")
Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 13
Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the MK2 Inhibitor ATI-450 in Healthy Subjects: A Placebo-Controlled, Randomized Phase 1 Study
Authors Gordon D, Hellriegel ET, Hope HR, Burt D, Monahan JB
Received 4 February 2021
Accepted for publication 12 May 2021
Published 10 June 2021 Volume 2021:13 Pages 123—134
DOI https://doi.org/10.2147/CPAA.S305308
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Arthur E. Frankel
David Gordon,1 Edward T Hellriegel,1 Heidi Rath Hope,2 David Burt,1 Joseph B Monahan2
1Research and Development, Aclaris Therapeutics, Inc., Wayne, PA, USA; 2Research and Development, Aclaris Therapeutics, Inc., and Confluence Discovery Technologies, Inc., St. Louis, MO, USA
Correspondence: David Gordon
Chief Medical Officer, Research and Development, Aclaris Therapeutics, Inc., 640 Lee Road, Suite 200, Wayne, PA, 19087, USA
Tel +1 484-540-6273
Email [email protected]
Purpose: ATI-450 is an oral, small-molecule inhibitor of the p38α mitogen-activated protein kinase (MAPK)/MAPK-activated protein kinase 2 (MK2) inflammatory signaling pathway. This phase 1, single and multiple ascending dose (SAD, MAD) study evaluated ATI-450 safety, tolerability, pharmacokinetics, and pharmacodynamics.
Patients and Methods: Healthy adults were randomly assigned to SAD (10, 30, 50, 100 mg; n=24) and MAD (10, 30, 50 mg twice daily [BID] for 7 days; n=24) cohorts of ATI-450 or placebo (n=14). Safety and tolerability were evaluated through clinical and laboratory assessments. Pharmacokinetic parameters were evaluated in plasma samples; pharmacodynamic assessments included quantification of cytokine levels (tumor necrosis factor α [TNF-α], interleukin [IL]-1β, IL-6, IL-8) and phosphorylation of the MK2 downstream substrate, heat shock protein 27 (p-HSP27).
Results: The most common adverse events were headache (10/48, 20.8%), dizziness (6/48, 12.5%), upper respiratory tract infection (3/48, 6.3%), and constipation (3/48, 6.3%). Pharmacokinetics were dose-proportional, with a terminal half-life of 9‒12 hours in the MAD cohorts on day 7. Dose- and concentration-dependent inhibition of ex vivo stimulated cytokines and target biomarker was observed. On day 7, patients in the 50 mg BID dose cohort recorded mean trough drug levels that were 1.4, 2.2, 2.3, and 2.4 times greater than the IC80 for TNF-α, IL-1β, IL-8, and p-HSP27, respectively. Mean Cmax was 3.5, 5.4, 5.6, and 6.0 times greater than the IC80 for TNF-α, IL-1β, IL-8, and p-HSP27, respectively. IL-6 inhibition > 50% was noted for part of the dosing interval.
Conclusion: ATI-450 was well tolerated at the doses investigated, exhibited dose- and time-independent (ie, linear) pharmacokinetics, and dose-related pharmacodynamic effects. These results support further study of ATI-450 in immunoinflammatory diseases in phase 2 trials.
Keywords: immunologic diseases, rheumatoid arthritis, serine-threonine kinases, protein kinase inhibitors, pharmacokinetics/pharmacodynamics
Introduction
The p38 mitogen-activated protein kinase (p38MAPK) signaling pathway has been a target for therapeutic intervention for inflammatory diseases because of its involvement in the regulation and expression of multiple cytokines (eg, tumor necrosis factor α [TNF-α], interleukin [IL] 1β, and IL-6) and other inflammatory signals.1,2 MAPK-activated protein kinase 2 (MK2) is a direct downstream substrate of p38MAPK, which has been recognized as a key driver of inflammation.3,4 Inflammatory cytokines activated through the p38MAPK/MK2 pathway include TNF-α, IL-1β, IL-6, and IL-8.4–9
Small-molecule inhibitors of p38MAPK have been evaluated in clinical trials for the treatment of inflammatory diseases; however, clinical development of these compounds has been limited by toxicity and lack of sustained efficacy.4,5,10,11 The clinical efficacy achieved with p38MAPK inhibitors has been generally disappointing. Of note, there is evidence in certain autoimmune diseases that p38MAPK inhibitors exhibit tachyphylaxis after multiple treatments, making sustained inhibition of the inflammatory response difficult to achieve in these therapeutic indications, such as rheumatoid arthritis.12,13 Tachyphylaxis may be explained by inhibition of the negative feedback and/or anti-inflammatory target substrates described above. Therefore, the approach of targeting key downstream kinase substrates of p38MAPK that specifically regulate inflammatory cytokines to potentially achieve greater specificity for blockade of pathological inflammation has gained interest.
ATI-450 (also known as CDD-450), a recently developed MK2 inhibitor, has a novel mechanism of action by which it targets the modified p38MAPK ATP binding pocket and juxtaposed MK2 formed upon the interaction between p38MAPK and MK2 (Supplementary Figure S1).5 The p38MAPK binding site on MK2 is located within the C terminus; thermodynamic studies have shown that the 2 proteins in the complex bind tightly, and deletion of the binding sequence in MK2 abrogates p38-dependent phosphorylation and activation of MK2.14 Following the formation of the p38MAPK-MK2 biomolecular complex, ATI-450 binds to the interface of the complex with significantly higher affinity than to either kinase alone, thereby selectively inhibiting p38MAPK phosphorylation of MK2 and locking MK2 in an inactive conformation.5 MK2 regulates inflammatory cytokine mRNA stability and translation through phosphorylation of downstream effectors including adenylate-uridylate–rich element-binding proteins (eg, tristetraprolin).15–17 Therefore, inhibition of p38αMAPK-MK2 blocks downstream MK2-mediated inflammatory actions.
Preclinical animal disease models have been used to predict the potential of ATI-450 (ie, CDD-450) as a treatment in several immune-mediated inflammatory diseases. In a rat streptococcal cell wall arthritis model for rheumatoid arthritis, ATI-450 showed joint protective effects and preserved bone mineral density.5 ATI-450 also was studied in a murine model of neonatal-onset multisystem inflammatory disease (NOMID), which is a severe form of a cryopyrin-associated periodic syndrome; NOMID and other cryopyrin-associated periodic syndromes comprise a spectrum of rare hereditary autoinflammatory disorders in which IL-1β and IL-18 are overproduced. A dramatic attenuation of disease was observed in this transgenic animal model, including a reduction in bone marrow levels of IL-1β in mice treated with ATI-450 relative to those treated with placebo.5 These preclinical results support clinical development of ATI-450 in humans for autoimmune and autoinflammatory diseases driven by the cytokines TNF-α, IL-1β, IL-6, and IL-8.
A first-in-human, phase 1, randomized, observer-blind, placebo-controlled study was conducted to evaluate the safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of ATI-450 in single and multiple ascending dose (SAD and MAD) cohorts in healthy subjects. In addition, the study included separate cohorts that were used to evaluate the PK of fed versus fasting administration and of co-administration with methotrexate; these findings will be reported separately because of content size limitations. Here, we report data from the SAD and MAD cohorts.
Methods
The study’s clinical protocol, informed consent forms, and amendments to all documents were reviewed and approved by Midlands Independent Review Board of Overland Park, Kansas. The protocol ATI-450-PKPD-101 was first approved on June 7, 2019 and there were 4 amendments, with the final protocol approved on November 6, 2019. Informed written consent was obtained from all subjects before any study-related procedures began. The study was conducted by PRA Health Sciences – Early Development Services (Lenexa, KS) in accordance with the principles of the Declaration of Helsinki and in compliance with the International Council for Harmonization E6 Guideline for Good Clinical Practice, and any applicable national and local laws and regulations. Consistent with guidelines, this phase 1 study was not submitted to a clinical trial register. The conduct of the study included the ability of subjects to opt out and subject deidentification and anonymization. No personal information was collected.
Study Design
PRA Health Sciences led the study at a clinical site in Lenexa, Kansas, and prepared the randomization scheme for all cohorts while maintaining blinding of observers associated with the study for the cohorts described here. PRA assigned randomization codes sequentially as healthy subjects became eligible for randomization after enrolling in the study. For each SAD dose cohort, subjects were randomly assigned to ATI-450 or placebo in an overall 6:2 ratio. For each MAD dose cohort, subjects were randomly assigned to ATI-450 or placebo in an overall 8:2 ratio.
Single Ascending Dose (SAD) Cohorts
Thirty-two subjects were randomly assigned to 1 of 4 cohorts to receive ATI-450 doses of 10, 30, 50, or 100 mg (n=6 per cohort) or placebo (n=2 per cohort). Each subject received a single oral dose of ATI-450 or placebo in the morning on an empty stomach. All subjects were admitted to the clinical study facility on day −1 and stayed until 48 hours after ATI-450 or placebo administration.
Multiple Ascending Dose (MAD) Cohorts
Thirty subjects were randomly assigned to 1 of 3 cohorts to receive ATI-450 doses of 10, 30, or 50 mg twice daily (BID) (n=8 per cohort) or placebo (n=2 per cohort). Subjects received each dose of ATI-450 or placebo on an empty stomach for 7 days, in the morning and the evening, with the final dose administered on the morning of day 7. Subjects were admitted to the clinical facility on day −1 and remained at the site until at least 72 hours after the morning dose on day 7.
Subjects
Subjects were healthy males or females between 18 and 55 years of age, inclusive; had a body mass index between 18 and 32 kg/m2, inclusive, with a minimum body weight of 50 kg; and tested negative for HIV-1 and HIV-2 antibodies, hepatitis B surface antigen, and hepatitis C virus antibody at screening. Female subjects were not pregnant or nursing and agreed to use 2 effective methods of contraception if heterosexually active and of childbearing potential.
Subjects were excluded from the study if they had a current acute or chronic disease or used medications that may have affected their safety or other study assessments within 2 weeks of admission.
Safety and Tolerability Assessments
Adverse events, clinical laboratory tests, vital signs, an electrocardiogram, and physical examination results were evaluated for each subject in both parts of the study. Holter monitoring data were collected for future analysis.
Blood Sampling and Bioanalysis
Two-milliliter blood samples were collected for PK analysis in plasma samples. For the SAD cohorts, blood was collected pre-dose and 0.5, 1, 2, 4, 6, 8, 12, 24, 36, and 48 hours post-dose. For the MAD cohorts, blood was collected on day 1 prior to the morning dose and 0.5, 1, 2, 4, 6, 8, and 12 hours post-dose (before the evening dose); before the morning dose on days 2 through 6; and on day 7, before the morning dose and 0.5, 1, 2, 4, 6, 8, 12, 13, 14, 24, 36, 48, and 72 hours after the morning dose.
Blood samples of 10 mL were collected for PD analysis. For the SAD cohorts, blood was collected pre-dose and 1, 12, and 24 hours post-dose. For the MAD cohorts, on day 1 blood was collected before the morning dose and 4 and 12 hours after the morning dose (prior to the evening dose); on day 7, blood was collected before the last dose and 4 and 12 hours after the last dose.
ATI-450 concentrations in plasma samples were determined by PRA Health Sciences Bioanalytical Laboratory (Lenexa, Kansas) using a validated ultra-performance liquid chromatography with tandem mass spectrometry method (details in Supplementary Information). The lower limit of quantification was 0.500 ng/mL.
PD bioanalysis (performed by Confluence Discovery Technologies, Inc. [St. Louis, Missouri]) of phosphorylated heat shock protein 27 (p-HSP27), IL-1β, IL-6, IL-8, and TNF-α levels in blood samples were analyzed under 3 conditions: (1) unstimulated, (2) ex vivo lipopolysaccharide–stimulated, and (3) ex vivo lipopolysaccharide–stimulated in the presence of exogenously added 10 μM ATI-450. Historical data show the percentage of maximum inhibition by ATI-450 achieved for a given analyte is consistent and therefore useful for normalization across samples. Additional details of the assays for analyzing p-HSP27 and cytokines are included in the Supplementary Information.
SAD cohort data were calculated for each subject set (pre-dose, 1 hour post-dose, and 12 hours post-dose on day 1, and 24 hours post-dose on day 2) as % day 1 pre-dose using the ex vivo lipopolysaccharide stimulation as the maximum signal and normalizing across the sample set using the day 1 ex vivo lipopolysaccharide plus 10 µM ATI-450 as maximum inhibition.
MAD cohort data were calculated for each subject set (pre-dose, 4 hours post-dose, and 12 hours post-dose on days 1 and 7) as % day 1 pre-dose using the ex vivo lipopolysaccharide stimulation as the maximum signal and normalizing across the sample set using the day 1 ex vivo lipopolysaccharide plus 10 µM ATI-450 as maximum inhibition.
Statistics
Pharmacokinetics
ATI-450 plasma concentrations and PK parameters were determined for all SAD and MAD cohorts. Noncompartmental analysis with Phoenix WinNonlin™, version 8.2 (Certara, Princeton, NJ) was used to determine the following PK parameters from the plasma concentration–time data for ATI-450: maximum observed plasma concentration (Cmax); time to maximum observed plasma concentration (tmax); area under the plasma concentration–time curve from time 0 through time t (AUC0-t), where t is the time of the last quantifiable concentration; apparent terminal elimination rate constant (λz); and associated half-life (t1/2). For the SAD cohorts, additional PK parameters determined were area under the plasma concentration–time curve from time 0 to infinity (AUC0-inf), apparent clearance (CL/F), and apparent volume of distribution (Vz/F). For the MAD cohorts, several additional PK parameters were determined, including area under the plasma concentration–time curve over the 12-hour dosing interval, tau (AUCtau); minimum observed plasma concentration in the dosing interval after multiple dosing, on day 7 (Cmin); average plasma concentration after multiple dosing, on day 7 (Cavg); measured concentration at the end of a dosing interval (Ctrough); accumulation ratio for Cmax and AUCtau (Rac, Cmax and Rac, AUCtau); apparent clearance at steady state (CLss/F); and apparent volume of distribution at steady state (Vss/F).
Pharmacodynamics
PD data analysis was conducted using Microsoft Excel 2010 (Microsoft, Redmond, WA), Meso Scale Discovery Workbench® analysis software (Rockville, MD), and Phoenix WinNonlin, version 8.1. For the PK/PD analysis in Phoenix WinNonlin, the ATI-450 plasma concentrations and the percentages of inhibition of production of lipopolysaccharide-stimulated p-HSP27, TNF-α, IL-1β, IL-6 and IL-8 levels in human whole blood from all SAD/MAD cohorts were simultaneously fit using an inhibitory Emax model (WinNonlin, model 104). The parameters estimated by the model for each biomarker were the baseline effect at zero concentration (E0), the maximum percent inhibition (Imax), and the ATI-450 plasma concentration at half maximal inhibition (IC50). The ATI-450 concentrations at 80% inhibition (IC80) were subsequently calculated from the IC50 estimates using the following equation: ICx = (x/100−x)*IC50, where x is the percent inhibition (ie, 80%). Additionally, multiples of the peak and trough concentrations of ATI-450 50 mg BID on day 7 to the IC50 and IC80 estimates for p-HSP27 and each cytokine were determined.
Sample Size Calculations
No prospective calculations of statistical power were made for sample size. Sample sizes of 32 subjects and 30 subjects for the SAD and MAD cohorts, respectively, were selected and are typical for a first-in-human study.
Results
Study Subjects
The first screening for subjects began on July 8, 2019, and the last follow-up visit was on December 10, 2019.
In the SAD cohorts, 32 subjects were enrolled, randomized, and included in the safety set. A total of 31 subjects completed the study; 1 subject in the 30 mg ATI-450 cohort withdrew from the study for non-safety reasons after receiving a single dose on day 1 and thus did not complete all study assessments. PK and PD analyses included the 24 subjects who received ATI-450. Most subjects in the SAD cohorts were female (84%; Table 1) and ages ranged from 18 to 51 years.
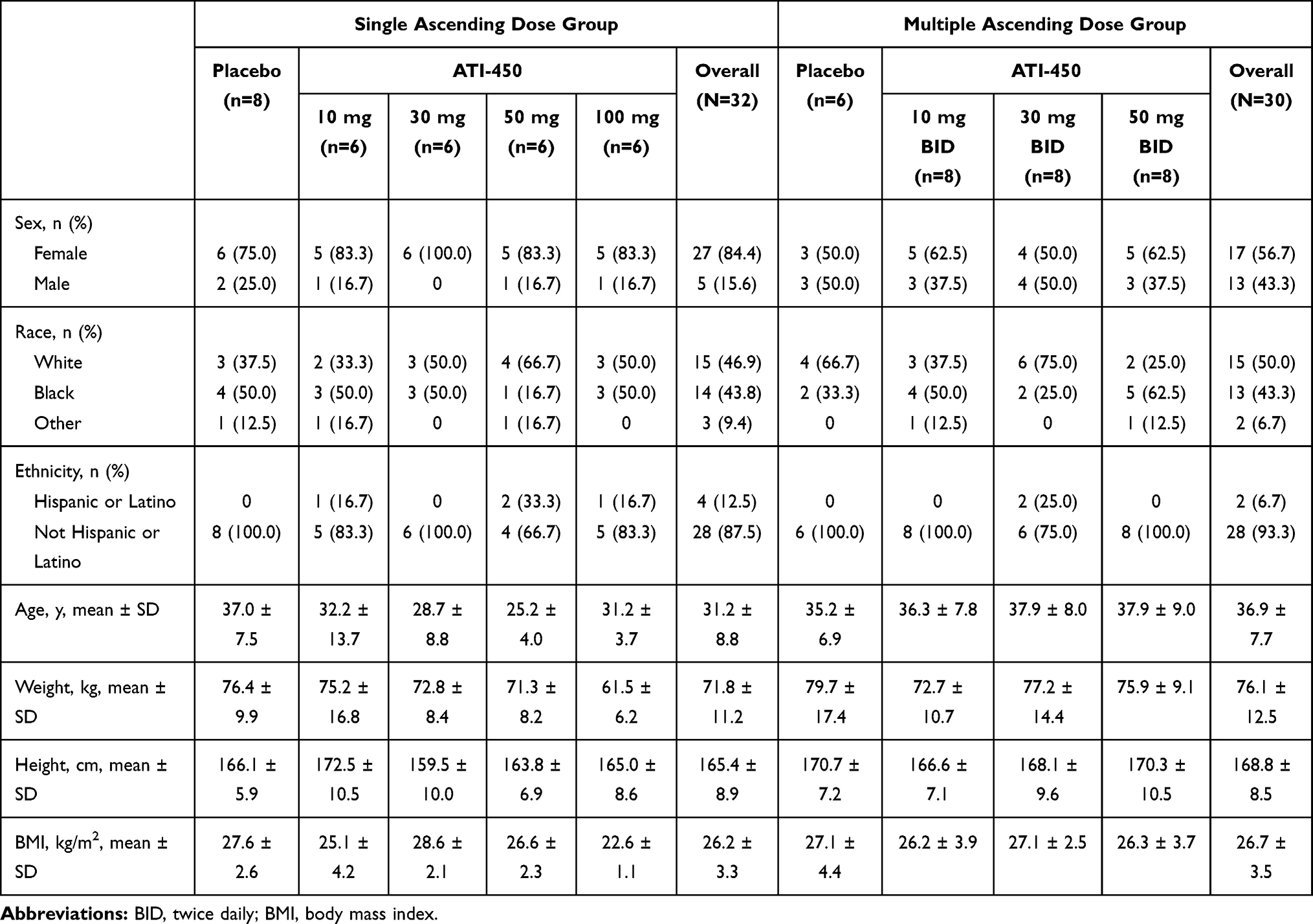 |
Table 1 Summary of Demographic Characteristics |
In the MAD cohorts, 30 subjects were enrolled, randomized, and completed the study, and all subjects were included in the safety set; the 24 subjects who received ATI-450 were included in the PK and PD sets. These cohorts were more evenly split between female and male subjects (57% female, 43% male) and ages ranged from 21 to 53 years.
Safety and Tolerability
No clinically significant findings in vital signs, electrocardiograms, or clinical laboratory values were observed. The most common treatment-emergent adverse events (TEAEs) among all study participants are shown in Table 2. In the SAD cohorts, 15 TEAEs were reported by 7 of 24 (29%) subjects who received ATI-450 and 6 TEAEs were reported by 4 of 8 (50%) subjects who received placebo. No deaths or serious adverse events (SAEs) and no discontinuations due to TEAEs occurred. All TEAEs were mild, transient, and resolved without sequelae by the end of the study. Four of 24 (17%) subjects who received ATI-450 reported TEAEs that were considered by the investigator to be related to the study drug (0 subjects in the 10 mg cohort; 2 [33%] subjects in the 30 mg cohort [1 event of nausea and 2 events each of headache and dizziness]; 1 [17%] subject in the 50 mg cohort [1 event of headache]; and 1 subject in the 100 mg cohort [1 event of dizziness]).
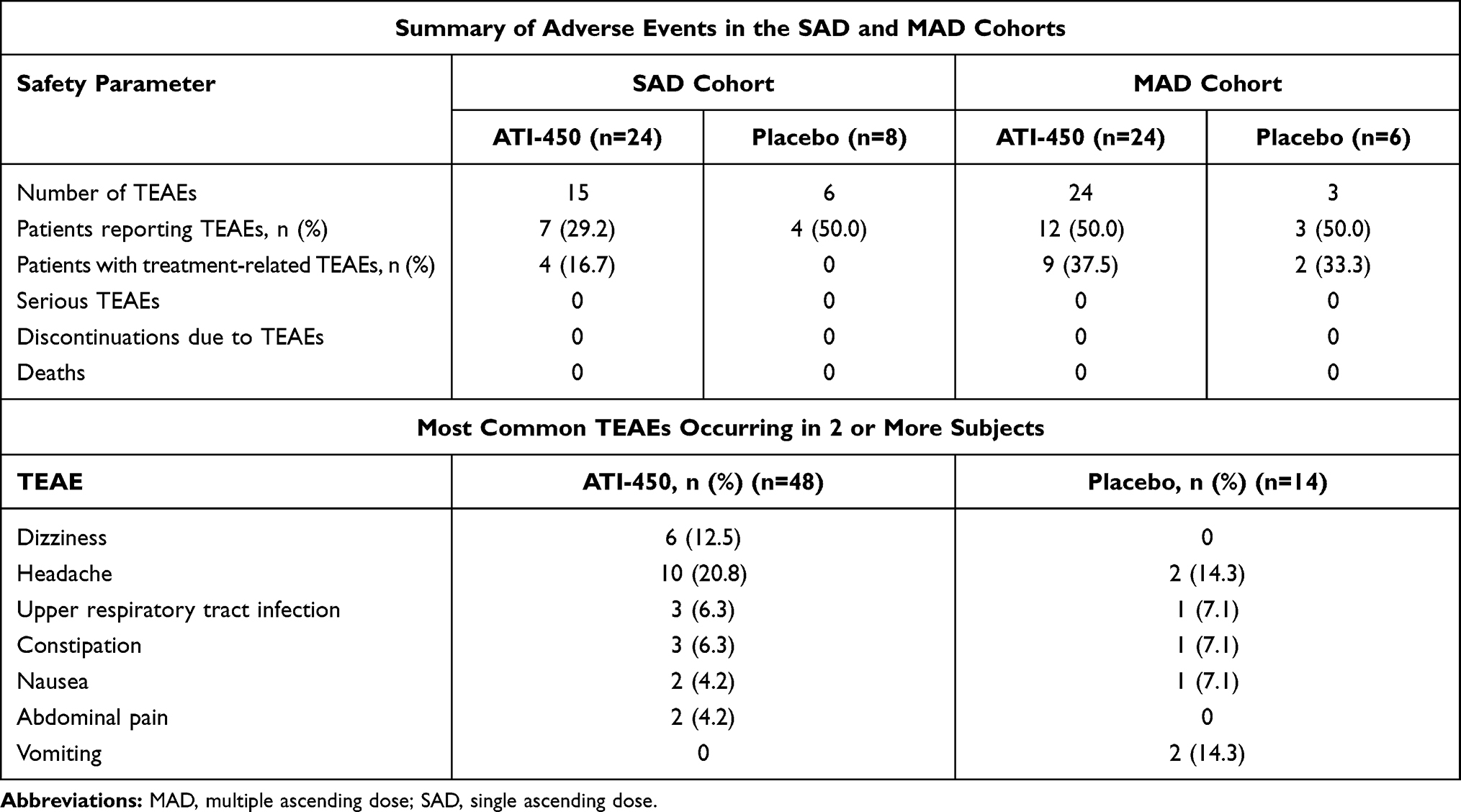 |
Table 2 Summary of Treatment-Emergent Adverse Events (TEAEs), Safety Set |
In the MAD cohorts, there were 24 TEAEs reported by 12 of 24 (50%) subjects who received ATI-450 and 3 TEAEs reported by 3 of 6 (50%) subjects who received placebo. As in the SAD cohorts, there were no deaths or SAEs and no subjects discontinued the study due to TEAEs; all TEAEs were mild, transient, and resolved without sequelae by the next visit. Nine of 24 (38%) subjects who received ATI-450 reported TEAEs that were considered by the investigator to be related to the study drug (1 of 8 [13%] subjects in the 10 mg BID cohort [1 event of headache], 5 of 8 [63%] subjects in the 30 mg BID cohort [1 event each of blurred vision, constipation, abdominal pain, diarrhea, arthralgia, dizziness, and oropharyngeal pain], and 3 of 8 [38%] subjects in the 50 mg BID cohort [1 event of headache and 2 events of dizziness]). In the placebo group, 2 of 6 (33%) subjects had TEAEs that were considered by the investigator to be related to the study drug (1 event each of vomiting and headache).
Laboratory changes were generally unremarkable. Dose-dependent reductions in neutrophils were observed at day 2 and day 5 with the maximum effect in the MAD cohort. The mean reduction at day 7 in the 50 mg BID group was 24% (3318.4 cells/μL at baseline versus 2514.4 cells/μL at day 7).
Pharmacokinetics
SAD Cohorts
Administration of ATI-450 at single doses of 10 mg, 30 mg, 50 mg, and 100 mg under fasting conditions resulted in ATI-450 post-dose mean plasma concentrations above the lower limit of quantification through 24 hours post-dose at 10 mg and through 48 hours post-dose at all other dose levels. ATI-450 mean plasma concentrations increased with increasing doses (Figure 1). Systemic exposure increased in a dose-proportional manner across the ATI-450 dose range from 10 to 100 mg, with approximately 10.8-, 12.6-, and 12.8-fold increases in Cmax, AUC0-t, and AUC0-inf, respectively (Table 3). Statistical analysis for dose proportionality using the power model is shown in Supplementary Table S1, where systemic exposure (Cmax, AUC0-t, and AUC0-inf) increased in proportion to ATI-450 dose after single dosing as indicated by the slope estimates near unity and the 90% CIs including unity. ATI-450 concentrations appeared rapidly in plasma, with median tmax ranging from 2 to 4 hours across the dose cohorts. Mean t1/2 of ATI-450 in plasma ranged from 8.5 to 11.2 hours. CL/F, tmax, and t1/2 values showed no clear relation with dose over the dose range of 10 to 100 mg. Across the dose cohorts, low inter-subject variability in Cmax, AUC0-inf, and AUC0-t was observed with coefficients of variation ranging from 12.7% to 28.1%.
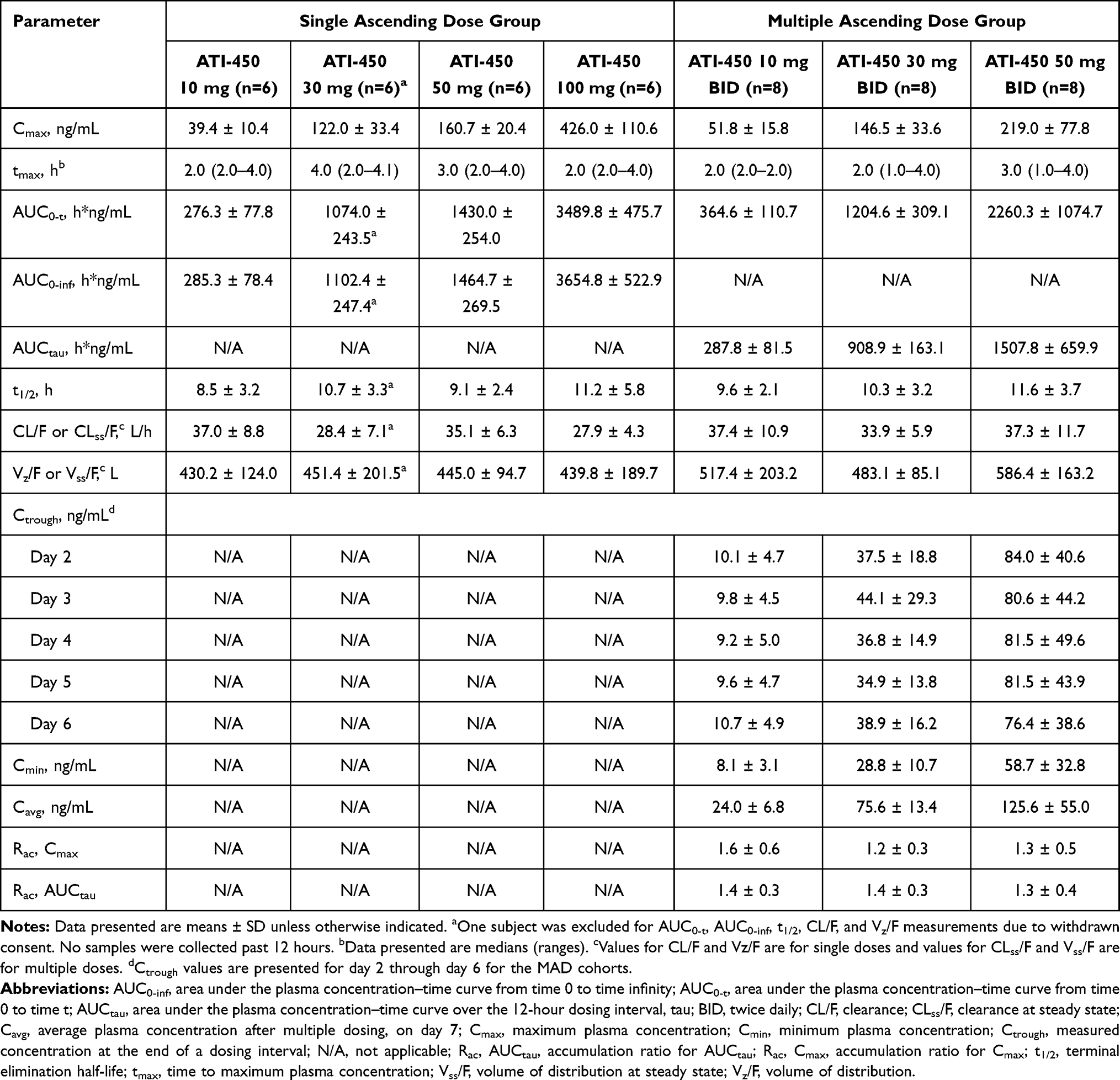 |
Table 3 ATI-450 Pharmacokinetic Parameters |
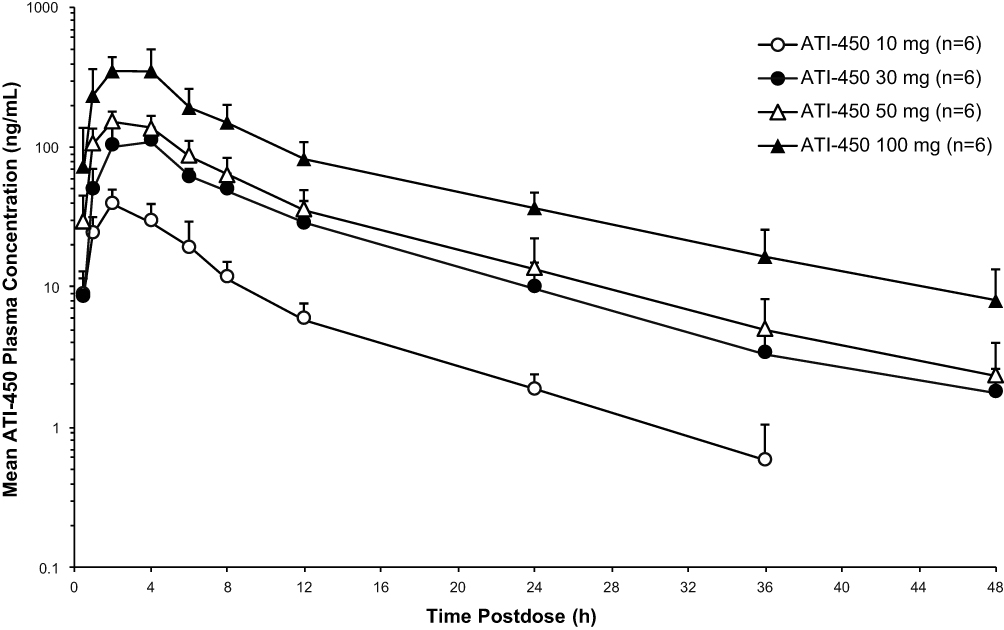 |
Figure 1 Mean (SD) plasma concentration–time profiles of ATI-450 after a single dose under fasted conditions, semi-log scale. Hour 0 is the pre-dose value. |
MAD Cohorts
Figure 2 shows the mean (SD) plasma concentration–time profiles of ATI-450 from 0 to 72 hours after the last dose on day 7, representing dose-proportional PK for the MAD cohorts, and Table 3 shows ATI-450 PK parameters on day 7 after multiple doses. The results obtained after the first dose on day 1 in the MAD cohorts were consistent with those obtained after single doses in the SAD cohorts (data not shown). Tmax appeared to be independent of dose, with median values ranging from 2 to 3 hours following the final dose administration on day 7. Mean t1/2 was similar across doses following repeated dose administration (from 9.6 to 11.6 hours). AUCtau and Cmax increased with escalating doses and there was slight accumulation after multiple doses. Over the dose range of 10 to 50 mg BID, Cmax and AUCtau increased 5.4- and 5.9-fold on day 1, respectively, and 4.2- and 5.2-fold on day 7, respectively. Steady state appeared to be reached by day 2 of administration at all dose levels based on the similarity in the mean Ctrough of ATI-450 on days 2 through 6. Mean accumulation of ATI-450 after 7 days of BID dosing ranged from 1.2- to 1.6-fold for Cmax and 1.3- to 1.4-fold for AUCtau. The mean values of t1/2, CL, and apparent volume of distribution after multiple doses were comparable to those after a single dose, indicating time-independent PK of ATI-450 after 7 days of BID administration. Statistical analysis of dose proportionality on day 7 using the power model is shown in Supplementary Table S2, where systemic exposure (Cmax, AUCtau) following BID administration of ATI-450 increased in proportion to dose over the 5-fold dose range as indicated by the slope estimates near unity and the 90% CIs including unity.
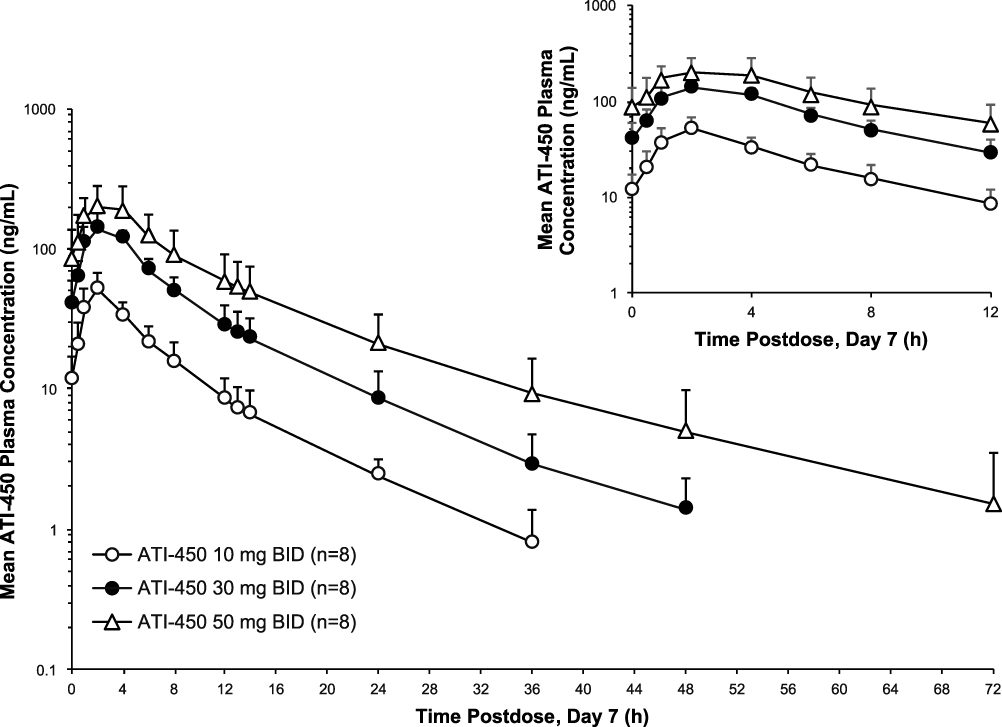 |
Figure 2 Mean (SD) plasma concentration–time profiles of ATI-450 after 7 days of BID dosing under fasted conditions, semi-log scale. Hour 0 is the pre-dose value. Abbreviation: BID, twice daily. |
Pharmacodynamics
A relationship between concentrations of each analyte and ATI-450 plasma levels was developed and the data were then fit to a nonlinear inhibitory Emax model, from which IC50 and IC80 concentrations were established. The p-HSP27 and cytokine inhibition parameters generated from the model are shown in Supplementary Table S3. Target modulation potency (inhibition of HSP27 phosphorylation) for ATI-450 was similar to potencies for inhibition of production of 3 of the 4 cytokines, TNF-α, IL-1β, and IL-8, but the potency for inhibition of IL-6 production was lower (less potent). Imax was greatest for p-HSP27, TNF-α, and IL-6, with greater than 96% inhibition, and lower for IL-1β (74%) and IL-8 (57%).
For the 50 mg BID dose MAD cohort, concentrations at Ctrough were in excess of the relative IC80 concentrations (1.4- to 2.4-fold) for the target biomarker p-HSP27 and for 3 of the 4 cytokines (TNF-α, IL-1β, and IL-8), but not for IL-6 (Table 4). TNF-α and IL-1β were inhibited by up to 92.7% and 83.0%, respectively, versus baseline. Plasma levels were greater than the IC50 for IL-6 for at least part of the dosing interval. The effects of ATI-450 dose on the relative concentration of each cytokine analyte and the biomarker p-HSP27 in the ex vivo stimulated assay, expressed as a percentage of pre-dose analyte levels (set to 100%) for the 10 mg, 30 mg, and 50 mg BID MAD cohorts on day 7, 4 hours post-dose and 12 hours post-dose, are shown in Figure 3. The 4-hour post-dose day 7 samples reflected approximate steady-state Cmax ATI-450 concentrations while the 12-hour post-dose day 7 samples reflected steady-state Ctrough concentrations of the drug. A marked dose-dependent reduction in concentration was observed for all 4 cytokines at the 4-hour time point and persisted through 12 hours. TNF-α, IL-1β, IL-8, and p-HSP27 all demonstrated a reduction in concentration that persisted for the entire dosing interval. Concentration-dependent and dose-dependent modulation of p-HSP27 and inhibition of the 4 cytokines analyzed were demonstrated.
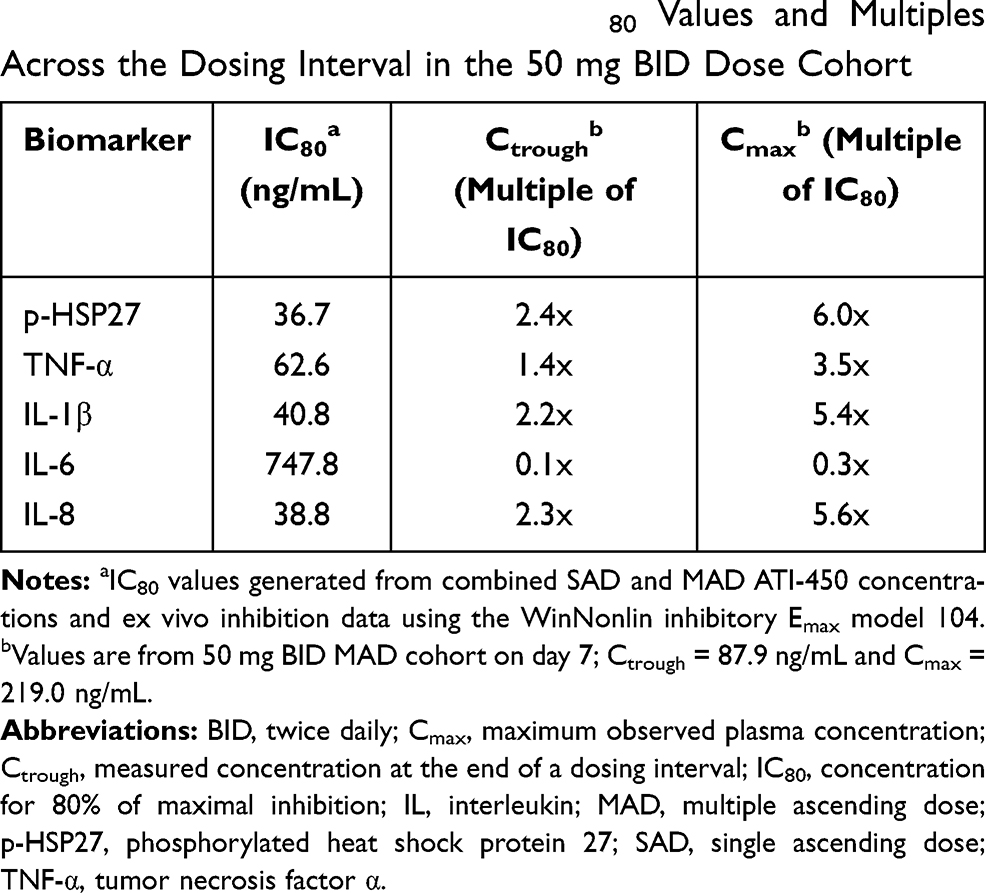 |
Table 4 Cytokine and Biomarker IC80 Values and Multiples Across the Dosing Interval in the 50 mg BID Dose Cohort |
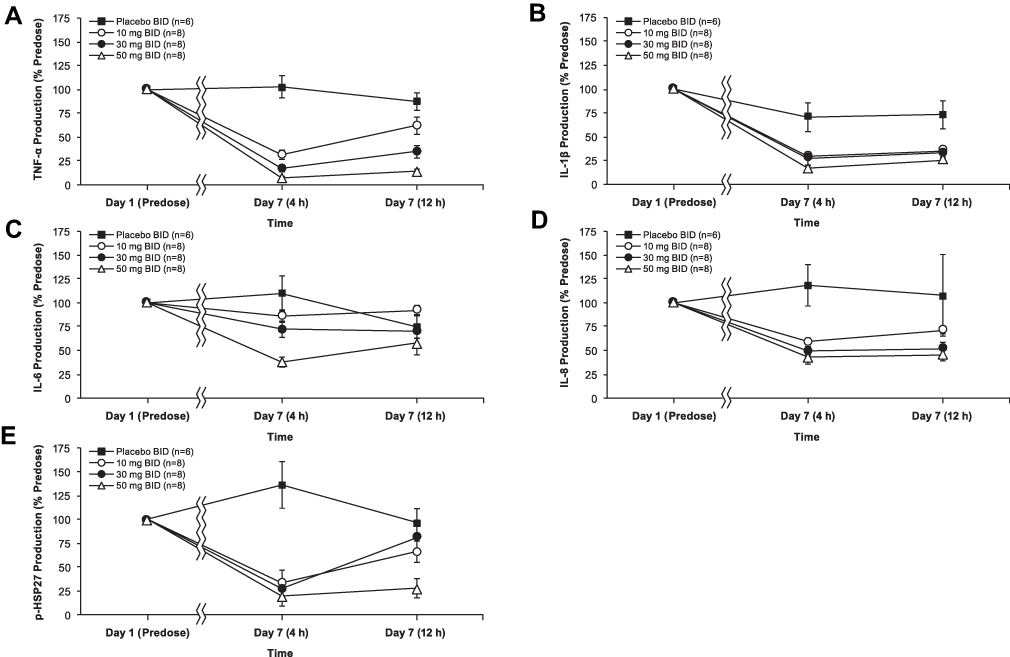 |
Figure 3 Mean (± SEM) cytokine and biomarker levels for (A) TNF-α, (B) IL-1β, (C) IL-6, (D) IL-8, and (E) p-HSP27 in the BID dosing cohorts comparing day 1 pre-dose values (set to 100%) with day 7 values 4 hours post-dose (approximate Cmax) and 12 hours post-dose (Ctrough). Abbreviations: BID, twice daily; Cmax, maximum observed plasma concentration; Ctrough, measured concentration at the end of a dosing interval; IL, interleukin; p-HSP27, phosphorylated heat shock protein 27; TNF, tumor necrosis factor. |
Discussion
A key challenge with global inhibition of p38MAPK is its ubiquitous expression and the broad effects it exerts on cellular physiology as a consequence of the more than 60 substrates that it regulates.4 Through phosphorylation of these substrates, p38MAPK regulates negative feedback loops and anti-inflammatory pathways, which are both involved in downregulating inflammation;4,18 thus, global blockade of p38MAPK pathways may inhibit both proinflammatory and anti-inflammatory pathways, limiting its potential efficacy in some disease states. Furthermore, several p38MAPK substrates are involved in the regulation of cellular function and inhibition of these proteins may result in toxicity.4
The novel approach of selectively inhibiting the p38MAPK/MK2 biomolecular complex instead of p38MAPK alone results in effective blockade of the proinflammatory axis while sparing the anti-inflammatory pathways, negative feedback substrates, and proteins that regulate general cellular function. This approach has the potential to generate safer compounds with greater and more sustained anti-inflammatory efficacy compared with global p38MAPK inhibitors,5 as direct targeting of MK2 has been largely unsuccessful.4
ATI-450 is an oral, small-molecule inhibitor of the p38α MAPK/MK2 inflammatory signaling pathway. In this study, ATI-450 was generally safe and well tolerated by healthy subjects over a range of single doses up to 100 mg and multiple doses up to 50 mg BID for 7 days. All TEAEs resolved without sequelae and there were no discontinuations due to adverse events. Dizziness occurred more frequently in the active arms but there was no clear dose response; the events were generally transient in nature and almost all resolved while on study drug. Dose-dependent reductions in neutrophils were observed and were consistent with the mechanism of action of ATI-450. PK parameters demonstrated that plasma concentrations of ATI-450 increased in a dose-proportional manner after administration of single or multiple ascending doses. Across all SAD and MAD cohorts, median tmax ranged from 2 to 4 hours and mean t1/2 ranged from approximately 9 to 12 hours with no clear relationship to dose. ATI-450 demonstrated concentration-dependent and dose-dependent modulation of the target biomarker p-HSP27 and inhibition of the production of TNF-α, IL-1β, IL-6, and IL-8.
ATI-450 presents a novel mechanism by which to inhibit MK2 and the proinflammatory cytokines activated by the p38MAPK pathway. As an orally administered drug, ATI-450 provides a potential alternative to injectable biologic medications that treat immune-mediated inflammatory diseases. ATI-450 has demonstrated efficacy in animal models for various immune-mediated inflammatory diseases and some types of cancer.5,19 Inhibition of TNF-α and IL-1β observed in healthy subjects in this study align with inhibition of the same cytokine biomarkers demonstrated in animal models for rheumatoid arthritis and cryopyrin-associated periodic syndromes.5
The PD responses in this study have been expressed as ratios of plasma concentrations of ATI-450 to the IC80 for each cytokine. This measurement assists in identification of the key biomarkers that are modulated in response to ATI-450 and allows for adjustment of ATI-450 dosing to maintain plasma concentrations that are above the IC80 for those biomarkers. ATI-450 demonstrated potent inhibition of 4 of the 5 biomarkers (p-HSP27, TNF-α, IL-1β, and IL-8). This inhibition was observed even at trough levels, as demonstrated in the 50 mg BID cohort, in which systemic drug concentrations in excess of the IC80 were achieved for p-HSP27, TNF-α, IL-1β, and IL-8 at Cmax (3.5x to 6.0x) and Ctrough (1.4x to 2.4x). The more modest inhibition by ATI-450 of IL-6 induction in healthy subjects observed in this study may not be indicative of response in patients with rheumatoid arthritis because it does not take into account the impact of inhibiting IL-1β on downstream IL-6 release. Studies evaluating the potency of ATI-450 in human whole blood stimulated with IL-1β demonstrated potency for inhibiting IL-6 that was comparable to that for TNF-α and IL-8 (data not shown).
Limitations of this study include the small number of participants who were healthy volunteers; further study in patients who have an inflammatory disease is required to adequately quantify clinical efficacy and safety. However, this is a limitation that is present in most phase 1 studies of new compounds and is well accepted as the first step to determining whether a new compound is worth investigating further.
ATI-450 was well tolerated at the doses investigated, exhibited dose- and time-independent (ie, linear) PK, and dose-related PD effects. Results of this study support the progression of ATI-450 into phase 2 development for the treatment of rheumatoid arthritis and other immune-mediated inflammatory diseases.
Data Sharing Statement
All data requests should be submitted to the corresponding author for consideration. Access to de-identified data may be granted following review.
Acknowledgments
Writing and editorial assistance was provided to the authors by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and was funded by Aclaris Therapeutics, Inc. The authors would like to thank the volunteers; clinical personnel; Aclaris CMC, Laboratory and Operational Team; and Data Management and Biostatistical staff. Also, the authors thank Daniel Dickerson, MD, the principal study investigator, and Marco Cardillo, CCRA, Madison Bangs, BA, Emma Huff, BS, Aparna Kaul, PhD, Anne Hildebrand, PhD, and Barry Burnette, PhD for their contributions to the conduct of the study and analysis.
Funding
This study was sponsored by Aclaris Therapeutics, Inc., Wayne, Pennsylvania, USA. Neither honoraria nor other form of payment was made for authorship. Writing and editorial assistance was provided to the authors by Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and was funded by Aclaris Therapeutics, Inc.
Disclosure
David Gordon, Joseph B Monahan, Edward T Hellriegel, Heidi Rath Hope and David Burt are employees and shareholders of Aclaris Therapeutics. Dr David Gordon has patents 63076689, 63126173, 63136080, 63136967, 63138672, 63128523, and 63140116 pending. Mr Edward T Hellriegel has patents 62986954, 62986955, 63076689, 63126173, 63136080, 63136967, 63138672, and 63140116 pending to Aclaris Therapeutics. Dr Joseph B Monahan has patents 8563558, 8507499, 9359300, 9365546, 9365547, 9056110, 9115089, 9636333, and 63136967 pending to Aclaris Therapeutics; patents 63138672, 63140116, 63136080, 63126173, 63076689, 62980955, 62986954, 63128523, and 63024160 issued to Aclaris Therapeutics.
References
1. Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol. 2004;4(4):372–377. doi:10.1016/j.coph.2004.03.009
2. Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67(7):909–916. doi:10.1136/ard.2007.074278
3. Kotlyarov A, Neininger A, Schubert C, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1(2):94–97. doi:10.1038/10061
4. Singh RK, Najmi AK, Dastidar SG. Biological functions and role of mitogen-activated protein kinase activated protein kinase 2 (MK2) in inflammatory diseases. Pharmacol Rep. 2017;69(4):746–756. doi:10.1016/j.pharep.2017.03.023
5. Wang C, Hockerman S, Jacobsen EJ, et al. Selective inhibition of the p38α MAPK-MK2 axis inhibits inflammatory cues including inflammasome priming signals. J Exp Med. 2018;215(5):1315–1325. doi:10.1084/jem.20172063
6. Muniyappa H, Das KC. Activation of c-Jun N-terminal kinase (JNK) by widely used specific p38 MAPK inhibitors SB202190 and SB203580: a MLK-3-MKK7-dependent mechanism. Cell Signal. 2008;20(4):675–683. doi:10.1016/j.cellsig.2007.12.003
7. Ma W, Lim W, Gee K, et al. The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J Biol Chem. 2001;276(17):13664–13674. doi:10.1074/jbc.M011157200
8. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14(21):6735–6741. doi:10.1158/1078-0432.CCR-07-4843
9. Murphy C, McGurk M, Pettigrew J, et al. Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin Cancer Res. 2005;11(11):4117–4127. doi:10.1158/1078-0432.CCR-04-1518
10. Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis. 2010;69(Suppl1):i77–82. doi:10.1136/ard.2009.119479
11. Rokosz LL, Beasley JR, Carroll CD, et al. Kinase inhibitors as drugs for chronic inflammatory and immunological diseases: progress and challenges. Expert Opin Ther Targets. 2008;12(7):883–903. doi:10.1517/14728222.12.7.883
12. De Buck S, Hueber W, Vitaliti A, et al. Population PK-PD model for tolerance evaluation to the p38 MAP kinase inhibitor BCT197. Pharmacometr Syst Pharmacol. 2015;4(12):691–700. doi:10.1002/psp4.12037
13. Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009;60(5):1232–1241. doi:10.1002/art.24485
14. Fiore M, Forli S, Manetti F. Targeting mitogen-activated protein kinase-activated protein kinase 2 (MAPKAPK2, MK2): medicinal chemistry efforts to lead small molecule inhibitors to clinical trials. J Med Chem. 2016;59(8):3609–3634. doi:10.1021/acs.jmedchem.5b01457
15. Hitti E, Iakovleva T, Brook M, et al. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26(6):2399–2407. doi:10.1128/MCB.26.6.2399-2407.2006
16. Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol. 2001;21(19):6461–6469. doi:10.1128/MCB.21.9.6461-6469.2001
17. Chrestensen CA, Schroeder MJ, Shabanowitz J, et al. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem. 2004;279(11):10176–10184. doi:10.1074/jbc.M310486200
18. Jones DS, Jenney AP, Joughin BA, Sorger PK, Lauffenburger DA. Inflammatory but not mitogenic contexts prime synovial fibroblasts for compensatory signaling responses to p38 inhibition. Sci Signal. 2018;11:520. doi:10.1126/scisignal.aal1601
19. Murali B, Ren Q, Luo X, et al. Inhibition of the stromal p38MAPK/MK2 pathway limits breast cancer metastases and chemotherapy-induced bone loss. Cancer Res. 2018;78(19):5618–5630. doi:10.1158/0008-5472.CAN-18-0234
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.