")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Scientific Rationale for Waiving Clinical Efficacy Testing of Biosimilars
Authors Niazi S
Received 27 June 2022
Accepted for publication 9 August 2022
Published 24 August 2022 Volume 2022:16 Pages 2803—2815
DOI https://doi.org/10.2147/DDDT.S378813
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Sarfaraz Niazi
Department of Pharmaceutical Sciences, College of Pharmacy, University of Illinois, Chicago, IL, USA
Correspondence: Sarfaraz Niazi, Email [email protected]
Abstract: After 18 years and the administration of billions of doses, there is little doubt about biosimilars’ safety and efficacy. Yet, only 14 molecules in the EU and 9 in the US are available as biosimilars, among the 200+ targets, due mainly to the high development cost attributed to clinical efficacy testing after extensive analytical assessment, nonclinical testing, and clinical pharmacology comparisons. So far, none of the hundreds of clinical efficacy testing has failed because it cannot fail due to its lack of sensitivity for multiple reasons, as argued in this paper. This analysis is unique since biosimilars are the first category of products that are put to comparative testing as if these were new biological drugs. Clinical efficacy testing used to overcome differences in the analytical, nonclinical, and clinical pharmacology comparisons can lead to the approval of unsafe products. Only recently the regulatory agencies have begun to talk about this risk and shown their willingness to waive these studies. However, a clear change in the regulatory guidelines is required to change the mindset of all biosimilar stakeholders to bring a pivotal change in the availability of affordable biosimilars.
Keywords: biosimilars, clinical efficacy testing, BPCIA, biosimilarity, FDA, EMA, clinical equivalence
Introduction
So far, 84 biosimilars have been approved in Europe,1 including hormones, interferons, colony-stimulating, antibodies, and necrosis factor inhibitors. In addition, the FDA has approved 38 products2 containing nine molecules with the same product classes except for parathyroid hormone and FSH; 98 products are approved in India,2,3 in Latin America, 4 40 in Australia, 5 and 26 in Canada.6
The European Public Assessment Report (EPAR) program of the EMA7 and the FDA Approved Drugs Database8 provide access to regulatory filings along with the details of the Agency’s communications with the developers to enable an understanding of the mindset of both the developers and the agencies. The reporting of safety and efficacy of biosimilars approved in the US are available in the FDA’s “Adverse Event Reporting System Database”9 and the European EudraVigilance: “The European Database of Suspected Drug Reaction Reports”10 provides current information by the product name and the type of adverse event and the history. While the safety and efficacy of new drugs are established in smaller clinical trials, the real-time market data confirms these attributes when millions of patients are exposed to the drug. The biosimilars with billions of doses administered have shown an impeccable record of safety and efficacy.
This review is based on the analysis of reported clinical trials conducted to support the registration of biosimilars in the US and EU, the changes in the regulatory guidance over the past 18 years, and recommendations to the regulatory agencies on how to improve the approval process. The developers are presented with scientific arguments to request clinical efficacy testing waivers.
Testing drugs in patients is the gold standard for developing new drugs. But for drugs whose efficacy and safety have already well-established, such testing is redundant as long as the products are similar at the molecular level. It works well for chemical drugs as there is no dispute about their chemical structure; for biological drugs, the structure is a variable requiring side-by-side comparisons with the reference product to ensure that the variability is comparable. Given the complexity of the structure, subtle differences may not be identified. Unlike chemical drugs, such subtle differences may result in safety issues, requiring additional testing of disposition kinetic, pharmacodynamic, and immunogenicity comparisons with the reference product. Animal testing is useless as most species do not have the receptors pivotal to the action of biological drugs, even though regulatory agencies continue to suggest this testing. However, it is certain that if a developer asks for animal testing waivers, these will be granted.
Biosimilars are put to clinical efficacy testing after meeting all other merits described above to assure that there are “no clinically meaningful differences” compared with the designated reference product.11 Many biosimilars have been approved with efficacy testing, but only if reliable pharmacodynamic parameters can be compared in clinical pharmacology studies in healthy subjects or patients.
The clinical efficacy trials constitute more than 70% of the cost of developing biosimilars, estimated to be around USD 150–300 million.12 If these studies could add any value to the assessment of safety and efficacy, these costs would be justified. However, being the least sensitive testing, such studies raise ethical concerns codified in the US 21 CFR 320.25(a)(13), the universal belief that “No unnecessary human testing should be performed.”13
Why do the regulatory agencies require or suggest clinical efficacy testing if these studies do not add value to the safety or efficacy evaluation? Until the era of generic chemical drugs, there were just branded drugs that had undergone extensive testing; the generic medicines that were chemically equivalent required a reduction in testing. In most cases, the efficacy testing was considered redundant. Yet, all generic drugs were required to demonstrate bioequivalence in healthy subjects until a few years later, it was realized that drugs with high solubility and high permeability do not need to be tested for bioequivalence leading to the allowance of waivers of bioequivalence testing. There is still a need for these guidelines to change, but that is a different topic. So, when the biosimilars arrived, the history was repeated. However, this time the issue was less on bioequivalence and more on efficacy since the molecular structure was not identical. Regulatory agencies are known to require testing often for an abundance of caution. However, we have clearly understood that clinical efficacy testing of biosimilars is useless because we are trying to find a needle in the haystack when there is no haystack or haystack. The main issue is the mindset of the stakeholders, particularly the prescribers, who are used to seeing such trials to gain confidence in a new product. It is difficult for many to visualize that comparative testing when the two products are supposed to be the same cannot fail because of study size constraints, arbitrary equivalence requirements, and the well-known dose-response variabilities. Since this is the first time we have faced this type of equivalence testing, it is taking longer to act. These walls of resistance are falling slowly, but they need to come down quickly to enable biosimilars to find their place in assuring the affordability of lifesaving drugs.
Background
If a product is chemically and biologically equivalent, it is correct to assume it will be clinically equivalent.14 Biosimilars have an analytically similar structure, the same dose, the same concentration (or strength), the same mode of action, and the same route of administration. However, they may have a different formulation, as described in 2019, the Biological Price Competition and Innovation Act.15 The regulatory plan to approve biosimilars assures it has “no clinically meaningful differences”11 with its designated reference product.
US
There are over 100 biosimilar programs enrolled with the FDA.16 To expedite the approval process, the FDA has taken several significant steps. The FDA has created two new guidelines, the extension of the Q&A presentations17 and the third revised draft guidance18 titled “New and Revised Draft Q&As on Biosimilar Development and the BPCI Act.” The FDA also updated The Purple Book FAQ section.18
The BPCIA states15 that the
Secretary may determine, in the Secretary’s discretion, that an element described in clause (i) (I) [the biosimilar testing] is unnecessary in an application submitted under this subsection.
The FDA has subtly implemented this change in its new biosimilar guidance. However, unlike the EMA, MHRA, or WHO, the FDA is bound by the BPCIA legislation that further states that
an application submitted under this subsection shall include information demonstrating that the biological product is biosimilar to a reference product based upon data derived from analytical studies, animal studies, and clinical studies.
The new education material includes the phrase “in addition to analytical studies, other studies that may be needed”, not shall be, as stated in the BPCIA.
Biosimilars “may be approved based on PK and PD biomarker data without a comparative clinical study with efficacy endpoint(s)”, said the FDA.19,20 These studies are more sensitive than the clinical efficacy testing, as demonstrated in the comparison of testing of clinical efficacy with endpoint(s) for GCSF;21 the FDA has approved filgrastim-aafi, filgrastim-sndz, filgrastim-ayow (author’s), pegfilgrastim-jmdb, pegfilgrastim-cbqv, pegfilgrastim-pbbk (author’s) and epoetin alfa-epbx without any efficacy testing. The FDA now fully acknowledges the role of PD markers in establishing biosimilarity.22–24
Comparison of biomarkers is more objective and more sensitive, and the FDA guidance and the “Biosimilars Action Plan” includes a selection of strategies to identify PD biomarkers.20 We can use multiple biomarkers from broader epigenome, transcriptome, and proteome choices. How should we test the drugs that do not offer pharmacodynamic markers? Suggestions are made on using a clinical efficacy study with reliable markers instead of efficacy, but these, too, are highly variable and do not reduce the testing burden. Remember, the FDA allowed waivers of bioequivalence testing after research papers were published to question the wisdom of such testing. The FDA should come up with a clear explanation for why such studies are redundant and unacceptable to demonstrate biosimilarity. Though developers can challenge such study requirements but inquire if the FDA finds any “residual uncertainty”, and if so, it would be resolved by conducting a single efficacy testing. The latter part is the weakest FDA position point.
Changes in the FDA guidelines based on approved legislation are impossible until the rules are changed, and that requires a lot of political work.
EU
In 2001, much of the EU’s directive-based legislation concerning the regulation of medicines was codified as Directive 2001/83/EC. The EMA has issued concept papers, draft guidance, and public scientific workshops. The EMA’s Committee for Medicinal Products for Human Use (CHMP) has also issued product class-specific guidance for recombinant erythropoietin, granulocyte-colony stimulating factor, recombinant human soluble insulin, low-molecular-weight heparins, somatropin, and recombinant interferon alfa.25 However, EMA has announced that they intend not to issue more specific biosimilar guidelines but instead prefer to give tailored advice on a case-by-case basis.25 This change in the EMA policy came from the FDA statements that such guidelines can misdirect the development of biosimilars.
The EMA has brought several changes to its guidelines; a decade ago, EMA would require efficacy testing even when there were reliable pharmacodynamic markers available; now, there is some harmony with the FDA, allowing waivers of some clinical efficacy testing of cytokines. Unlike the FDA, where a legislative change is required, it is relatively easier for the EMA to make such changes, though they still have to go through various committee discussions.
WHO
The World Health Organization (WHO) is not a regulatory authority, but it is mandated to support regulatory authorities in its 194 Member States. The WHO guidelines on evaluating biosimilars26 suggest to the National Regulatory Agencies (NRAs) the principles for approving biosimilars.
In 2019, the WHO Expert Committee on Biological Standardization (ECBS) considered that a more tailored and potentially reduced clinical data package might be acceptable in cases where the available scientific evidence supported this. In addition, the committee endorsed the review of current scientific evidence to consider updating the Guidelines to provide more flexibility and clarity. Thus, the WHO reviewed scientific evidence and experience to identify issues/cases for further reducing non-clinical and clinical data. The progress was reported to the committee in 2020 (72nd and 73rd).27 It has resulted in additional suggestions on evaluating biosimilar monoclonal antibodies (mAbs) and an expanded Q&A document.
In April 2022, the WHO published28 a revised guideline based on the 22 comments received. While the newest guideline and suggestions made by the WHO represent the views of global regulatory agencies, it still falls short of adding clarity to the need for clinical efficacy testing. Many misconceptions like using non-inferiority testing, not allowing entry of biosimilars until after the safety of the reference product are established in market analysis, using diverse sources of the reference product, and possibly using clinical efficacy testing to justify analytical differences.
The WHO statement,
It may also be prudent not to waive the efficacy and safety study when the reference product has common or unpredictable serious adverse effects that cannot be merely explained by exaggerated pharmacological action
is a classic example of the misconceptions. Unfortunately, it is not likely that the 194 NRAs will agree on a significant change because of their mindset about the value of such testing, not realizing the uniqueness of this testing and why it does not serve any purpose.
UK
The MHRA, which broke off from EMA after Brexit, took several years to finalize its biosimilar guideline on 14 May 2022: “although each biosimilar development needs to be evaluated on a case-by-case basis, it is considered that, in most cases, a comparative efficacy trial may not be necessary if sound scientific rationale supports this approach. Therefore, a well-argued justification for the absence of an efficacy trial should be appended to CTD Module 1 of the submitted application.29 This guideline has taken a clearer and more definite approach to the issues of animal testing and clinical efficacy evaluation. The UK made this decision since there are no multiple vested interests in the EMA or WHO, nor the legislative hurdles as in the US. It is indeed a remarkable advance in the regulatory plan that should be adopted by all agencies.
ROW
Most countries follow the guidelines discussed above; however, the WHO members are more inclined to follow them unless they are more affluent, like Saudi Arabia, where the FDA/EMA guidelines apply. Many countries treat biosimilars like generic chemical products, with no clinical testing. In most cases, clinical testing is suggested for a fixed number of patients. Recently, the concept of biological API (active pharmaceutical ingredient) has risen, where companies import the drug substance and finish it locally.
Action Plan
In the early days of biosimilar registrations, it was taken for granted by the developers that proof of clinical efficacy was required, regardless of the nature of the product. However, the text of the BPCIA:15
If there is residual uncertainty about biosimilarity after conducting structural analyses, functional assays, animal testing, human PK and PD trials, and the clinical immunogenicity assessment, the sponsor should then consider what additional clinical data may be needed to address that uncertainty (section VII.D.3) adequately
had provided hints for alternate possibilities that were ignored. The term “additional clinical data” need not come from an efficacy trial, as clarified by the FDA in its “Biosimilar Action Plan.”15 This could well be another PK/PD study. Thus, after completing an analytical assessment, nonclinical testing, and clinical pharmacology trials, developers should inquire with the FDA or EMA if any “residual uncertainty” will call for additional clinical data submission. Instead, the developers were ready to conduct these studies to market their products as new biological drugs.30
EMA also states,
generally, clinical data aim to address slight differences shown at previous steps and to confirm the comparable clinical performance of the biosimilar and the reference product.
This statement creates confusion about how to define “slight difference.” An immunogenic response can trigger with slight modifications, leading to ADAs that might reduce the product’s effectiveness. Therefore, using clinical efficacy data to support differences in “slight differences” or “residual uncertainty” is inappropriate because there is no basis for this justification.31
Clinical Pharmacology Trials
Clinical testing at the pharmacology stage is pivotal to demonstrating biosimilarity. These trials are like bioequivalence testing. Even though biological drugs are administered mainly by intravenous and other parenteral routes, bioequivalence testing is needed to extend the analytical assessment. In this testing, we learn how the body views the molecule and how the molecules interact with the body. There can be differences in the disposition kinetics profile in many cases where these drugs are administered subcutaneously. This confirms that the best tool to establish biosimilarity is through PK/PD trials. The developers have often conducted multiple clinical pharmacology trials to establish immunogenicity profiles. The data reported in clinicaltrials.gov for Phase 1 and 2 studies list 370 studies32 as developers have reported numerous trials in the regulatory filings. In some instances, these trials were conducted in patients to avoid the risk of inducing immunogenicity in the healthy population, where the pharmacokinetic or pharmacodynamic responses are disease-dependent or where the ADA is a serious concern, as in the case of adalimumab33 or idarucizumab.34 Antidrug antibodies (ADAs), and more particularly, neutralizing antibodies (NAbs), can impact efficacy because capturing antibodies reduces the available dose. The immune complexes, size, and structure depend on ratio, concentration, and the epitope.35
The FDA has recognized the role of ADAs in assessing the immunogenicity of insulins and now allows waiver since immunogenicity does not alter the pharmacokinetic profile of insulins. The FDA36 guidance on Clinical Immunogenicity Considerations for Biosimilar and Interchangeable Insulin waives immunogenicity testing if it does not change the PK profile; the guidance associated with insulins should apply to other products is no PK dependence on immunogenicity. But this will require filing a petition to the FDA to secure waivers. Additionally, in vitro immunogenicity testing plans are under development, leading to the possibility that no immunogenicity testing will be required at the clinical level.
While the PK/PD studies represent a smaller cost of clinical trials, these can also be reduced significantly37 in a parallel design to assess all parameters. Novel PK modeling methods apply best to biological drugs because of their receptor binding properties and tissue distribution relevance. PK modeling using the distribution volume as a function of time is a novel approach that can bring relevance to receptor binding profile comparisons.37–40 The Biosimilars Action Plan of the FDA also suggests in silico modeling.
Clinical Efficacy Trials
The “Biologics Price Competition and Innovation Act” (BPCIA) of 200915 describes when clinical trials are required:
(cc) a clinical study or trials (including the assessment of immunogenicity and pharmacokinetics or pharmacodynamics) that are sufficient to demonstrate safety, purity, and potency in one or more appropriate conditions of use for which the reference product is licensed and intended to be used and for which licensure is sought for the biological product.
The EMA presents its position on testing immunogenicity using alternate methods as follows:41
… ongoing consideration should be given to the use of emerging technologies (novel in silico, in vitro, and in vivo models), which might be used as tools during development or for the first estimation of risk for clinical immunogenicity. For example, in vitro assays based on innate and adaptive immune cells could help reveal cell-mediated responses.
Most of these guidelines make a mistake, suggesting that the nature and extent of clinical evidence required should depend on the similarity shown by physicochemical assays and PK/PD profiles. The fact is that there is no degree of similarity since all testing is done under an acceptance protocol–a study either meets or does not meet the requirement. Next, no data are available to correlate the differences observed with the clinical response. For example, it is impossible to extrapolate an immune response to differences in physicochemical properties, and so is the efficacy since the biological drugs have a wide dose-response relationship. Conducting more extensive clinical testing does little to add to the value of a smaller clinical trial.
Indications
Biosimilars are allowed extrapolation of all indications of the reference product. However, if clinical efficacy testing is suggested to overcome a residual uncertainty, can a single study for one indication resolve the uncertainty, even if the mode of action is the same? Regulatory agencies have no answer. For example, TNF-alfa blockers can be tested in a smaller psoriasis study while receiving approval for rheumatoid arthritis indication. Should a biosimilar be tested in all indications? This is where the logic of evaluating biosimilars fails.
Interchangeability
Only the US has two classes of biosimilars; standard biosimilars that have “no clinically meaningful difference” with its reference product; and an upgraded version, interchangeable biosimilars when the standard biosimilars are further tested in a three-cycle switching and interchanging with the reference product to assure that there is no reduced efficacy and no enhanced immunogenicity, to allow the pharmacist to substitute them at the dispensing level if the State laws allow. The first interchangeable products approved by the FDA are adalimumab, ranibizumab, and insulin glargine,42, which will have a 12-month exclusivity before another product is given the interchangeable status.
There is no scientific argument to support this biosimilar classification, presenting one of the weakest points in the FDA guideline. However, removing this category of a biosimilar will require a legislative change as advised by the author to the US Congress.43
Statistical Model
There are two types of clinical trials; equivalence and non-inferiority. Both FDA and EMA discourage non-inferiority trials because of the likelihood of approving a more effective biosimilar that assumingly has higher side effects since the two are proportional to most biological drugs.44 However, it is not uncommon for large pharma to conduct non‐inferiority testing, planning to convince the prescribers that their product is superior, as reported for Abasaglar and Lusduna (insulin glargine) and Truxima, Rituzena, and Blitzima.45 This observation shows how biosimilar developers are trying to prove the superiority of their products when it is a sign of inferiority. This should be discouraged and disallowed if these data are submitted for the approval of a product. Unfortunately, not much can be done if these studies are conducted aside from the regulatory submission.
The statistical analysis should be prespecified, as with other clinical research protocols. Equivalence margins are essential for the test decision and must be predetermined because biosimilar trials are equivalence (or non-inferiority). Since there are many different examined indications in effectiveness studies and the permissible difference must indicate a difference that is not clinically significant and is disease-specific, it is impossible to utilize a uniform margin as is the case in bioequivalence trials. Equivalence margins in effectiveness trials typically vary from 0.3% to 15%, and in certain trials, up to 45% with no justification. In specific EPAR files, no margins were disclosed because there are no rules or guidelines. In non-inferiority trials, it is noteworthy that the margin structure is frequently created to maintain at least 50% of the treatment benefit.50
Bayes Probability
What is the probability of a product being clinically equivalent if the efficacy testing meets the acceptance criteria? The Bayes hypothesis46 allows this calculation based on three factors: 1-β = probability of a “true positive”, ie, correctly rejecting the null hypothesis, also known as the study power that is set at 80%; α = probability of a Type I error, known as a “false positive”, that is set at 5%.; and, the probability of biosimilars being equivalent. Figure 1 shows the calculated Bayesian probability vs equivalence expected of a biosimilar. Even if the biosimilars are equivalent only 10% of the time, the likelihood that a clinical efficacy will report as meeting equivalence is 64% of the time. At the probability of 90%, the study will conclude a product is a biosimilar when it is not.
 |
Figure 1 Equivalence as Bayes probability based on the α=0.05 and β=0.2. |
Study Size
The study size is calculated based on the limitations described above for Bayesian testing and on the intersubjective variability in a comparator mode.47 The study sizes are much smaller in a placebo-controlled trials than in a comparator trial, as evidenced by the analysis presented in Box 1, listing the number of subjects enrolled in biosimilar clinical efficacy trials of biosimilars. The number of enrollees for the reference product was chosen for one indication and for the smallest size to demonstrate that a placebo-controlled study need not be large enough to provide the required study power. In addition, studies not intended for US or EU submission are identified in Box 1. None of these studies failed; the likelihood that the large sample size will reveal any clinical relevance between the biosimilar candidate and the reference product is minuscule.
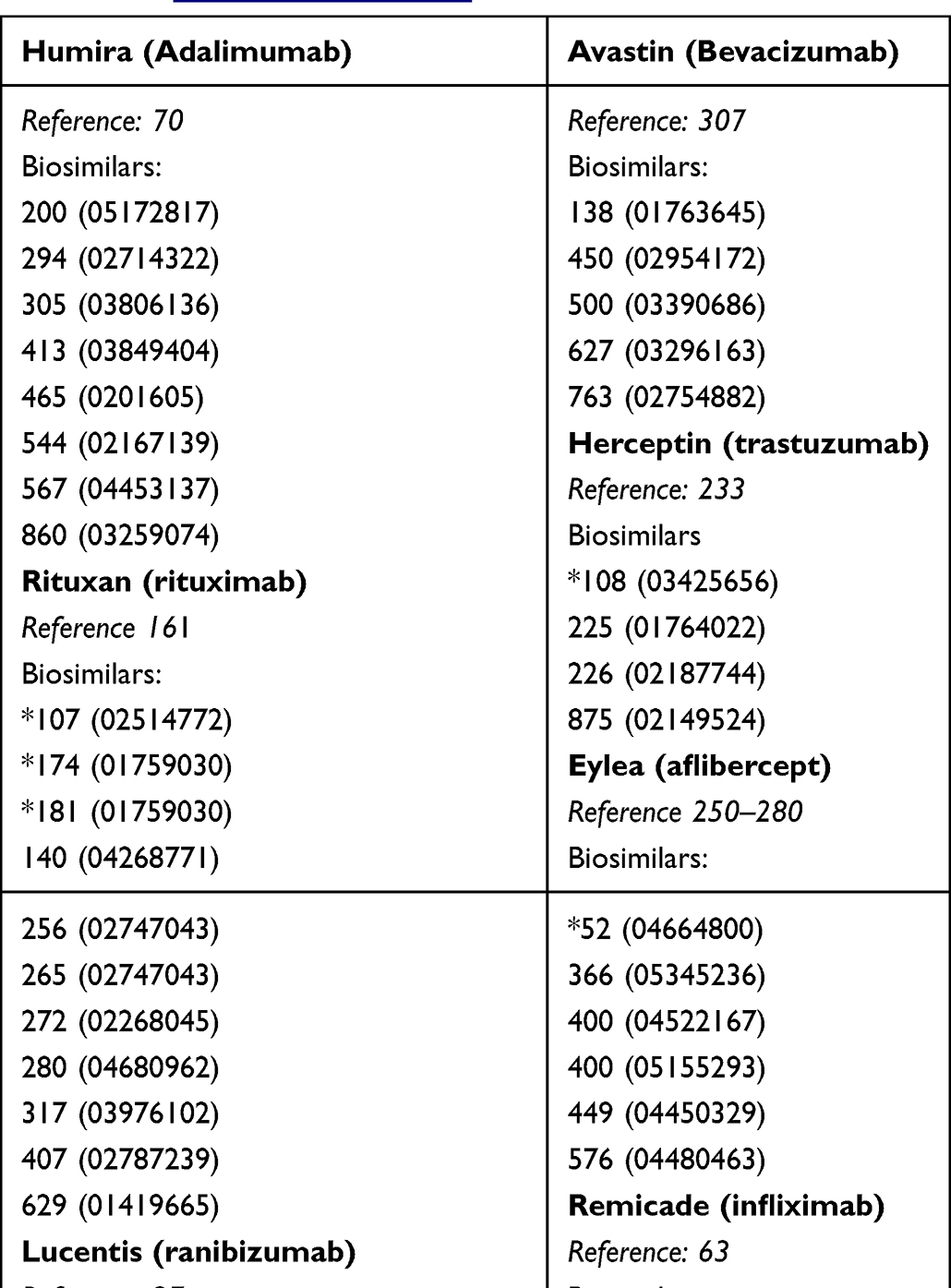 |
Box 1 Number of Subjects Enrolled in Clinical Efficacy Testing of Biosimilars (www.clinicaltrials.gov) |
The study size calculations are further complicated by unique disease situations. As an example, there were 222 patients enrolled on H0649g48 (trastuzumab reference) with a goal of 200 patients. Of these 223 patients, 213 patients were treated with Herceptin. Ten patients did not receive Herceptin for multiple reasons. Of those patients treated with Herceptin, 117 missed 253 doses for various reasons, including 15 for death and 11 for refusing to take the drug.
Current listings on clinicaltrials.gov provide a broad view of the status of biosimilar clinical trials. There are 58 completed clinical efficacy trials and 38 ongoing clinical efficacy trials. None of the completed trials failed to meet the acceptance criteria.
Insensitivity and Lack of Specificity
Since clinical efficacy testing does not have sufficient sensitivity, other alternate options to confirm the safety and efficacy include:
- Using established PD parameters such as hematocrit for erythropoietin or WBC for filgrastim products to compare the efficacy, and thus no comparative clinical efficacy testing is required. Similarly, PD parameters in any product can be presented as an alternative to efficacy testing. FDA suggests that “biosimilars may be approved based on PK and PD biomarker data without a comparative clinical study with efficacy endpoint(s)” to reduce the time cost significantly when tested in healthy subjects20 while providing a highly reliable evaluation. When testing filgrastim biosimilars, the area under the curve of the neutrophil profile is found to be more sensitive, as it is a more objective test.22 The FDA has approved filgrastim, pegfilgrastim, and erythropoietin biosimilars without requiring clinical efficacy data. The developers can now be confident that no efficacy studies will be necessary if suitable PD biomarkers are available for these and other products; using multiple biomarkers provides a stronger argument for establishing biosimilarity.49,50 PD markers are also a major topic in the FDA’s Biosimilars Action Plan.20
- The long half-life of the antibody is due to the Fc-FcRn interaction, which is essential for preventing antibody catabolism and enhancing antibody retention in the body. In addition, IgG antibodies can activate the host immune system by engaging with FcRs expressed on diverse effector cells. These molecular characteristics significantly impact therapeutic antibodies’ PK and PD characteristics, giving them several therapeutic benefits such as a long half-life, high potency, and little off-target toxicity.51 Since the reaction is not always linear, and a comparative PK profile will serve as a more accurate indicator of efficacy than a real-time efficacy.
- ADA formation is a process influenced by the interaction of factors related to the antibody itself (eg, non-human sequence, glycosylation, impurities, aggregation), the patient (eg, genetic factors, immune status), or the drug’s route and frequency of administration, is a significant source of variability for antibody clearance.52 ADAs can have various impacts, such as neutralizing the drug, changing the PK/PD profiles of the antibodies, lowering the effectiveness of the treatment, or occasionally even precipitating severe adverse immunological reactions.52–55 Despite their significant impact and incidence, ADA’s molecular mechanisms are only partially known. The human immune system’s recognition of the non-human antibody components as “non-self” was the leading cause of ADA development. Although most currently licensed antibodies are humanized or fully human with few to no non-human components, ADA occurrences were nevertheless often seen among antibodies and populations.56–58 Making accurate forecasts of ADA prevalence at the individual level remains difficult, creating ambiguity when forecasting antibody PK.59
- The in vitro models are more valuable for establishing equivalency than the PK/PD tests. Different mechanisms, such as neutralizing pathogenic antigens, blocking signaling pathways, or inducing effector functions, are used by antibodies to provide pharmacological effects.;60 In vitro techniques can compare most of these features. The extrapolation of the indications is further validated and supported by these tests.
Conclusion
The sensitive analytical methods to characterize the molecular structure, correlate binding properties with structure, and establish a robust similarity of the critical quality attributes are of greatest significance in evaluating the safety and efficacy of biosimilars. While analytical assessment plays a pivotal role, any deviations found are difficult to justify using any other study, including animal pharmacology/toxicology, PK/PD, and clinical efficacy testing. It is expected that developers will use newer new testing methodologies that are far more sensitive to finding differences between two products than possible in any number of clinical efficacy testing. For example, a 2019 meta-analysis61 showed that the 38 biosimilars met the comparative clinical efficacy. However, two showed differences in immunogenicity that could have been identified if the analytical assessment had been conducted using the current methods; these two products were approved more than a decade ago.
In the early days of biosimilar submissions, proof of clinical efficacy was always required as if they were branded new medications. The language in the BPCIA states,
If there is residual uncertainty about biosimilarity after conducting structural analyses, functional assays, animal testing, human PK and PD trials, and the clinical immunogenicity assessment, the sponsor should then consider what additional clinical data may be needed to address that uncertainty (section VII.D.3) adequately
was overlooked by the majority of developers. It’s possible that the additional clinical study, as proposed in the FDA’s Biosimilar Action Plan, will be further in silico pharmacokinetic experiments rather than a clinical efficacy study. Therefore, developers should ask about the “residual uncertainty before proposing any extra study.” Dr. Janet Woodcock, a recent acting commissioner of the FDA, has the ideal recommendation for this strategy: “Why should we put people through all these different experiments only to check a box?” The FDA has recently questioned this idea of real-time testing, claiming that clinical efficacy testing is “broken.”62 Following the 21st Century Cure Act, new digital technologies and real-world evidence (RWE) are necessary.63
The EMA has begun working on a pilot clinical trials program to advise how clinical testing might be minimized or avoided for biosimilars.31 Their first pilot exercise, however, was a failure due to the erroneous data that the developers provided.
The development of more precise technologies for assessing bioequivalence, such as protein mass spectrometry, which is 10 million times more sensitive than it was a decade ago, is acknowledged by the EMA (EMA). Therefore, it is widely believed that comparative clinical trials are unreliable tools for determining the similarity of biological agents. It means that the tests were too insensitive to detect any difference or the clinical efficacy studies for the items were unnecessary because they were biosimilar.64
The arguments mentioned above are becoming entrenched inside regulatory organizations. Although, for example, the FDA claims that the present system for evaluating clinical efficacy is “broken”,65 The 21st Century Cure Act states that these outdated practices must be replaced with the incorporation of new digital technology and real-world evidence (RWE).63 Also, Biosimilar efficacy testing requires entirely different Master Protocols to make clinical testing relevant.
The FDA has started to doubt the logic of conventional clinical trial designs. “I feel the clinical trial system is flawed”, Janet Woodcock, head of FDA’s CDER, stated during a panel discussion for industry on November 14, 2018. It does not, in my opinion, serve patients’ best interests. In 2017, Woodcock published papers demonstrating why the current understanding of what clinical trials do is fallacious.66 Accordingly, she asks, “Why should we put patients through all these different trials just to check a box.”
The FDA’s Biosimilar Action Plan states,
Creating information resources and development tools for sponsors of biosimilar applications. This includes tools such as in silico models and simulations to correlate pharmacokinetic and pharmacodynamic responses with PD markers that are readily available for several biologic products
that can replace the need for additional clinical trials.67 In addition, many cytokines could qualify for a waiver of comparative efficacy trials, and the FDA is now developing validation protocols for using PD markers to obviate additional clinical trials.68 Note: additional clinical trials mean trials beyond the PK/PD testing, generally efficacy testing, though these could be further PK/PD trials.
The EMA (EMA) has started work on a pilot clinical trial program to provide recommendations on how to reduce or do away with clinical testing in the development of biosimilars. The EMA has begun work on a prototype clinical trials program to guide biosimilars on how clinical testing can be minimized or eliminated.31 However, because of the inaccurate data that the developers provided, their first pilot exercise was unsuccessful. Furthermore, EMA recognizes the development of more accurate technology for evaluating bioequivalence, such as protein mass spectrometry, which is 10 million times more sensitive now than it was a decade ago. Therefore, comparative clinical trials are now considered unreliable for establishing the similarity between biological agents.
The licensure of biosimilars incurs high costs and delays due to clinical safety and efficacy investigations. If these trials could bring substantial value, their use can be justified. However, as long as a product passes the other tests, no biosimilars get rejected based on clinical efficacy and safety assessment. This indicates that the products were biosimilar or that the tests were too insensitive to identify differences.64 This testing is no longer relevant in any scenario. Meta-analyses have repeatedly demonstrated that biosimilars have consistently demonstrated identical efficacy, especially in the case of oncology medications.69
More significant benefits of the change in the assessment of biosimilars will come to the developing world; most ROW agencies follow the FDA or EMA, at least in its spirit of regulatory compliance. They are always too conservative to make any changes and, for reasons well-understood, always like to have clinical proof of efficacy, even if the evidence is just a token. An excellent example of this comes from the CDSCO70 Indian guidance just states that efficacy studies must be conducted in 100 patients; there is no rationale, but this waste continues. I foresee a large wave of new biosimilars entering the market once the major agencies accept the recommendations. These have already been adopted by at least one major agency, the MHRA.
If a biosimilar product is well-characterized and demonstrates a highly similar clinical pharmacology profile, it should be safe and effective. Adding an efficacy study that is much less sensitive to identify is superfluous, and the risk that the developers might use these studies to justify critical differences. The regulatory agencies should take bold steps like the MHRA has done and remove efficacy testing as a requirement for approval. For the rest of the stakeholders, it will take a significant change in their mindset. But it will make biosimilars safer and more affordable. There is a dire need for the harmonization of biosimilars approval guidelines to remove many misconceptions, both at the regulatory agency and developer levels.71
Disclaimer
The author is not advising any legal violations when suggesting how to secure the rights to intellectual property.
Funding
This research received no external funding.
Disclosure
The author declares no conflict of interest. The author is also a patent law practitioner in the US.
References
1. EMA. Biosimilar Medicines Overview. Available from: https://www.ema.europa.eu/en/human-regulatory/overview/biosimilar-medicines-overview.
2. FDA. Biosimilar product information. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-product-information.
3. Jeremias S. Part 1: India works on guidelines for biological products; 2020. Available from: https://www.centerforbiosimilars.com/view/india-works-on-its-guidelines-for-biological-products.
4. Similar biotherapeutic products approved in Latin America; 2019. Available from: https://www.gabionline.net/biosimilars/general/Similar-biotherapeutic-products-approved-and-marketed-in-Latin-America.
5. Biosimilars approved in Australia; 2021. Available from: https://www.gabionline.net/biosimilars/general/Biosimilars-approved-in-Australia.
6. Biosimilars are approved in Canada. Available from: https://www.gabionline.net/biosimilars/general/biosimilars-approved-in-canada.
7. EMA. EPAR documents. Available from: https://www.ema.europa.eu/en/medicines/field_ema_web_categories%253Aname_field/Human/ema_group_types/ema_medicine.
8. FDA. Regulatory documents. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm.
9. FDA. FDA Adverse Events Reporting System (FAERS) Public Dashboard; 2022. Available from: https://fis.fda.gov/sense/app/95239e26-e0be-42d9-a960-9a5f7f1c25ee/sheet/7a47a261-d58b-4203-a8aa-6d3021737452/state/analysis.
10. EudraVigilence-European database of suspected adverse drug reaction reports. Available from: https://www.adrreports.eu/en/index.html.
11. FDA. Biosimilar development, review, and approval. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval.
12. Chen Y, Monnard A, da Silva JS. An inflection point for biosimilars, McKinsey & Co; 2021. Available from: https://www.mckinsey.com/industries/life-sciences/our-insights/an-inflection-point-for-biosimilars.
13. US Congress Public Law 98-417. Abbreviated new drug applications. Available from: https://www.govinfo.gov/content/pkg/STATUTE-98/pdf/STATUTE-98-Pg1585.pdf.
14. FDA. 21CFR314.3 Application for FDA approval to market a new drug. Available from: www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=314.3.
15. US Congress. Biological price competition and innovation act. Available from: https://www.congress.gov/bill/111th-congress/house-bill/3590.
16. FDA-TRACK. Center for drug evaluation & research—pre-approval safety review—biosimilars dashboard. Available from: https://www.fda.gov/about-fda/fda-track-agency-wide-program-performance/fda-track-center-drug-evaluation-research-pre-approval-safety-review-biosimilars-dashboard.
17. FDA. Questions and answers on biosimilar development and the bpci act guidance for industry; 2021. Available from: https://www.fda.gov/media/119258/download.
18. FDA. Purple Book. Available from: https://purplebooksearch.fda.gov/faqs#5.
19. FDA Guidance. Scientific considerations in demonstrating biosimilarity to a reference product. FDA. Available from: https://www.fda.gov/media/82647/download.
20. FDA. Biosimilars action plan. Available from: https://www.fda.gov/media/114574/download.
21. Li L, Ma L, Schrieber SJ, et al. Quantitative relationship between AUEC of absolute neutrophil count and duration of severe neutropenia for G-CSF in breast cancer patients. Clin Pharmacol Ther. 2018;104:742–748. doi:10.1002/cpt.991
22. Li J, Florian J, Campbell E, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology trials. Clin Pharmacol Ther. 2020;107:40–42. doi:10.1002/cpt.1653
23. FDA Guidance. scientific considerations in demonstrating biosimilarity to a reference product. Available from: https://www.fda.gov/media/82647/download.
24. FDA Guidance. clinical pharmacology data to demonstrate biosimilarity to a reference product. Available from: https://www.fda.gov/media/88622/download.
25. EMA. Human Regulatory. Biosimilars. Available from: https://www.ema.europa.eu/en/human-regulatory/research-development/scientific-guidelines/multidisciplinary/multidisciplinary-biosimilar#-product-specific-biosimilar-guidelines-section.
26. WHO Expert Committee on Biological Standardization. Annex 2. Guidelines on evaluation of similar biotherapeutic products (SBPs) WHO technical report series no 977 Geneva: World Health Organization; 2013 Available from: https://cdn.who.int/media/docs/defaultsource/biologicals/trs_977_annex_2.pdf?sfvrsn=e2389a69_3&download.
27. WHO. 72nd and 73rd report: WHO TRS N°1030; 2020. Available from: https://www.who.int/publications/i/item/9789240024373.
28. WHO. Guideline on the evaluation of biosimilars. Available from: https://www.who.int/publications/m/item/guidelines-on-evaluation-of-biosimilars.
29. MHRA. Final guidance on biosimilars. Available from: https://www.gov.uk/government/publications/guidance-on-the-licensing-of-biosimilar-products/guidance-on-the-licensing-of-biosimilar-products.
30. Niazi SK. The coming of age of biosimilars: a personal perspective. Biologics. 2022;2(2):107–127. doi:10.3390/biologics2020009
31. EMA. Tailored scientific advice for biosimilars development. Available from: https://www.ema.europa.eu/en/documents/report/tailored-scientific-advice-biosimilar-development-report-experience-pilot-2017-2020_en.pdf.
32. ClinicalTrials.gov Biosimilar Trials. Available from: https://www.clinicaltrials.gov/ct2/results?term=biosimilar&age_v=&gndr=&type=&rslt=&phase=0&phase=1&Search=Apply.
33. Puri A, Niewiarowski A, Arai Y, et al. Pharmacokinetics, safety, tolerability and immunogenicity of FKB327, a new biosimilar medicine of Adalimumab/Humira, in healthy subjects. Br J Clin Pharmacol. 2017;83(7):1405–1415. doi:10.1111/bcp.13245
34. Khalilieh S, Hussain A, Montgomery D, et al. Effect of tildrakizumab(MK-3222), a high affinity, selective anti-IL23p19 monoclonal anti-body, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br J Clin Pharmacol. 2018;84(10):2292–2302. doi:10.1111/bcp.13670
35. Hoffmann E, Jordan G, Lauer M, et al. Generation, characterization, and quantitative bioanalysis of Drug/Anti-drug antibody immune complexes to facilitate dedicated in vivo studies. Pharm Res. 2019;36(9):129. doi:10.1007/s11095-019-2661-0
36. FDA. Clinical immunogenicity considerations for biosimilar and interchangeable insulin products. https://www.fda.gov/media/133014/download.
37. Niazi S. Testimony to the US FDA. Available from: https://downloads.regulations.gov/FDA-2019-P-1236-0003/attachment_1.pdf.
38. Niazi S. Volume of distribution as a function of time. J Pharm Sci. 1976;65(3):452–454. doi:10.1002/jps.2600650339
39. Wesolowski CA, Wesolowski MJ, Babyn PS, et al. Time-varying apparent volume of distribution and drug half-lives following intravenous bolus injections. PLoS One. 2016;11(7):e0158798. doi:10.1371/journal.pone.0158798
40. Colburn WA. A time-dependent volume of distribution term used to describe linear concentration-time profiles. J Pharmacokinet Biopharm. 1983;11:389–400. doi:10.1007/BF01058957
41. EMA. Scientific guideline for immunogenicity assessment of therapeutic proteins. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf.
42. FDA. First Interchangeable product. Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-first-interchangeable-biosimilar-insulin-product-treatment-diabetes.
43. Niazi S. No two classes of biosimilars: urgent advice to the US Congress and the FDA. J Clin Pharm Ther. 2022;47:1–10. doi:10.1111/jcpt.13612
44. FDA. Guidance for industry non-inferiority clinical trials. Draft Guidance. Available from: https://www.fda.gov/media/78504/download.
45. Mielke J, Jilma B, Jones B, et al. An update on the clinical evidence that supports biosimilar approvals in Europe. Br J Clin Pharmacol. 2018;84(7):1415–1431. doi:10.1111/bcp.13586
46. Bayesian Calculator. Available from: http://psych.fullerton.edu/mbirnbaum/bayes/bayescalc.htm.
47. Charan J, Biswas T. How to calculate sample size for different study designs in medical research? Indian J Psychol Med. 2013;35(2):121–126. doi:10.4103/0253-7176.116232
48. FDA. Drugs@FDA: FDA-Approved Drugs; Trastuzumab. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=103792.
49. FDA guidance. Clinical pharmacology data to demonstrate biosimilarity to a reference product. Available from: https://www.fda.gov/media/88622/download.
50. Van Der Graaf PH, Gabrielsson J. Pharmacokinetic–pharmacodynamic reasoning in drug discovery and early development. Futur Med Chem. 2009;1:1371–1374. doi:10.4155/fmc.09.124
51. Zhou L, Hoofring SA, Wu Y, et al. Stratification of antibody-positive subjects by antibody level reveals an impact of immunogenicity on pharmacokinetics. AAPS J. 2012;15:30–40. doi:10.1208/s12248-012-9408-8
52. Bendtzen K, Geborek P, Svenson M, et al. Individualized monitoring of drug bioavailability and immunogenicity in rheumatoid arthritis patients treated with the tumor necrosis factor α inhibitor infliximab. Arthritis Rheum. 2006;54:3782–3789. doi:10.1002/art.22214
53. Vaisman-Mentesh A, Rosenstein S, Yavzori M, et al. Molecular landscape of anti-drug antibodies reveals the mechanism of the immune response following treatment with TNFα Antagonists. Front Immunol. 2019;10:2921. doi:10.3389/fimmu.2019.02921
54. Nelson AL, Dhimolea E, Reichert JM, et al. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi:10.1038/nrd3229
55. Ternant D, Aubourg A, Magdelaine-Beuzelin C, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30:523–529. doi:10.1097/FTD.0b013e318180e300
56. Wade JR, Parker G, Kosutic G, et al. Population pharmacokinetic analysis of certolizumab pegol in patients with Crohn’s disease. J Clin Pharmacol. 2015;55:866–874. doi:10.1002/jcph.491
57. Brandse JF, Mould D, Smeekes O, et al. A real-life population pharmacokinetic study reveals factors associated with clearance and immunogenicity of infliximab in inflammatory bowel disease. Inflamm Bowel Dis. 2017;23:650–660. doi:10.1097/MIB.0000000000001043
58. Casteele NV, Mould DR, Coarse J, et al. Accounting for pharmacokinetic variability of certolizumab pegol in patients with Crohn’s disease. Clin Pharmacokinet. 2017;56:1513–1523. doi:10.1007/s40262-017-0535-3
59. Gill KL, Machavaram KK, Rose RH, et al. potential sources of inter-subject variability in monoclonal antibody pharmacokinetics. Clin Pharmacokinet. 2016;55:789–805. doi:10.1007/s40262-015-0361-4
60. Pyzik M, Rath T, Lencer WI, et al. The architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194:4595–4603. doi:10.4049/jimmunol.1403014
61. Schiestl M, Ranganna G, Watson K, et al. The path towards a tailored clinical biosimilar development. BioDrugs. 2020;34:297–306. doi:10.1007/s40259-020-00422-1
62. Brennan J. FDA’s woodcock says the clinical trial system is broken. regulatory affairs professionals society. Available from: https://www.raps.org/regulatory-focus%E2%84%A2/news-articles/2017/9/fda-s-woodcock-the-clinical-trials-system-is-broken.
63. FDA. 21st century cures act. Available from: https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act.
64. Niazi SK. Biosimilars: a futuristic fast-to-market advice to developers. Expert Opin Biol Ther. 2022;22:149–155. doi:10.1080/14712598.2022.2020241
65. Zachary B. Regulatory affairs professionals society. Available from: https://www.raps.org/regulatory-focus%E2%84%A2/news-articles/2017/9/fda-s-woodcock-the-clinical-trials-system-is-broken.
66. Dunn A. FDA’s Woodcock: “The clinical trial system is broken”. BioPharma Dive; 2018. Available from: https://www.biopharmadive.com/news/fdas-woodcock-the-clinical-trial-system-is-broken/542698.
67. Joubert MK, Deshpande M, Yang J, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One. 2016;11(8):e0159328. doi:10.1371/journal.pone.0159328
68. Moore TJ, Mouslim MC, Blunt JL, et al. Assessment of availability, clinical testing, and US review of biosimilar biologic products. JAMA Intern Med. 2021;181:52–60. doi:10.1001/jamainternmed.2020.3997
69. Bloomfield D, D’Andrea E, Nagar S, et al. Characteristics of clinical trials evaluating biosimilars in the treatment of cancer: a systematic review and meta-analysis. JAMA Oncol. 2022;8(4):537–545. doi:10.1001/jamaoncol.2021.7230
70. Central Drugs Standard Control Organization, India. Guidelines on Similar Biologics: Regulatory Requirements for Marketing Authorization in India, 2016. Available from: https://cdsco.gov.in/opencms/resources/UploadCDSCOWeb/2018/UploadAlertsFiles/BiosimilarGuideline2016.pdf.
71. Niazi SK. Biosimilars: harmonizing the approval guidelines. Biologics. 2022;2:171–195. doi:10.3390/biologics2030014
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.