")
Back to Journals » Journal of Inflammation Research » Volume 8
Differential inflammatory response to Helicobacter pylori infection: etiology and clinical outcomes
Authors White J, Winter J, Robinson K
Received 20 May 2015
Accepted for publication 10 July 2015
Published 13 August 2015 Volume 2015:8 Pages 137—147
DOI https://doi.org/10.2147/JIR.S64888
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jonathan Richard White,1 Jody Anne Winter,2 Karen Robinson1
1NIHR Biomedical Research Unit in Gastrointestinal and Liver Diseases at Nottingham University Hospitals NHS Trust and The University of Nottingham, Nottingham, 2Interdisciplinary Biomedical Research Centre, School of Science and Technology, Nottingham Trent University, Nottingham, UK
Abstract: The bacterial pathogen Helicobacter pylori commonly colonizes the human gastric mucosa during early childhood and persists throughout life. The organism has evolved multiple mechanisms for evading clearance by the immune system and, despite inducing inflammation in the stomach, the majority of infections are asymptomatic. H. pylori is the leading cause of peptic ulcer disease and gastric cancer. However, disease outcomes are related to the pattern and severity of chronic inflammation in the gastric mucosa, which in turn is influenced by both bacterial and host factors. Despite over 2 decades of intensive research, there remains an incomplete understanding of the circumstances leading to disease development, due to the fascinating complexity of the host–pathogen interactions. There is accumulating data concerning the virulence factors associated with increased risk of disease, and the majority of these have pro-inflammatory activities. Despite this, only a small proportion of those infected with virulent strains develop disease. Several H. pylori virulence factors have multiple effects on different cell types, including the induction of pro- and anti-inflammatory, immune stimulatory, and immune modulatory responses. The expression of multiple virulence factors is also often linked, making it difficult to assess the meaning of their effects in isolation. Overall, H. pylori is thought to usually modulate inflammation and limit acute damage to the mucosa, enabling the bacteria to persist. If this delicate balance is disturbed, disease may then develop.
Keywords: Helicobacter pylori, inflammation, mucosal immunity, peptic ulcer disease, gastric cancer
Introduction
Barry Marshall and Robin Warren were the first to isolate a spiral bacterium, now known as Helicobacter pylori, from inflamed mucosal tissue of the human stomach.1 In most cases, the infection is asymptomatic. The severity and type of disease depend on the characteristics of the colonizing strain and how it interacts with the host to cause chronic inflammation. Many of the main H. pylori virulence factors have multiple effects on different cell types and may have both pro- and anti-inflammatory activities. It is therefore necessary to assess the relative importance and net effects of these factors in order to understand the circumstances leading to disease development.
H. pylori infection
H. pylori has coevolved with humans over the last 60,000 years.2 It typically first colonizes the gastric mucosa during early childhood and persists lifelong in the absence of effective eradication treatment.3 It is estimated that approximately 50% of the world’s population is colonized, although the prevalence differs between countries. Developing countries have a much higher infection rate than developed countries, and this is thought to be due to differences in living conditions and the use of antibiotics, especially in childhood.4 Globally, H. pylori prevalence is declining. In the US, approximately 10% of individuals under the age of 20 are infected compared to 40% over 60 years of age.5 This higher rate of H. pylori infection seen with increasing age is not due to acquisition of the infection at a later age, but a birth cohort effect.
H. pylori is found almost exclusively in humans. Other Helicobacter species are occasionally found in humans and these are thought to be acquired from domestic pets. The exact route of infectious transmission is not clear, but person-to-person transmission is likely to be a combination of fecal–oral and oral–oral routes. H. pylori strains are usually isolated from gastric biopsy tissue, but it is also possible for the bacterium to be isolated from saliva, gastric reflux fluid, and vomitus.6
Consequences of H. pylori infection
Acute infection
Acute infectious symptoms (such as nausea, halitosis, dyspepsia, and malaise) are experienced by most infected adults but the symptoms are variable. These tend to resolve within 2 weeks. Supporting evidence for the above is mainly from cases of deliberate ingestion. When examined histologically, acute infection is accompanied by severe gastritis, characterized by infiltration of neutrophils and inflammatory cells with marked persistent lymphocyte penetration. A reduction in stomach acid secretion also occurs simultaneously.7 It is unknown whether children suffer similar symptoms or whether histological features are concordant.8
Chronic infection and disease outcome
Chronic H. pylori infection leads to local inflammation of the gastric mucosa (gastritis). Disease risk increases with the level of inflammation, but the pattern of inflammation determines the disease outcome. Host genetic factors, bacterial virulence, environmental factors, and age of infection all influence the distribution of resulting gastritis.8 These complex and only partially understood interactions are thought to explain why only 15% of infected individuals develop disease in their lifetime.9
The most common and serious complications of H. pylori infection include peptic ulcer disease, distal gastric adenocarcinoma, and primary gastric mucosa associated lymphoid tissue (MALT) lymphoma. Other conditions associated with H. pylori infection include dyspepsia, atrophic gastritis, iron deficiency anemia, and idiopathic thrombocytopenia purpura. In contrast, epidemiological evidence also suggests a protective association between H. pylori infection and disorders such as gastroesophageal reflux disease (GERD), esophageal adenocarcinoma, inflammatory bowel disease, multiple sclerosis, and asthma.10,11
Peptic ulceration
Peptic ulcers are breaks in the lining of the duodenal or gastric mucosa, most commonly caused by H. pylori and nonsteroidal anti-inflammatory drugs. Peptic ulcer disease is associated with significant mortality and complications include hemorrhage and perforation. H. pylori eradication heals existing ulcers and prevents their recurrence.12
H. pylori is the causative agent in over 75% of duodenal ulcer cases. Antral-predominant inflammation leads to increased gastric acid output (Figure 1). Gastric metaplasia of the duodenal epithelium then permits H. pylori to colonize and cause inflammation, which may lead to duodenal ulceration. H. pylori is also the leading cause of gastric ulcers, which develop in patients with pangastritis. Here the acid output is normal or reduced, thus preventing the development of duodenal ulcers, but gastric ulcers may develop. Premalignant lesions and gastric adenocarcinoma may also arise.2,13,14
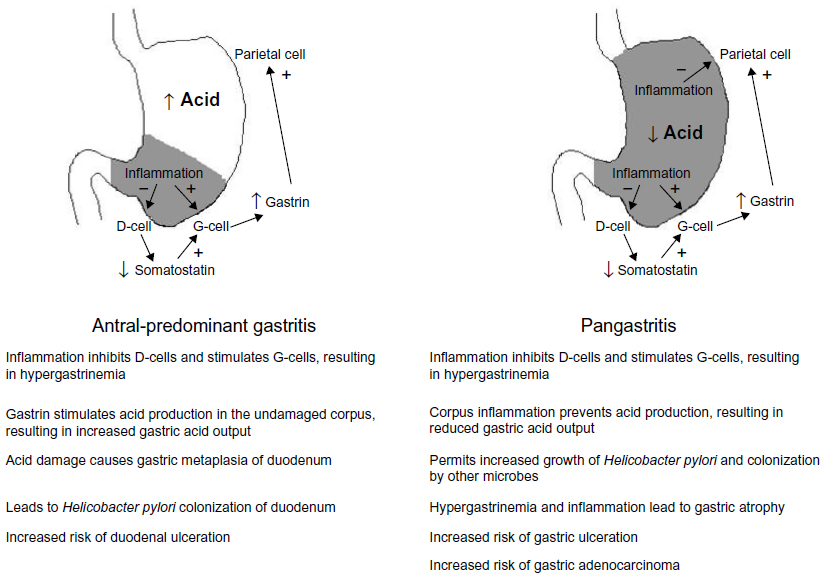 | Figure 1 The effect of gastritis pattern on gastric acid production and associations with duodenal and gastric disease. |
Gastric adenocarcinoma
Gastric cancer is ranked the fifth most common malignancy worldwide with an estimated 100,000 new cases per year.15 Most cases are found in Asia, with over two-thirds occurring in the People’s Republic of China.16 Gastric cancer is the third most common cause of cancer-related deaths, since initial diagnosis is usually at a late stage.17,18 It can be divided into two subtypes depending on the location: cardia (arising from the gastroesophageal junction) and noncardia (arising from the distal stomach). Cardia gastric cancers share risk factors with esophageal adenocarcinoma, Barrett’s esophagus, obesity, and GERD.15 Noncardia gastric cancer is strongly associated with H. pylori, and it is thought that up to 89% may be attributed to the infection. Thus H. pylori has been classified as a human carcinogen.19 The lifetime risk of an H. pylori-infected individual developing gastric cancer is 1%–2%.14,20
There are two histological types of gastric adenocarcinoma: intestinal and diffuse. The intestinal type develops gradually, following a stepwise progression driven by inflammation. H. pylori infection of the normal gastric mucosa leads to a state of chronic gastritis, which later leads to atrophic gastritis (characterized by gland loss and infiltration of inflammatory cells), intestinal metaplasia (where gastric epithelial cells are replaced with those of an intestinal type), dysplasia (neoplasia confined to epithelial cells), and finally adenocarcinoma.21 The diffuse type usually affects younger patients and is not associated with intestinal metaplasia. Although thought to be triggered by H. pylori infection, the exact mechanism is not known.17 H. pylori eradication has been shown to reduce the incidence of atrophic gastritis but does not result in a reduction in gastric cancer incidence unless achieved before the appearance of premalignant changes.22
MALT lymphoma
H. pylori colonization is strongly linked to MALT lymphoma.23 Due to the rarity of this condition, the exact number of individuals coinfected with H. pylori is not known but the condition occurs in less than 1% of those who are colonized. Low-grade B-cell MALT lymphomas normally regress following H. pylori eradication treatment.24
Host response to H. pylori and its association with disease risk
H. pylori elicits a strong immune response, stimulating the expression of cytokines and chemokines from gastric epithelial cells. These factors attract neutrophils, macrophages, dendritic cells (DCs), natural killer (NK) cells, and lymphocytes,8,25 and induce the release of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Gastric carcinogenesis is associated with ROS/RNS-mediated DNA damage, silencing of tumor suppressor genes via DNA methylation, histone epigenetic modification, and epithelial–mesenchymal transition.26 The level and nature of the immune response varies and this affects the risk of disease development.27
Interactions of H. pylori with gastric epithelium
The surface of the gastric mucosa is covered by protective mucus consisting of a cell-associated layer (predominantly MUC1) and secreted mucin (mainly MUC5AC).28 This layer has a profound impact on H. pylori adhesion to the gastric mucosa. H. pylori interacts with mucin fucosylated Lewisb blood group antigen moieties via the BabA adhesin.29 During gastritis, there is an increase in sialylated mucin structures such as sialyl-Lewisx and sialyl-Lewisa, and these bind to the adhesin SabA.30 Recently, the LabA adhesin was identified as binding a motif on MUC5AC.31 The mucus layer is also important for H. pylori motility; the organism reduces its viscosity in order to move through it.32 Mucins also have natural antibiotic activity against the bacterium,33 and H. pylori binding to MUC1 induces multiple effects on host cells including the modulation of inflammation.34
Innate immunity and inflammation
Pattern recognition receptors (PRRs) expressed by gastric epithelial cells interact with H. pylori and activate inflammatory gene expression. These molecules, which include the toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), recognize pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide (LPS), flagellins, and cell wall peptides.35 Some H. pylori PAMPs are modified to weakly activate PRRs, since its tetra-acetylated LPS is poorly recognized by TLR4, and the FlaA flagellin binds much less efficiently to TLR5.36 Unlike other bacteria, TLR2 appears to be the main receptor for H. pylori LPS, but TLR2 is also activated by other components (Table 1).37 Polymorphisms in TLR1, TLR2, TLR4, TLR5, and TLR9 genes have been associated with more severe gastritis and increased risk of premalignant pathology.38,39
 | Table 1 Major virulence factors and their effects on inflammation and the immune response |
Interaction of the cytotoxin-associated gene pathogenicity island (cagPAI) encoded type IV secretion system (T4SS) with gastric epithelial cells results in the transfer of soluble peptidoglycan components into the cytoplasm, NOD1 activation, and pro-inflammatory gene expression.40 The largest NLR subfamily includes the NLRPs, which are the scaffolding proteins of inflammasomes. NLRPs interact with adaptor proteins leading to the activation of caspase-1, which controls the maturation of inflammatory cytokines such as IL-1β and IL-18.41 There is increased expression of these factors and other NLRP3-related molecules in infected gastric tissue.39,42
Autophagy, the pathway for breakdown and removal of damaged cellular components, is an important homeostatic mechanism which regulates inflammatory signaling.43 Dysregulation of autophagy has been reported to result in increased production of ROS and DNA damage. This leads to accumulations of damaged organelles, changes in cell metabolism, and carcinogenesis.44 H. pylori-mediated induction of autophagy has been reported;45,46 however, more virulent H. pylori isolates rapidly downregulate autophagy in gastric epithelial and monocytic cells lines.47 Characterization of the H. pylori B128 7.13 strain, which causes gastric cancer in Mongolian gerbils, revealed a mutation in a peptidoglycan deacetylase gene (pgdA). This led to reduced autophagy in vitro and cancer development in animals.48
Secreted antimicrobial peptides are produced in response to H. pylori. Elevated levels of human beta defensin 2 (hBD2), hBD3, hBD4, adrenomedullin, angiogenin, alpha defensins 1, 2, and 3, and the human cationic antimicrobial peptide 18 (LL-37) are present in the gastric mucosa of H. pylori-infected patients and/or infected human gastric epithelial cells in vitro.49–54
As a consequence of H. pylori interactions with the epithelium, pro-inflammatory chemokines and cytokines, including IL-8, IL-1β, tumor necrosis factor alpha (TNFα), IL-6, IL-12, CCL2-5, CCL20, and CXCL1-3, are upregulated in the infected gastric mucosa.55,56 Gene polymorphisms resulting in increased expression of pro-inflammatory cytokines (IL-6, IL-8, TNFα, IL-1β), or reduced expression of anti-inflammatory cytokines (IL-10), are associated with higher risk of disease.27,57,58 The presence of chemokines leads to the recruitment of immune cells, including neutrophils, macrophages, DCs, NK cells, and lymphocytes.2 Neutrophils contribute to gastritis by secreting inflammatory cytokines and releasing tissue damaging factors from neutrophilic granules. They also phagocytose bacteria, and within the phagolysosomes the bacteria are exposed to bactericidal factors, including myeloperoxidase and matrix metalloproteinases which degrade cell walls and proteins, and ROS and RNS, which induce DNA damage. H. pylori prevents the oxidative burst and can survive intracellularly within neutrophils.59 Helicobacter-infected neutrophil-depleted mice appear to be colonized at the same densities as normal mice.60 These data imply that neutrophils may play a lesser role in protective immunity, but contribute to mucosal damage.
Macrophage-depleted mice have a significantly reduced H. pylori gastritis severity.61 Both M1 and M2 macrophages are present in the infected gastric mucosa.62 M1 macrophages secrete pro-inflammatory cytokines and nitric oxide and have potent bactericidal activity compared with M2 macrophages, which promote cell proliferation and tissue repair.63 H. pylori is able to survive phagocytosis by macrophages, since it induces the fusing together of phagosomes to form megasomes without lysosomal fusion.64 The megasomes provide a protected intracellular niche and may even contribute to the persistence of infection.65 H. pylori is also able to neutralize the released ROS via catalase activity, and arginase production by the bacteria inhibits nitric oxide production.66,67 Chronic exposure to ROS and RNS, however, results in host cell DNA damage and favors cancer development.
Mast cells are also present at higher frequencies in the H. pylori-infected human gastric mucosa.68 The role of these cells has not been widely studied, but they may be involved in tissue repair, inflammation, and vaccine-mediated clearance of the infection.69
DCs in H. pylori-infected gastric tissue tend to be of a myeloid type (mDCs) and express DC-SIGN and high levels of HLA-DR, but are semi-mature and tolerogenic.70–75 Together with the DC response, macrophage-derived cytokines also have an important influence on the development and balance of the adaptive immune response.76 It has recently been shown that both human gastric epithelial cells and gastric mucosal DCs produce retinoic acid (RA), an important factor that regulates inflammation. When infected with H. pylori, however, mucosal RA production is impaired, leading to increased inflammation and possibly resulting in incresed risk of peptic ulceration and gastric carcinogenesis.77
Despite recent interest in invariant lymphoid and NK cell populations there is very little data on these in the context of H. pylori infection. NKT cells are more abundant in the infected gastric mucosa, and a larger NK cell population was detected in the peripheral blood of infected donors.78,79 How these cell types contribute to disease is not understood; however, NK cell-derived perforin and granzymes may cause damage to host cells. NK cells respond to incubation with H. pylori or its secreted products by secreting inflammatory cytokines such as interferon-gamma (IFNγ) and TNFα.80
Adaptive immunity
Strong IgG and IgA antibody responses are present in H. pylori-infected individuals and these may trigger autoimmunity.66 Molecular mimicry by H. pylori induces antibodies that react with host antigens in the gastric mucosa, such as the parietal cell H+, K+-ATPase.81 Such autoreactive antibodies are frequently present in the serum of infected patients, and these may increase local inflammation and damage in the stomach or elsewhere.82
H. pylori infection induces a vigorous T-cell response, which includes both CD4+ and CD8+ cells. The gastric mucosa of infected humans and mice contains increased numbers of CD8+ cells and these contribute to inflammation and disease.83 More is known about the CD4+ T-helper (Th) response. The main Th subsets induced by H. pylori infection are pro-inflammatory Th17 and Th1 and anti-inflammatory regulatory T-cell (Treg) populations; however, Th2 and Th22 responses have also been reported.84–88 Th-derived cytokines orchestrate the host response, having an impact on H. pylori-induced inflammation and immunity, as well as playing an important role in determining H. pylori-associated disease risk.
Th1 cells secrete cytokines IFNγ and TNFα, which stimulate macrophages to secrete further pro-inflammatory factors and have more bactericidal activity.89 Th17 cells secrete IL-17A, IL-17F, IL-21, and IL-22, and stimulate the expression of antimicrobial peptides, ROS, RNS, and chemokines. This leads to increased inflammation and neutrophil recruitment.90 H. pylori-induced expression of B-cell activating factor of TNF family (BAFF) by macrophages is important for the differentiation of Th17 cells.76 In H. pylori-infected mice, a Th17 response is observed in addition to the Th1 response, leading to more severe gastritis.91
In the infected human and mouse gastric mucosa, the severity of gastritis correlates with the number of Th1 and Th17 cells.92–94 Although a strong Th1 response may contribute to carcinogenesis, there is evidence that a high Th1 response leads to a better prognosis for gastric cancer patients due to stronger antitumor immunity.95 On the other hand, high-level Th17 and Th22 responses are associated with gastric cancer progression and poor survival, possibly due to the role of their cytokines in angiogenesis and tumor invasiveness.96 Gastric Th1 cells from the antrum of patients with peptic ulcer disease provide help for B-cell antibody isotype switching, induce epithelial cells to express higher levels of MHC class II, and also have H. pylori-specific cytolytic activity.94 They are proposed to contribute to disease via cytotoxicity against antigen-presenting epithelial cells, and may also promote autoimmune reactions such as in autoimmune gastritis and gastric MALT lymphoma.81,94 T-cell clones from patients with MALT lymphoma, however, are commonly Th0 rather than Th1 types. These have a markedly reduced cytotoxic activity against B-cells and an impaired ability to induce apoptosis in T-cells. This may explain the unchecked B-cell expansion in MALT lymphoma.97
H. pylori has multiple mechanisms for directing the immune system away from a pro-inflammatory T-cell response and toward a suppressive Treg response.98 Increased numbers of Tregs are observed in the gastric mucosa and peripheral blood of H. pylori-infected patients, and peptic ulceration is more frequently found in those with reduced Treg numbers in their gastric mucosa.56,84,92,99 Tregs may act by secreting cytokines such as IL-10 and transforming growth factor beta to modulate inflammation, or they may act via contact-mediated mechanisms.100 H. pylori influences DCs to promote the differentiation of naïve T-cells into Tregs. Such responses are reported to protect against extra-gastric immune and inflammatory conditions including asthma and inflammatory bowel disease.74,101
In addition to Treg induction, H. pylori utilizes many other mechanisms to modulate the immune and inflammatory response. Several virulence factors have anti- as well as pro-inflammatory functions (Table 1), and expression of B7-H1 is upregulated in gastric epithelial cells during H. pylori infection. Interaction with this molecule suppresses T-cell activity.102
Virulence factors and inflammation
H. pylori produces numerous virulence factors, many of which are highly polymorphic, phase variable, genetically linked, and/or have diverse and sometimes opposing functions. This diversity, together with the complexity of the host immune response, makes it difficult to define clearly the relative roles of individual virulence factors in H. pylori-mediated inflammation and disease. Pro- and anti-inflammatory influences of some of the best-studied H. pylori virulence factors are briefly summarized in this section.
The cag pathogenicity island and CagA
The cagPAI is a 40 kb horizontally transmitted segment of DNA. It encodes a T4SS, with CagL at the tip of the needle-like structure which binds to α5β1 integrin on host cells.103 CagA, an immunodominant 120-145 kDa protein, is injected into cells through the T4SS together with peptidoglycan peptides. This process activates NF-κB, triggering the secretion of pro-inflammatory cytokines and chemokines, most notably IL-8. Once inside the host cell, CagA is rapidly tyrosine phosphorylated at its EPIYA (Glu-Pro-Ile-Tyr-Ala) motifs by Src kinases and then interacts with the SHP-2 cellular phosphatase. This ultimately leads to cytoskeletal changes via actin rearrangement.104 Unphosphorylated CagA also interacts with numerous targets inside the host cell including the tight junction protein ZO-1 (causing tight junction disruption) and E-cadherin (disrupting E-cadherin/β-catenin complexes to promote β-catenin mediated upregulation of genes with oncogenic potential).105,106 Taken together, cagPAI activity drives a scattering/elongation, or “hummingbird”, phenotype and pro-inflammatory responses in gastric epithelial cells. However, cagPAI-mediated NF-κB activation also downregulates the expression of the antimicrobial and pro-inflammatory defensin hBD1, and the activation of SHP-2 by CagA prevents EGFR-mediated expression of hBD-3.53,107 Downregulation of these β-defensins may help promote the persistence of CagA-positive H. pylori strains.
The cagPAI may be present fully, partially, or not at all. Strains with a functional cag T4SS are strongly associated with increased gastric cancer risk. The cagA gene sequence is itself polymorphic. EPIYA motifs may be categorized as EPIYA-A, B, C, or D depending on their flanking sequences, with EPIYA-A, B, and C found in Western CagA types and EPIYA-A, B, and D found in East Asian CagA. A larger number of EPIYA-C motifs or the presence of an EPIYA-D increases interactions with SHP-2, and is associated with a higher risk of intestinal metaplasia and gastric cancer.108 Strains lacking CagA may induce inflammation via other cagPAI-dependent mechanisms. If the T4SS is functional, peptidoglycan peptides enter the cell and activate NOD1-mediated signaling.40 Additionally, interaction of CagL with the α5β1 integrin is sufficient to activate NF-κB and induce IL-8 expression.109
Vacuolating cytotoxin (VacA)
Virtually all H. pylori strains possess the vacA gene but it is highly polymorphic, with two alternative allelic variants for the signal (s1/s2), intermediate (i1/i2), and mid- (m1/m2) regions. The mid-region plays a role in host cell binding, and m1 forms are able to bind a wider range of cell types than m2. s2 and i2 VacA have reduced activity compared to the s1 and i1 variants.110,111 VacA is a pore-forming toxin, originally named for its ability to induce vacuolation in gastric epithelial cells in vitro. A myriad of other functions have also been attributed to it, including the induction of epithelial cell apoptosis, autophagy, and inhibition of T-cell activation (Table 1).46,112,113
The vacA s1 and i1 alleles are associated with increased risk of peptic ulceration, atrophy, and gastric adenocarcinoma, but genetic linkage between these alleles and the presence of cagA makes it difficult to determine with certainty the contribution of each individual factor.111 There is also functional linkage between VacA and CagA, for example, VacA induces apoptosis in gastric epithelial cells, but CagA blocks this activity and can also prevent VacA gaining access into host cells.114,115 This may protect the host cell to which the bacterium has adhered, while allowing continued VacA-mediated disruption of more distant cells. Conversely, VacA inhibits the induction of the hummingbird phenotype by CagA.116 Recently, VacA and another secreted H. pylori protein, γ-glutamyl transferase have been shown to tolerize DCs, promoting Treg responses and protecting against asthma in a mouse model.117,118 Since both s1i1m1 and s2i2m2 VacA can tolerize DCs, this anti-inflammatory function may be one reason for the otherwise unexplained maintenance of apparently nonfunctional type 2 toxin variants in the H. pylori genome.
DupA and tfs4
H. pylori genomes contain regions of low GC content and high diversity, known as “plasticity zones”. The number and contents of PZs vary between strains, and several PZ-specific genes are associated with disease. Of these, one of the best studied is the duodenal ulcer-promoting gene, dupA. The tfs4 gene cluster comprises dupA and other vir homologues which are thought to encode a type IV secretion system.119
Although dupA was initially identified as a duodenal ulcer-promoting virulence factor, numerous subsequent conflicting studies have left the role of dupA in disease unclear. This is likely due to the requirement for other components of the tfs4 to produce a functional type IV secretion system, making dupA alone an imperfect marker.120,121 The presence of dupA in clinical H. pylori isolates is associated with increased IL-8 levels in the antrum of infected individuals.122,123
H. pylori neutrophil-activating protein (HP-NAP)
HP-NAP is a highly conserved dodecameric 150 kDa protein, named for its ability to stimulate endothelial adhesion and production of oxygen radicals by neutrophils. The protein is also a neutrophil chemottractant and stimulates these cells to produce pro-inflammatory cytokines and chemokines. Since neutrophil infiltration is a dominant characteristic of H. pylori gastritis, HP-NAP may play a central role in H. pylori-associated disease. HP-NAP may also associate with the outer membrane of intact bacteria and play a role in binding to host mucin carbohydrates.124,125
Adhesins
Adherent bacteria might be expected to induce stronger inflammatory responses than nonadherent bacteria. H. pylori possesses several major adhesins including the blood group antigen binding adhesin (BabA), sialic acid binding adhesin (SabA), and OipA.
BabA is expressed by a subset of H. pylori strains, and it binds to difucosylated Leb blood group antigens on host epithelial cells.29 BabA may facilitate close association with the epithelium for delivery of other virulence factors, and indeed babA2-positive strains are associated with increased gastric mucosal granulocyte infiltration and IL-8 expression.126
sabA is a phase-variable gene that may be switched “on” or “off”. SabA allows H. pylori to adhere to sialylated Lewis antigens, which are present during gastritis.30 BabA plays the major role in bacterial adhesion soon after colonization, and SabA becomes the predominant adhesin once chronic inflammation is established. Colonization with SabA-producing strains is associated with increased risk of gastric cancer, atrophy, and intestinal metaplasia; however, there is a negative association between SabA expression and neutrophil infiltration.127
OipA is another phase-variable adhesin, and it has several other putative functions including the induction of actin stress fiber formation and IL-8 production by epithelial cells. OipA shares some activities and host cell signaling pathways with the cagPAI, and IL-8 expression may be induced synergistically. There is also OipA-specific signaling, however. While “on” OipA is associated with increased risk of duodenal ulcer and gastric cancer, defining the relative roles of cagPAI and oipA is not straightforward because oipA “on” strains are also likely to be cagPAI-positive.128,129
Conclusion
H. pylori infection strongly stimulates gastric mucosal inflammation and both the innate and acquired immune response. The usual consequence of H. pylori infection is chronic asymptomatic gastritis, probably because the bacteria have adapted to evade and suppress the immune response. The inflammatory response is important in the development of gastric adenocarcinoma; however, there is growing evidence that other aspects of the local and systemic response are also central to disease pathogenesis. It may ultimately be possible to develop prognostic tests based on these parameters, along with bacterial virulence types, to predict who is at risk of developing gastric cancer. However, since many of the major virulence factors have both pro- and anti-inflammatory activities, further research is necessary to gain a complete understanding of the circumstances leading to disease occurrence.
Acknowledgments
KR’s research is supported by the National Institute for Health Research (NIHR), through the Biomedical Research Unit in Gastrointestinal and Liver Diseases at Nottingham University Hospitals NHS Trust and the University of Nottingham. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1(8390):1311–1315. | |
Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009; 119(9):2475–2487. | |
Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113(3):321–333. | |
Peleteiro B, Bastos A, Ferro A, Lunet N. Prevalence of Helicobacter pylori infection worldwide: a systematic review of studies with national coverage. Dig Dis Sci. 2014;59(8):1698–1709. | |
Grad YH, Lipsitch M, Aiello AE. Secular trends in Helicobacter pylori seroprevalence in adults in the United States: evidence for sustained race/ethnic disparities. Am J Epidemiol. 2012;175(1):54–59. | |
Goodman KJ, Correa P. Transmission of Helicobacter pylori among siblings. Lancet. 2000;355(9201):358–362. | |
Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. 1987;82(3):192–199. | |
Robinson K, Argent RH, Atherton JC. The inflammatory and immune response to Helicobacter pylori infection. Best Pract Res Clin Gastroenterol. 2007;21(2):237–259. | |
Atherton JC. The pathogenesis of Helicobacter pylori-induced gastro-duodenal diseases. Annu Rev Pathol. 2006;1:63–96. | |
Robinson K. Helicobacter pylori-mediated protection against extra-gastric immune and inflammatory disorders: the evidence and controversies. Diseases. 2015;3(2):34–55. | |
Arnold IC, Hitzler I, Muller A. The immunomodulatory properties of Helicobacter pylori confer protection against allergic and chronic inflammatory disorders. Front Cell Infect Microbiol. 2012;2:10. | |
Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374(9699):1449–1461. | |
Malfertheiner P. The intriguing relationship of Helicobacter pylori infection and acid secretion in peptic ulcer disease and gastric cancer. Dig Dis. 2011;29(5):459–464. | |
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19(3):449–490. | |
Colquhoun A, Arnold M, Ferlay J, Goodman KJ, Forman D, Soerjomataram I. Global patterns of cardia and non-cardia gastric cancer incidence in 2012. Gut. 2015. Epub 2015 Mar 6. | |
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. | |
Peek RM Jr, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2(1):28–37. | |
Herrero R, Park JY, Forman D. The fight against gastric cancer – the IARC Working Group report. Best Pract Res Clin Gastroenterol. 2014;28(6):1107–1114. | |
Plummer M, Franceschi S, Vignat J, Forman D, de Martel C. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer. 2015;136(2):487–490. | |
Kuipers EJ. Review article: exploring the link between Helicobacter pylori and gastric cancer. Aliment Pharmacol Ther. 1999;13(Suppl 1):3–11. | |
Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol. 1995;19(Suppl 1):S37–S43. | |
Malfertheiner P, Fry LC, Monkemuller K. Can gastric cancer be prevented by Helicobacter pylori eradication? Best Pract Res Clin Gastroenterol. 2006;20(4):709–719. | |
Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet. 1991;338(8776):1175–1176. | |
Parsonnet J, Isaacson PG. Bacterial infection and MALT lymphoma. N Engl J Med. 2004;350(3):213–215. | |
Sundquist M, Quiding-Jarbrink M. Helicobacter pylori and its effect on innate and adaptive immunity: new insights and vaccination strategies. Expert Rev Gastroenterol Hepatol. 2010;4(6):733–744. | |
Na HK, Woo JH. Helicobacter pylori induces hypermethylation of CpG islands through upregulation of DNA methyltransferase: possible involvement of reactive oxygen/nitrogen species. J Cancer Prev. 2014; 19(4):259–264. | |
Macarthur M, Hold GL, El-Omar EM. Inflammation and cancer II. Role of chronic inflammation and cytokine gene polymorphisms in the pathogenesis of gastrointestinal malignancy. Am J Physiol Gastrointest Liver Physiol. 2004;286(4):G515–G520. | |
Dunne C, Dolan B, Clyne M. Factors that mediate colonization of the human stomach by Helicobacter pylori. World J Gastroenterol. 2014; 20(19):5610–5624. | |
Ilver D, Arnqvist A, Ogren J, et al. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279(5349):373–377. | |
Mahdavi J, Sonden B, Hurtig M, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002; 297(5581):573–578. | |
Rossez Y, Gosset P, Boneca IG, et al. The lacdiNAc-specific adhesin LabA mediates adhesion of Helicobacter pylori to human gastric mucosa. J Infect Dis. 2014;210(8):1286–1295. | |
Bansil R, Celli JP, Hardcastle JM, Turner BS. The influence of mucus microstructure and rheology in Helicobacter pylori infection. Front Immunol. 2013;4:310. | |
Fukuda M, Kawakubo M, Ito Y, Kobayashi M, Lee H, Nakayama J. Assay of human gastric mucin as a natural antibiotic against Helicobacter pylori. Methods Enzymol. 2006;415:164–179. | |
Park YS, Guang W, Blanchard TG, Kim KC, Lillehoj EP. Suppression of IL-8 production in gastric epithelial cells by MUC1 mucin and peroxisome proliferator-associated receptor-gamma. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G765–G774. | |
Smith SM. Role of Toll-like receptors in Helicobacter pylori infection and immunity. World J Gastrointest Pathophysiol. 2014; 5(3):133–146. | |
Salama NR, Hartung ML, Muller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nat Rev Microbiol. 2013;11(6):385–399. | |
Smith SM, Moran AP, Duggan SP, et al. Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol. 2011;186(4):2462–2471. | |
El-Omar EM, Ng MT, Hold GL. Polymorphisms in Toll-like receptor genes and risk of cancer. Oncogene. 2008;27(2):244–252. | |
Castano-Rodriguez N, Kaakoush NO, Mitchell HM. Pattern-recognition receptors and gastric cancer. Front Immunol. 2014;5:336. | |
Viala J, Chaput C, Boneca IG, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5(11):1166–1174. | |
Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol. 2015;25(5):308–315. | |
Jiang J, Liu S, Luo J, et al. [The expressions of NLRP3 inflammasome and its downstream molecules in the mouse model of Helicobacter pylori infection]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2013;29(8):785–788. Chinese. | |
Joven J, Guirro M, Marine-Casado R, Rodriguez-Gallego E, Menendez JA. Autophagy is an inflammation-related defensive mechanism against disease. Adv Exp Med Biol. 2014;824:43–59. | |
Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. | |
Raju D, Hussey S, Ang M, et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology. 2012;142(5):1160–1171. | |
Terebiznik MR, Raju D, Vazquez CL, et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy. 2009;5(3):370–379. | |
Castano-Rodriguez N, Kaakoush NO, Goh KL, Fock KM, Mitchell HM. Autophagy in Helicobacter pylori infection and related gastric cancer. Helicobacter. Epub 2015 Feb 9. | |
Suarez G, Romero-Gallo J, Piazuelo MB, et al. Modification of Helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res. 2015;75(8):1749–1759. | |
Isomoto H, Mukae H, Ishimoto H, et al. Elevated concentrations of alpha-defensins in gastric juice of patients with Helicobacter pylori infection. Am J Gastroenterol. 2004;99(10):1916–1923. | |
Allaker RP, Kapas S. Adrenomedullin and mucosal defence: interaction between host and microorganism. Regul Pept. 2003;112(1–3):147–152. | |
Hase K, Murakami M, Iimura M, et al. Expression of LL-37 by human gastric epithelial cells as a potential host defense mechanism against Helicobacter pylori. Gastroenterology. 2003;125(6):1613–1625. | |
Boughan PK, Argent RH, Body-Malapel M, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem. 2006;281(17):11637–11648. | |
Patel SR, Smith K, Letley DP, et al. Helicobacter pylori downregulates expression of human beta-defensin 1 in the gastric mucosa in a type IV secretion-dependent fashion. Cell Microbiol. 2013;15(12):2080–2092. | |
Otte JM, Neumann HM, Brand S, Schrader H, Schmidt WE, Schmitz F. Expression of beta-defensin 4 is increased in human gastritis. Eur J Clin Invest. 2009;39(2):126–138. | |
Peek RM Jr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest. 1995;73(6):760–770. | |
Cook KW, Letley DP, Ingram RJ, et al. CCL20/CCR6-mediated migration of regulatory T cells to the Helicobacter pylori-infected human gastric mucosa. Gut. 2014;63(10):1550–1559. | |
Ramis IB, Vianna JS, Goncalves CV, von Groll A, Dellagostin OA, da Silva PE. Polymorphisms of the IL-6, IL-8 and IL-10 genes and the risk of gastric pathology in patients infected with Helicobacter pylori. J Microbiol Immunol Infect. Epub 2015 Mar 24. | |
El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with pro-inflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124(5):1193–1201. | |
Allen LA, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005; 174(6):3658–3667. | |
Ismail HF, Fick P, Zhang J, Lynch RG, Berg DJ. Depletion of neutrophils in IL-10(-/-) mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter. J Immunol. 2003;170(7):3782–3789. | |
Kaparakis M, Walduck AK, Price JD, et al. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect Immun. 2008;76(5):2235–2239. | |
Quiding-Jarbrink M, Raghavan S, Sundquist M. Enhanced M1 macrophage polarization in human Helicobacter pylori-associated atrophic gastritis and in vaccinated mice. PLoS One. 2010;5(11):e15018. | |
Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs functional differentiation. Front Immunol. 2014;5:514. | |
Schwartz JT, Allen LA. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol. 2006;79(6):1214–1225. | |
Allen LA. The role of the neutrophil and phagocytosis in infection caused by Helicobacter pylori. Curr Opin Infect Dis. 2001;14(3):273–277. | |
Wilson KT, Crabtree JE. Immunology of Helicobacter pylori: insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology. 2007;133(1):288–308. | |
Gobert AP, McGee DJ, Akhtar M, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A. 2001;98(24):13844–13849. | |
Bamba N, Nakajima S, Andoh A, et al. Stem cell factor expressed in human gastric mucosa in relation to mast cell increase in Helicobacter pylori-infected gastritis. Dig Dis Sci. 2002;47(2):274–282. | |
Velin D, Bachmann D, Bouzourene H, Michetti P. Mast cells are critical mediators of vaccine-induced Helicobacter clearance in the mouse model. Gastroenterology. 2005;129(1):142–155. | |
Drakes ML, Czinn SJ, Blanchard TG. Regulation of murine dendritic cell immune responses by Helicobacter felis antigen. Infect Immun. 2006;74(8):4624–4633. | |
Algood HM, Gallo-Romero J, Wilson KT, Peek RM Jr, Cover TL. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol Med Microbiol. 2007;51(3):577–586. | |
Bimczok D, Clements RH, Waites KB, et al. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol. 2010;3(3):260–269. | |
Khamri W, Walker MM, Clark P, et al. Helicobacter pylori stimulates dendritic cells to induce interleukin-17 expression from CD4+ T lymphocytes. Infect Immun. 2010;78(2):845–853. | |
Oertli M, Sundquist M, Hitzler I, et al. DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest. 2012;122(3):1082–1096. | |
Rizzuti D, Ang M, Sokollik C, et al. Helicobacter pylori inhibits dendritic cell maturation via interleukin-10-mediated activation of the signal transducer and activator of transcription 3 pathway. J Innate Immun. 2015;7(2):199–211. | |
Munari F, Fassan M, Capitani N, et al. Cytokine BAFF released by Helicobacter pylori-infected macrophages triggers the Th17 response in human chronic gastritis. J Immunol. 2014;193(11):5584–5594. | |
Bimczok D, Kao JY, Zhang M, et al. Human gastric epithelial cells contribute to gastric immune regulation by providing retinoic acid to dendritic cells. Mucosal Immunol. Epub 2014 Sep 24. | |
O’Keeffe J, Gately CM, O’Donoghue Y, Zulquernain SA, Stevens FM, Moran AP. Natural killer cell receptor T-lymphocytes in normal and Helicobacter pylori-infected human gastric mucosa. Helicobacter. 2008;13(6):500–505. | |
Rudnicka K, Matusiak A, Miszczyk E, Rudnicka W, Tenderenda M, Chmiela M. Immunophenotype of peripheral blood natural killer cells and IL-10 serum levels in relation to Helicobacter pylori status. APMIS. 2013;121(9):806–813. | |
Yun CH, Lundgren A, Azem J, et al. Natural killer cells and Helicobacter pylori infection: bacterial antigens and interleukin-12 act synergistically to induce gamma interferon production. Infect Immun. 2005;73(3):1482–1490. | |
D’Elios MM, Appelmelk BJ, Amedei A, Bergman MP, Del Prete G. Gastric autoimmunity: the role of Helicobacter pylori and molecular mimicry. Trends Mol Med. 2004;10(7):316–323. | |
Smyk DS, Koutsoumpas AL, Mytilinaiou MG, Rigopoulou EI, Sakkas LI, Bogdanos DP. Helicobacter pylori and autoimmune disease: cause or bystander. World J Gastroenterol. 2014;20(3):613–629. | |
Figueiredo Soares T, Aguiar Rocha G, Camargos Rocha AM, et al. Differences in peripheral blood lymphocyte phenotypes between Helicobacter pylori-positive children and adults with duodenal ulcer. Clin Microbiol Infect. 2007;13(11):1083–1088. | |
Robinson K, Kenefeck R, Pidgeon EL, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57(10):1375–1385. | |
Serelli-Lee V, Ling KL, Ho C, et al. Persistent Helicobacter pylori specific Th17 responses in patients with past H. pylori infection are associated with elevated gastric mucosal IL-1beta. PLoS One. 2012;7(6):e39199. | |
Enarsson K, Lundgren A, Kindlund B, et al. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin Immunol. 2006;121(3):358–368. | |
Zhuang Y, Cheng P, Liu XF, et al. A pro-inflammatory role for Th22 cells in Helicobacter pylori-associated gastritis. Gut. Epub 2014 Aug 18. | |
Buzelli JN, Chalinor HV, Pavlic DI, et al. IL33 is a stomach alarmin that initiates a skewed Th2 response to injury and infection. Cell Mol Gastroenterol Hepatol. 2015;1(1):203–221. | |
Popova A, Kzhyshkowska J, Nurgazieva D, Goerdt S, Gratchev A. Pro- and anti-inflammatory control of M-CSF-mediated macrophage differentiation. Immunobiology. 2011;216(1–2):164–172. | |
Wilke CM, Bishop K, Fox D, Zou W. Deciphering the role of Th17 cells in human disease. Trends Immunol. 2011;32(12):603–611. | |
Shi Y, Liu XF, Zhuang Y, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184(9):5121–5129. | |
Serrano C, Wright SW, Bimczok D, et al. Downregulated Th17 responses are associated with reduced gastritis in Helicobacter pylori-infected children. Mucosal Immunol. 2013;6(5):950–959. | |
Hitzler I, Kohler E, Engler DB, Yazgan AS, Muller A. The role of Th cell subsets in the control of Helicobacter infections and in T cell-driven gastric immunopathology. Front Immunol. 2012;3:142. | |
D’Elios MM, Manghetti M, De Carli M, et al. T helper 1 effector cells specific for Helicobacter pylori in the gastric antrum of patients with peptic ulcer disease. J Immunol. 1997;158(2):962–967. | |
Chang WJ, Du Y, Zhao X, Ma LY, Cao GW. Inflammation-related factors predicting prognosis of gastric cancer. World J Gastroenterol. 2014;20(16):4586–4596. | |
Liu T, Peng L, Yu P, et al. Increased circulating Th22 and Th17 cells are associated with tumor progression and patient survival in human gastric cancer. J Clin Immunol. 2012;32(6):1332–1339. | |
D’Elios MM, Amedei A, Manghetti M, et al. Impaired T-cell regulation of B-cell growth in Helicobacter pylori-related gastric low-grade MALT lymphoma. Gastroenterology. 1999;117(5):1105–1112. | |
Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138(3):1046–1054. | |
Rad R, Brenner L, Bauer S, et al. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology. 2006;131(2):525–537. | |
Ai TL, Solomon BD, Hsieh CS. T-cell selection and intestinal homeostasis. Immunol Rev. 2014;259(1):60–74. | |
Luther J, Owyang SY, Takeuchi T, et al. Helicobacter pylori DNA decreases pro-inflammatory cytokine production by dendritic cells and attenuates dextran sodium sulphate-induced colitis. Gut. 2011;60(11):1479–1486. | |
Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176(5):3000–3009. | |
Kwok T, Zabler D, Urman S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449(7164):862–866. | |
Tegtmeyer N, Wessler S, Backert S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011;278(8):1190–1202. | |
Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science. 2003;300(5624):1430–1434. | |
Franco AT, Israel DA, Washington MK, et al. Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci U S A. 2005;102(30):10646–10651. | |
Bauer B, Pang E, Holland C, Kessler M, Bartfeld S, Meyer TF. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe. 2012;11(6):576–586. | |
Peek RM Jr, Fiske C, Wilson KT. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol Rev. 2010; 90(3):831–858. | |
Gorrell RJ, Guan J, Xin Y, et al. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol. 2013;15(4):554–570. | |
Atherton JC, Cao P, Peek RM, Jr., Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270(30):17771–17777. | |
Rhead JL, Letley DP, Mohammadi M, et al. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology. 2007;133(3):926–936. | |
Kim IJ, Blanke SR. Remodeling the host environment: modulation of the gastric epithelium by the Helicobacter pylori vacuolating toxin (VacA). Front Cell Infect Microbiol. 2012;2:37. | |
Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301(5636):1099–1102. | |
Oldani A, Cormont M, Hofman V, et al. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009;5(10):e1000603. | |
Akada JK, Aoki H, Torigoe Y, et al. Helicobacter pylori CagA inhibits endocytosis of cytotoxin VacA in host cells. Dis Model Mech. 2010; 3(9–10):605–617. | |
Argent RH, Thomas RJ, Letley DP, Rittig MG, Hardie KR, Atherton JC. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med Microbiol. 2008;57(Pt 2):145–150. | |
Oertli M, Noben M, Engler DB, et al. Helicobacter pylori gamma-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc Natl Acad Sci U S A. 2013; 110(8):3047–3052. | |
Engler DB, Reuter S, van Wijck Y, et al. Effective treatment of allergic airway inflammation with Helicobacter pylori immunomodulators requires BATF3-dependent dendritic cells and IL-10. Proc Natl Acad Sci U S A. 2014;111(32):11810–11815. | |
Fischer W, Windhager L, Rohrer S, et al. Strain-specific genes of Helicobacter pylori: genome evolution driven by a novel type IV secretion system and genomic island transfer. Nucleic Acids Res. 2010;38(18):6089–6101. | |
Jung SW, Sugimoto M, Shiota S, Graham DY, Yamaoka Y. The intact dupA cluster is a more reliable Helicobacter pylori virulence marker than dupA alone. Infect Immun. 2012;80(1):381–387. | |
Shiota S, Matsunari O, Watada M, Hanada K, Yamaoka Y. Systematic review and meta-analysis: the relationship between the Helicobacter pylori dupA gene and clinical outcomes. Gut Pathog. 2010;2(1):13. | |
Lu H, Hsu PI, Graham DY, Yamaoka Y. Duodenal ulcer promoting gene of Helicobacter pylori. Gastroenterology. 2005;128(4):833–848. | |
Hussein NR, Argent RH, Marx CK, Patel SR, Robinson K, Atherton JC. Helicobacter pylori dupA is polymorphic, and its active form induces pro-inflammatory cytokine secretion by mononuclear cells. J Infect Dis. 2010;202(2):261–269. | |
de Bernard M, D’Elios MM. The immune modulating activity of the Helicobacter pylori HP-NAP: friend or foe? Toxicon. 2010;56(7):1186–1192. | |
Satin B, Del Giudice G, Della Bianca V, et al. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J Exp Med. 2000;191(9):1467–1476. | |
Rad R, Gerhard M, Lang R, et al. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J Immunol. 2002; 168(6):3033–3041. | |
Yamaoka Y, Ojo O, Fujimoto S, et al. Helicobacter pylori outer membrane proteins and gastroduodenal disease. Gut. 2006;55(6):775–781. | |
Yamaoka Y, Graham DY. Helicobacter pylori virulence and cancer pathogenesis. Future Oncol. 2014;10(8):1487–1500. | |
Yamaoka Y, Kikuchi S, el-Zimaity HM, Gutierrez O, Osato MS, Graham DY. Importance of Helicobacter pylori oipA in clinical presentation, gastric inflammation, and mucosal interleukin 8 production. Gastroenterology. 2002;123(2):414–424. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.