of older adults with bipolar disorder. Int Journal Geriatr Psychiatry. 2015;30(7):710–719. | ||
Grande I, Hidalgo-Mazzei D, Nieto E, et al. Asenapine prescribing patterns in the treatment of manic in- and outpatients: Results from the MANACOR study. Eur Psychiatry. 2015;30(4):528–534. | ||
Cazorla P, Zhao J, Mackle M, Szegedi A. Asenapine effects on individual Young Mania Rating Scale items in bipolar disorder patients with acute manic or mixed episodes: a pooled analysis. Neuropsychiatr Dis Treat. 2013;9:409–413. | ||
Szegedi A, Zhao J, McIntyre RS. Early improvement as a predictor of acute treatment outcome in manic or mixed episodes in bipolar-1 disorder: a pooled, post hoc analysis from the asenapine development program. J Affect Disord. 2013;150(3):745–752. | ||
Azorin JM, Sapin C, Weiller E. Effect of asenapine on manic and depressive symptoms in bipolar I patients with mixed episodes: results from post hoc analyses. J Affect Disord. 2013;145(1):62–69. | ||
Berk M, Tiller JW, Zhao J, Yatham LN, Malhi GS, Weiller E. Effects of asenapine in bipolar I patients meeting proxy criteria for moderate-to-severe mixed major depressive episodes: a post hoc analysis. Journal Clin Psychiatry. 2015;76(6):728–734. | ||
McIntyre RS, Cohen M, Berk M, Zhao J, Weiller E. DSM-5 mixed specifier for manic episodes: evaluating the effect of depressive features on severity and treatment outcome using asenapine clinical trial data. J Affect Disord. 2013;150(2):378–383. | ||
Szegedi A, Zhao J, van Willigenburg A, Nations KR, Mackle M, Panagides J. Effects of asenapine on depressive symptoms in patients with bipolar I disorder experiencing acute manic or mixed episodes: a post hoc analysis of two 3-week clinical trials. BMC Psychiatry. 2011;11:101. | ||
McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine for long-term treatment of bipolar disorder: a double-blind 40-week extension study. J Affect Disord. 2010;126(3):358–365. | ||
Merck Sharpe and Dohme Corporation. Efficacy and Safety of Asenapine Treatment for Pediatric Bipolar Disorder. Available from: https://clinicaltrials.gov/ct2/show/NCT01244815?term=asenapine+pediatric+bipolar&rank=1. NLM identifier: NCT01244815. Accessed July 16, 2015. | ||
Merck Sharpe and Dohme Corporation. Extension Study of Asenapine (NCT01244815) for Pediatric Bipolar Disorder. Available from: https://clinicaltrials.gov/ct2/show/NCT01349907?term=asenapine+pediatric+bipolar&rank=2. NLM identifier: NCT01349907. Accessed July 16, 2015. | ||
U.S. Food and Drug Administration [database on the Internet]. New Pediatric Labeling Information Database – Detail: Asenapine. Available from: http://www.accessdata.fda.gov/scripts/sda/sdDetailNavigation.cfm?sd=labelingdatabase&id=14F7A847BCD30979E053564DA8C00FF5&rownum=15. Accessed July 16, 2015. | ||
Young AH, Altamura AC, Gonzalez-Pinto AM, Millet B, Wiedemann K. Use of asenapine in clinical practice for the management of bipolar mania. J Psychopharmacol. 2013;27(4 Suppl):3–13. | ||
Gao K, Mackle M, Cazorla P, Zhao J, Szegedi A. Comparison of somnolence associated with asenapine, olanzapine, risperidone, and haloperidol relative to placebo in patients with schizophrenia or bipolar disorder. Neuropsychiatr Dis Treat. 2013;9:1145–1157. | ||
Citrome L. Asenapine review, part II: clinical efficacy, safety and tolerability. Expert Opin Drug Saf. 2014;13(6):803–830. | ||
Taylor V, MacQueen G. Associations between bipolar disorder and metabolic syndrome: A review. J Clin Psychiatry. 2006;67(7):1034–1041. | ||
Vancampfort D, Vansteelandt K, Correll CU, et al. Metabolic syndrome and metabolic abnormalities in bipolar disorder: a meta-analysis of prevalence rates and moderators. Am J Psychiatry. 2013;170(3):265–274. | ||
McIntyre RS. Asenapine: a review of acute and extension phase data in bipolar disorder. CNS Neurosci Ther. 2011;17(6):645–648. | ||
Kemp DE, Zhao J, Cazorla P, et al. Weight change and metabolic effects of asenapine in patients with schizophrenia and bipolar disorder. J Clin Psychiatry. 2014;75(3):238–245. | ||
McIntyre RS, Cohen M, Zhao J, Alphs L, Macek TA, Panagides J. Asenapine versus olanzapine in acute mania: a double-blind extension study. Bipolar Disord. 2009;11(8):815–826. | ||
Chapel S, Hutmacher MM, Haig G, et al. Exposure-response analysis in patients with schizophrenia to assess the effect of asenapine on QTc prolongation. J Clin Pharmacol. 2009;49(11):1297–1308. | ||
Peuskens J, Pani L, Detraux J, De Hert M. The effects of novel and newly approved antipsychotics on serum prolactin levels: a comprehensive review. CNS Drugs. 2014;28(5):421–453. | ||
Shahid M, Walker GB, Zorn SH, Wong EH. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol. 2009;23(1):65–73. | ||
Caresano C, Di Sciascio G, Fagiolini A, et al. Cost-effectiveness of asenapine in the treatment of patients with bipolar I disorder with mixed episodes in an Italian context. Adv Ther. 2014;31(8):873–890. | ||
Sawyer L, Azorin JM, Chang S, et al. Cost-effectiveness of asenapine in the treatment of bipolar I disorder patients with mixed episodes. J Med Econ. 2014;17(7):508–519. | ||
Michalak EE, Guiraud-Diawara A, Sapin C. Asenapine treatment and health-related quality of life in patients experiencing bipolar I disorder with mixed episodes: post-hoc analyses of pivotal trials. Curr Med Res Opin. 2014;30(4):711–718. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.