Back to Journals » Drug Design, Development and Therapy » Volume 20
Synthesis and Biological Evaluation of Imidazopyridine-Isatin Hybrids as Inhibitors of Leishmania major Growth
Authors El Hassab MA, Eldehna WM ![]() , Elsayed ZM
, Elsayed ZM ![]() , Elbadawi MM, Negmeldin AT, Balaha M, Nasralla SN, Monir R, Ibrahim TM, Abe M, Tawfik HO
, Elbadawi MM, Negmeldin AT, Balaha M, Nasralla SN, Monir R, Ibrahim TM, Abe M, Tawfik HO ![]() , Bekhit AA
, Bekhit AA ![]() , Hemeda LR
, Hemeda LR
Received 27 September 2025
Accepted for publication 12 December 2025
Published 24 March 2026 Volume 2026:20 570670
DOI https://doi.org/10.2147/DDDT.S570670
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Muzammal Hussain
Mahmoud A El Hassab,1 Wagdy M Eldehna,2 Zainab M Elsayed,3 Mostafa M Elbadawi,2 Ahmed T Negmeldin,4,5 Marwa Balaha,2,6 Sherry N Nasralla,7 Rehan Monir,8 Tamer M Ibrahim,2 Manabu Abe,9 Haytham O Tawfik,10 Adnan A Bekhit,7,11 Loah R Hemeda12
1Department of Medicinal Chemistry, Faculty of Pharmacy, King Salman International University (KSIU), South Sinai, Egypt; 2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt; 3Scientific Research and Innovation Support Unit, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt; 4Department of Pharmaceutical Sciences, College of Pharmacy and Thumbay Research Institute for Precision Medicine, Gulf Medical University, Ajman, United Arab Emirates; 5Department of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 6Department of Pharmacy, “G. d’Annunzio” University of Chieti-Pescara, Chieti, Italy; 7Pharmacy Program, Allied Health Department, College of Health and Sport Sciences, University of Bahrain, Salmaniya, Bahrain; 8Clinical Biochemistry Department, College of Medicine, King Khalid University, Asir, Saudi Arabia; 9Department of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, Hiroshima, Japan; 10Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Tanta University, Tanta, Egypt; 11Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Alexandria University, Alexandria, Egypt; 12Department of Medicinal Chemistry, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, Egypt
Correspondence: Wagdy M Eldehna, Email [email protected] Ahmed T Negmeldin, Email [email protected]
Introduction: More than 20 different species of Leishmania cause leishmaniasis, a multifaceted disease that ranges from mild cutaneous lesions to fatal visceral forms.
Methods: Sixteen novel hybrids were designed by covalently linking an imidazo[1,2-a]pyridine core to an isatin scaffold via a hydrazide linker, and their in vitro anti-promastigote, anti-amastigote, cytotoxicity, and molecular docking profiles were evaluated, with folate-rescue assays used to assess the anti-folate mechanism.
Results: Several compounds showed potent anti-leishmanial activity compared to miltefosine, with the lead compound 5b exhibiting strong anti-promastigote (IC50 = 0.84 ± 0.06 μM) and anti-amastigote (IC50 = 1.28 ± 0.18 μM) activities, significantly surpassing miltefosine. Folic and folinic acids reversed the activity of 5b, confirming an anti-folate mechanism similar to that of the Lm-PTR1 inhibitor trimethoprim. The most active compounds showed higher selectivity indices (SI = 12.74) than miltefosine (SI = 438.03), and docking against Lm-PTR1 provided a plausible explanation for their potency, while in silico predictions indicated favorable drug-likeness and pharmacokinetic properties.
Conclusion: These findings highlight a promising new chemotype targeting the leishmanial folate pathway and expand the chemical diversity available for anti-leishmanial drug development.
Keywords: neglected tropical diseases, leishmaniasis, molecular modeling, anti-folate mechanism, imidazo[1,2-a]pyridine, isatin
Introduction
Over 12 million individuals worldwide suffer from leishmaniasis, a neglected tropical disease that causes severe morbidity and over 50,000 deaths yearly. New anti-leishmanial medications are desperately needed, as evidenced by the paucity of research and control efforts despite the disease’s worldwide effect.1,2 The internal parasite Leishmania, which is spread by phlebotomine sandflies, is the cause of the illness. After infecting macrophages, the parasite develops into amastigotes, which proliferate and avoid immune clearance, causing tissue damage. Depending on the species and host immunological response, the primary clinical manifestations include visceral leishmaniasis (VL), mucocutaneous (MCL), diffuse cutaneous (DCL), and cutaneous (CL).3,4
Dihydrofolate reductase (DHFR) and thymidylate synthase (TS) are essential enzymes in the folate metabolic pathway, which the parasite depends on for growth. Drug resistance can result from pteridine reductase 1 (PTR1) compensating for DHFR inhibition, while DHFR facilitates DNA synthesis by converting dihydrofolate (DHF) to tetrahydrofolate (THF).5,6 Pentavalent antimonials, amphotericin B, miltefosine, pentamidine, and paromomycin are examples of current anti-leishmanial medications that have drawbacks such as toxicity, decreased efficacy, resistance, and high cost.7–9 These difficulties highlight the need for novel oral drugs that are more economical, efficient, and safe.
There have been reports of pyridine derivatives having anti-leishmanial properties. As an illustration, triazolopyridyl ketones (compounds I–II, Figure 1) shown strong micromolar action against various Leishmania species promastigotes.10,11 The sulfone metabolite of fexinidazole (III, Figure 1) is one example of an imidazole derivative that has shown effectiveness against intracellular amastigotes.12 Numerous pharmacologically active substances, such as antifungal, anticancer, and antileishmanial drugs (IV–VII, Figure 1), contain fused imidazo[1,2-a]pyridine scaffolds with low cytotoxicity and action against both promastigote and amastigote stages.13–15
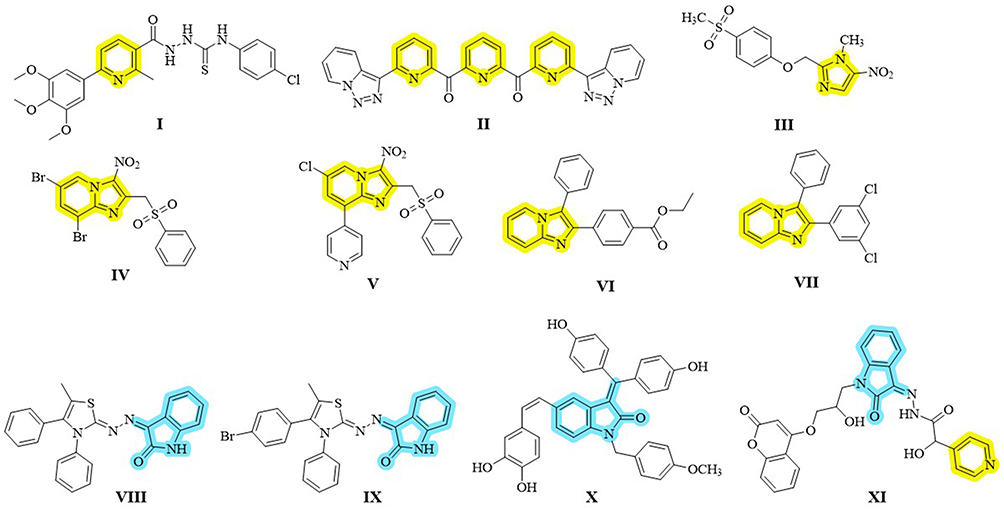 |
Figure 1 Selected heterocyclic derivatives as anti-leishmanial agents: pyridine (I–II), imidazole (III), and fused imidazo[1,2-a]pyridine (IV–VII) highlighted in yellow, and isatin-based derivatives (VIII–XI) with or without DHFR inhibitory activity highlighted in blue. |
Significant therapeutic promise, including anti-leishmanial properties, has also been demonstrated by heterocyclic compounds generated from isatin (indoline-dione). Targeting DHFR and exhibiting low micromolar potency against promastigotes and intracellular amastigotes, a few derivatives (VIII–XI, Figure 1) demonstrated improved action and selectivity.16–31
Building on these results, we postulated that by integrating the essential pharmacophoric characteristics of both scaffolds, covalently joining imidazo[1,2-a]pyridine and isatin via a hydrazide linker might produce hybrids with increased activity and selectivity. This led to the design and synthesis of a novel set of molecular hybrids (5a-h, 8a-d, 11a-d, Figure 2). In order to maximize interactions with the DHFR-TS and PTR1 targets, structural changes were made at the C2-position of imidazopyridine as well as at the C5/C7 and N1 of the isatin nucleus. This design approach is summarized in Figure 2.
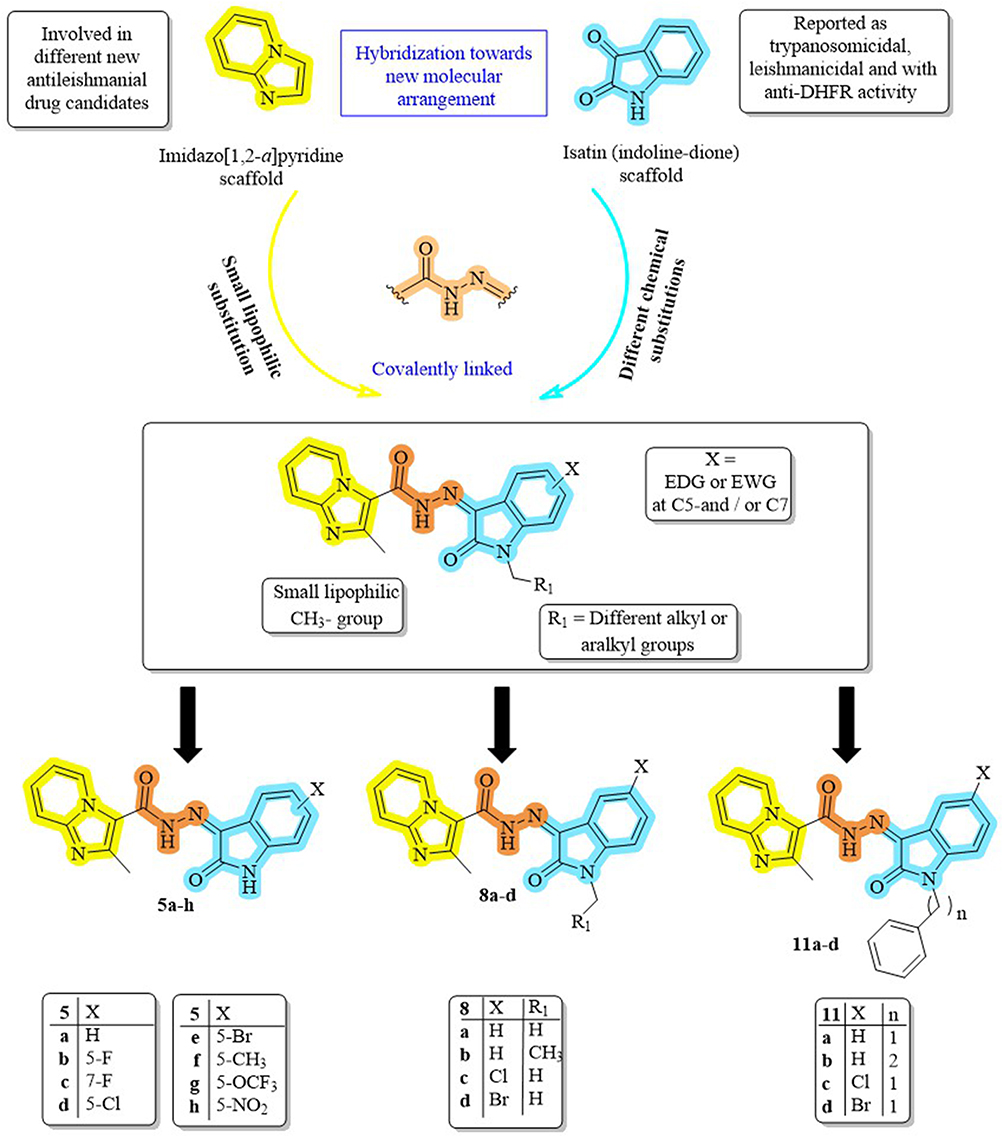 |
Figure 2 Structural planning of the newly synthesized compounds. |
Materials and Methods
Chemistry
An Electrothermal IA 9000 device that was not calibrated was used to detect melting points. All synthesized compounds’ high-resolution mass spectrometry (HR-TOF-ESI-MS) data were obtained using a JEOL JMS-700 device (Tokyo, Japan). At Tokushima Bunri University’s Faculty of Pharmaceutical Science in Japan, Bruker 400 MHz spectrometers were used to record the 1H and 13C NMR spectra. Coupling constants (J) are expressed in Hz, and chemical shifts (δ) in ppm. Using an iodine–potassium spray and a chloroform/methanol (9.5:0.5, v/v) mobile phase, the reaction progress was tracked by TLC on silica gel 60 F254 aluminum sheets (Merck). The preparation of compounds 2,32 3,32 7a-d,33 and 10a-d34 was done in accordance with previously published protocols. Supplementary Information contains the characterization information for the recently synthesized compounds.
General Procedure for the Synthesis of Target Compounds (5a-h, 8a-d, and 11a-d)
A solution of hydrazide 3 (48 mg, 0.25 mmol) in 10 mL of absolute ethanol with five drops of glacial acetic acid was made in a 25 mL round-bottom flask. Alkylated isatins 7a-d or 10a-d (0.25 mmol) and other isatins 4a-h were added to this solution. TLC was used to track the reaction mixture’s progress during its 10-hour reflux. Filtration was used to gather the precipitated solid, which was then dried and purified by recrystallization from an ethanol/DMF combination.
(Z)-2-Methyl-N’-(2-oxoindolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (5a)
Yield: 90% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.40 (s, 1H, NH of hydrazide), 11.29 (s, 1H, NH of isatin), 9.39 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.69 (dt, J = 8.9, 1.2 Hz, 1H, Aromatic-H), 7.59 (dt, J = 7.4, 1.0 Hz, 1H, Aromatic-H), 7.57–7.52 (m, 1H, Aromatic-H), 7.37 (td, J = 7.7, 1.3 Hz, 1H, Aromatic-H), 7.19 (td, J = 6.9, 1.3 Hz, 1H, Aromatic-H), 7.10 (td, J = 7.6, 1.0 Hz, 1H, Aromatic-H), 6.96 (dt, J = 7.8, 0.9 Hz, 1H, Aromatic-H), 2.78 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 320.11420 and Observed 320.11401. Microanalysis (%) Theoretical (Observed) for C17H13N5O2: C, 63.94 (64.17); H, 4.10 (4.11); N, 21.93 (22.00)%.
(Z)-N’-(5-fluoro-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (5b)
Yield: 76% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.44 (s, 1H, NH of hydrazide), 11.32 (s, 1H, NH of isatin), 9.40 (d, J = 7.0 Hz, 1H, Aromatic-H), 7.72 (s, 1H, Aromatic-H), 7.59 (s, 1H, Aromatic-H), 7.42 (dd, J = 8.0, 2.7 Hz, 1H, Aromatic-H), 7.24 (ddd, J = 11.6, 6.5, 2.4 Hz, 2H, Aromatic-H), 6.98 (dd, J = 8.6, 4.2 Hz, 1H, Aromatic-H), 2.80 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 338.10478 and Observed 338.10468. Microanalysis (%) Theoretical (Observed) for C17H12FN5O2: C, 60.53 (60.29); H, 3.59 (3.57); N, 20.76 (20.81)%.
(Z)-N’-(7-Fluoro-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (5c)
Yield: 70% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.41 (s, 1H, NH of hydrazide), 11.84 (s, 1H, NH of isatin), 9.41 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.73 (dt, J = 9.0, 1.2 Hz, 1H, Aromatic-H), 7.60–7.56 (m, 1H, Aromatic-H), 7.47 (dd, J = 7.5, 0.9 Hz, 1H, Aromatic-H), 7.33 (ddd, J = 10.5, 8.4, 1.0 Hz, 1H, Aromatic-H), 7.24–7.20 (m, 1H, Aromatic-H), 7.16–7.13 (m, 1H, Aromatic-H), 2.81 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 338.10478 and Observed 338.10458. Microanalysis (%) Theoretical (Observed) for C17H12FN5O2: C, 60.53 (60.77); H, 3.59 (3.58); N, 20.76 (20.70)%.
(Z)-N’-(5-Chloro-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (5d)
Yield: 82% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.39 (s, 1H, NH of hydrazide), 11.41 (s, 1H, NH of isatin), 9.42 (d, J = 6.9 Hz, 1H, Aromatic-H), 7.73 (d, J = 8.9 Hz, 1H, Aromatic-H), 7.61–7.55 (m, 2H, Aromatic-H), 7.44 (dd, J = 8.3, 2.2 Hz, 1H, Aromatic-H), 7.22 (t, J = 6.8 Hz, 1H, Aromatic-H), 7.00 (d, J = 8.3 Hz, 1H, Aromatic-H), 2.81 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+/[M+H+2]+ Theoretical 354.07523/356.07228 and Observed 354.07513/356.07205. Microanalysis (%) Theoretical (Observed) for C17H12ClN5O2: C, 57.72 (57.95); H, 3.42 (3.40); N, 19.80 (19.77)%.
(Z)-N’-(5-Bromo-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (5e)
Yield: 82% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.43 (s, 1H, NH of hydrazide), 11.41 (s, 1H, NH of isatin), 9.41 (s, 1H, Aromatic-H), 7.57 (s, 2H, Aromatic-H), 7.21 (s, 2H, Aromatic-H), 6.85 (s, 2H, Aromatic-H), 2.80 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+/[M+H+2]+ Theoretical 398.02471/400.02267 and Observed 398.02460/400.02240. Microanalysis (%) Theoretical (Observed) for C17H12BrN5O2: C, 51.27 (51.04); H, 3.04 (3.01); N, 17.59 (17.64)%.
(Z)-2-Methyl-N’-(5-methyl-2-oxoindolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (5f)
Yield: 76% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.43 (s, 1H, NH of hydrazide), 11.33 (s, 1H, NH of isatin), 9.41 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.97 (s, 1H, Aromatic-H), 7.73 (dt, J = 2.5, 1.2 Hz, 1H, Aromatic-H), 7.60–7.57 (m, 1H, Aromatic-H), 7.44 (d, J = 1.7 Hz, 1H, Aromatic-H), 7.24 (dt, J = 4.1, 2.1 Hz, 1H, Aromatic-H), 6.87 (d, J = 7.9 Hz, 1H, Aromatic-H), 2.80 (s, 3H, CH3), 2.31 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 334.12985 and Observed 334.12955. Microanalysis (%) Theoretical (Observed) for C18H15N5O2: C, 64.86 (65.07); H, 4.54 (4.55); N, 21.01 (20.98)%.
(Z)-2-Methyl-N’-(2-oxo-5-(trifluoromethoxy)indolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (5g)
Yield: 69% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.36 (s, 1H, NH of hydrazide), 11.46 (s, 1H, NH of isatin), 9.40 (d, J = 6.9 Hz, 1H, Aromatic-H), 7.70 (d, J = 8.9 Hz, 1H, Aromatic-H), 7.59–7.48 (m, 2H, Aromatic-H), 7.39 (d, J = 8.5 Hz, 1H, Aromatic-H), 7.21 (t, J = 6.8 Hz, 1H, Aromatic-H), 7.05 (d, J = 8.6 Hz, 1H, Aromatic-H), 2.80 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 404.09650 and Observed 404.09628. Microanalysis (%) Theoretical (Observed) for C18H12F3N5O3: C, 53.60 (53.38); H, 3.00 (3.00); N, 17.36 (17.33)%.
(Z)-2-Methyl-N’-(5-nitro-2-oxoindolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (5h)
Yield: 75% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.24 (s, 1H, NH of hydrazide), 11.52 (s, 1H, NH of isatin), 9.15 (s, 1H, Aromatic-H), 9.04 (d, J = 2.2 Hz, 1H, Aromatic-H), 8.32 (dd, J = 8.7, 2.2 Hz, 1H, Aromatic-H), 7.72 (d, J = 8.9 Hz, 1H, Aromatic-H), 7.59–7.54 (m, 1H, Aromatic-H), 7.18 (t, J = 7.1 Hz, 1H, Aromatic-H), 7.11 (d, J = 8.6 Hz, 1H, Aromatic-H), 2.74 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 365.09928 and Observed 365.09909. Microanalysis (%) Theoretical (Observed) for C17H12N6O4: C, 56.05 (55.84); H, 3.32 (3.34); N, 17.57 (17.60)%.
(Z)-2-Methyl-N’-(1-methyl-2-oxoindolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (8a)
Yield: 79% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.30 (s, 1H, NH of hydrazide), 9.38 (d, J = 6.8 Hz, 1H, Aromatic-H), 7.67 (d, J = 8.6 Hz, 1H, Aromatic-H), 7.60 (d, J = 7.4 Hz, 1H, Aromatic-H), 7.53 (t, J = 7.9 Hz, 1H, Aromatic-H), 7.43 (t, J = 7.8 Hz, 1H, Aromatic-H), 7.16 (dt, J = 15.7, 7.3 Hz, 3H, Aromatic-H), 3.23 (s, 3H, CH3), 2.80 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 334.12985 and Observed 334.12964. Microanalysis (%) Theoretical (Observed) for C18H15N5O2: C, 64.86 (65.11); H, 4.54 (4.56); N, 21.01 (21.07)%.
(Z)-N’-(1-ethyl-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (8b)
Yield: 86% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.30 (s, 1H, NH of hydrazide), 9.40 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.68 (dt, J = 8.9, 1.2 Hz, 1H, Aromatic-H), 7.61 (dd, J = 7.5, 1.2 Hz, 1H, Aromatic-H), 7.54 (ddd, J = 8.9, 6.8, 1.3 Hz, 1H, Aromatic-H), 7.44 (td, J = 7.8, 1.3 Hz, 1H, Aromatic-H), 7.22–7.13 (m, 3H, Aromatic-H), 3.81 (q, J = 7.2 Hz, 2H, CH2), 2.80 (s, 3H, CH3), 1.22 (t, J = 7.2 Hz, 3H, CH3). 13C NMR δ 160.80, 157.77, 148.55, 146.84, 142.82, 136.43, 131.87, 129.00, 128.37, 123.56, 121.01, 119.95, 116.92, 114.77, 114.26, 110.45, 40.67, 34.54, 17.29, 13.08. High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 348.14550 and Observed 348.14526. Microanalysis (%) Theoretical (Observed) for C19H17N5O2: C, 65.69 (65.90); H, 4.93 (4.95); N, 20.16 (20.09)%.
(Z)-N’-(5-Chloro-1-methyl-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (8c)
Yield: 69% (Yellowish powder); Melting point: > 300°C. High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 368.09088 and Observed 368.09079. Microanalysis (%) Theoretical (Observed) for C18H14ClN5O2: C, 58.78 (60.01); H, 3.84 (3.85); N, 19.04 (18.99)%.
(Z)-N’-(5-Bromo-1-Methyl-2-Oxoindolin-3-Ylidene)-2-Methylimidazo[1,2-A]pyridine-3-Carbohydrazide (8d)
Yield: 68% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.32 (s, 1H, NH of hydrazide), 9.43 (d, J = 6.8 Hz, 1H, Aromatic-H), 7.68 (d, J = 7.9 Hz, 1H, Aromatic-H), 7.60 (d, J = 7.9 Hz, 1H, Aromatic-H), 7.48 (d, J = 9.4 Hz, 1H, Aromatic-H), 7.20 (dt, J = 23.2, 10.8 Hz, 3H, Aromatic-H), 3.26 (s, 3H, CH3), 2.83 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+/[M+H+2]+ Theoretical 412.04036/414.03832 and Observed 412.04022/414.03806. Microanalysis (%) Theoretical (Observed) for C18H14BrN5O2: C, 52.44 (52.15); H, 3.42 (3.40); N, 16.99 (17.02)%.
(Z)-N’-(1-Benzyl-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (11a)
Yield: 79% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.31 (s, 1H, NH of hydrazide), 9.42 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.70 (dt, J = 8.9, 1.2 Hz, 1H, Aromatic-H), 7.64 (dd, J = 7.6, 1.2 Hz, 1H, Aromatic-H), 7.55 (ddd, J = 8.9, 6.9, 1.3 Hz, 1H, Aromatic-H), 7.41–7.33 (m, 5H, Aromatic-H), 7.31–7.27 (m, 1H, Aromatic-H), 7.20 (td, J = 6.9, 1.3 Hz, 1H, Aromatic-H), 7.15 (td, J = 7.6, 0.9 Hz, 1H, Aromatic-H), 7.05 (d, J = 7.9 Hz, 1H, Aromatic-H), 5.03 (s, 2H, CH2), 2.84 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 410.16115 and Observed 410.16092. Microanalysis (%) Theoretical (Observed) for C24H19N5O2: C, 70.40 (70.17); H, 4.68 (4.69); N, 17.10 (17.06)%.
(Z)-2-Methyl-N’-(2-oxo-1-phenethylindolin-3-ylidene)imidazo[1,2-a]pyridine-3-carbohydrazide (11b)
Yield: 65% (Yellowish powder); Melting point: > 300°C. High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 424.17680 and Observed 424.17673. Microanalysis (%) Theoretical (Observed) for C25H21N5O2: C, 70.91 (71.12); H, 5.00 (4.98); N, 16.54 (16.59)%.
(Z)-N’-(1-Benzyl-5-chloro-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (11c)
Yield: 80% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.29 (s, 1H, NH of hydrazide), 9.44 (dt, J = 7.0, 1.2 Hz, 1H, Aromatic-H), 7.73 (dt, J = 8.9, 1.2 Hz, 1H, Aromatic-H), 7.64 (d, J = 2.2 Hz, 1H, Aromatic-H), 7.58 (ddd, J = 8.7, 6.8, 1.3 Hz, 1H, Aromatic-H), 7.45 (dd, J = 8.4, 2.2 Hz, 1H, Aromatic-H), 7.39–7.30 (m, 5H, Aromatic-H), 7.24–7.21 (m, 1H, Aromatic-H), 7.08 (d, J = 8.4 Hz, 1H, Aromatic-H), 5.05 (s, 2H, CH2), 2.85 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+ Theoretical 444.12218 and Observed 444.12195. Microanalysis (%) Theoretical (Observed) for C24H18ClN5O2: C, 64.94 (65.19); H, 4.09 (4.10); N, 15.78 (15.83)%.
(Z)-N’-(1-Benzyl-5-bromo-2-oxoindolin-3-ylidene)-2-methylimidazo[1,2-a]pyridine-3-carbohydrazide (11d)
Yield: 74% (Yellowish powder); Melting point: > 300°C. 1H NMR δ (ppm): 13.24 (s, 1H, NH of hydrazide), 9.42 (d, J = 7.0 Hz, 1H, Aromatic-H), 7.69 (d, J = 6.0 Hz, 2H, Aromatic-H), 7.56 (t, J = 8.7 Hz, 2H, Aromatic-H), 7.39–7.27 (m, 5H, Aromatic-H), 7.20 (t, J = 7.2 Hz, 1H, Aromatic-H), 7.00 (d, J = 8.5 Hz, 1H, Aromatic-H), 5.03 (s, 2H, CH2), 2.83 (s, 3H, CH3). High-Resolution MS (ESI) m/z: [M+H]+/[M+H+2]+ Theoretical 488.07166/490.06962 and Observed 488.07184/490.06976. Microanalysis (%) Theoretical (Observed) for C24H18BrN5O2: C, 59.03 (58.88); H, 3.72 (3.70); N, 14.34 (14.38)%.
Biological Evaluation
In vitro Anti-Leishmanial Activity
Both promastigote and amastigote forms of L. major strain were used for the in vitro evaluation. All experiments were conducted as reported earlier.35–39 Axenic amastigotes were produced applying the method described by Teixeira et al40 Briefly, L. major parasites were cultured in tissue flasks containing RPMI- 1640 (Gibco, Invitrogen, Co., UK) and supplemented with 10% heat inactivated fetal calf serum (HIFGs), 100 IU penicillin (Sigma), 100 mg/mL streptomycin (Sigma) and 1% L-glutamine (Sigma). Different test compounds were dissolved in DMSO to a final concentration of 1 mg/mL. Fresh complete media was used to prepare threefold serial dilutions of the standard solutions, the test compounds and the reference drug miltefosine in appropriate concentrations. 100 mL culture media of 3×106 promastigotes of L. major were seeded into each well of a 96 well flat bottom plate. Various concentrations of the newly synthesized compounds (10, 3.33, 1.11, 0.37, 0.12 and 0.04 mg/mL) were added to the promastigote culture media. Wells containing only promastigotes were regarded as positive controls, while negative control wells contained 1% DMSO and the culture media alone. After 24 hours, 10 mL of alamar blue was added to each well, followed by measurement of fluorescence intensity (at wavelengths of 540 and 630 nm) using ELISA plate reader after 48 hours.
For evaluation of anti-amastigote activity, the test compounds were serially diluted in a 96-well microtitre plate to a final test concentration of 0.04–10 mg/mL in 50 mL culture medium. This was followed by addition of 50 mL suspensions containing 2×103 cells/mL amastigotes to each well. The plates were then incubated at 31◦C in a humidified environment under 5% CO2 72 hours. Following an incubation period of 68 hours, 10 mL of fluorochrome resazurin solution was added to each well. Absorbance of the resulting mixture was measured, after a total incubation time of 72 hours, using 37 Victor 3 Multilabel Counter, at excitation and emission wavelengths of 530 nm and 590 nm respectively. Finally, the IC50 value for each compound was determined from sigmoidal dose-response curves using the software Graphpad prism 3.0 (GraphPad Software, San Diego, CA, USA); results were expressed as mean ± SD of triplicate experiments with each test concentration measured in duplicates.
Reversal of the Anti-Leishmanial Activities
All operations were carried out in accordance with published protocols, and an in vitro promastigote growth assay was carried out using previously known methodology.27
In vitro Cytotoxicity Testing
The most active compounds were evaluated at concentrations ranging from 0 to 100 μM in 96-well plates, each containing 1×105 cells per well. Plates were incubated for 72 h at 37 °C under 95% humidity and 5% CO2. All experiments were conducted according to previously published methodologies.41 African green monkey Kidney (Vero cells) was obtained from Holding Company for Biological Products & Vaccines (VACSERA, Dokki, Giza, Egypt).
Physicochemical Factors and Pharmacokinetic Properties in silico Forecasts
Virtual platforms were used to predict the pharmacokinetic profiles, drug-likeness, and chemical and physicochemical properties. For a thorough evaluation of drug-likeness, ADMET-related characteristics, and possible toxicity hazards, ADMETlab 3.0, an online program, was utilized.42,43
Molecular Docking
The molecular docking investigations were conducted using AutoDock Vina and MGLTools, and the results were visualized using Discovery Studio. AutoDock Vina predicts how tiny molecules will attach to their targets using the AMBER force field, a united-atom scoring function, and a global optimization method based on a gradient-based local search genetic algorithm.44–47 The Protein Data Bank provided the X-ray crystal structures of Leishmania major PTR1 (PDB ID: 6RXC) and DHFR-TS (PDB ID: 1J3K), both of which were co-crystallized with inhibitors. MGLTools was used to prepare ligands and proteins and convert them to PDBQT format. The coordinates of the co-crystallized ligands were used to determine the active sites of both DHFR-TS and L. major PTR1, with a grid box size of 22 × 22×22 Å along the x, y, and z axes. RMSD values of 0.75 Å for DHFR-TS and 0.69 Å for L. major PTR1 were obtained by redocking the reference compounds into their corresponding binding sites in order to validate the docking methodology. Docking scores and binding interactions derived from 2D interaction diagrams were used to assess the final results.
Results and Discussion
Chemistry
The synthetic strategies for obtaining the sixteen final compounds (5a-h, 8a-d, and 11a-d) are outlined in Schemes 1 and 2. In Scheme 1, pyridin-2-amine 1 was heated under reflux with ethyl 2-chloro-3-oxobutanoate in abs. EtOH to furnish ethyl 2-methylimidazo[1,2-a]pyridine-3-carboxylate 2 (Figure 3), which is subjected to hydrazinolysis to produce the key intermediate 2-methylimidazo[1,2-a]pyridine-3-carbohydrazide 3.32,48–51 The terminal amino of the key intermediate 3 was subsequently condensed with various ketone moieties of isatin analogues 4a-h through refluxing in glacial acetic acid to yield the desired compounds 5a-h.
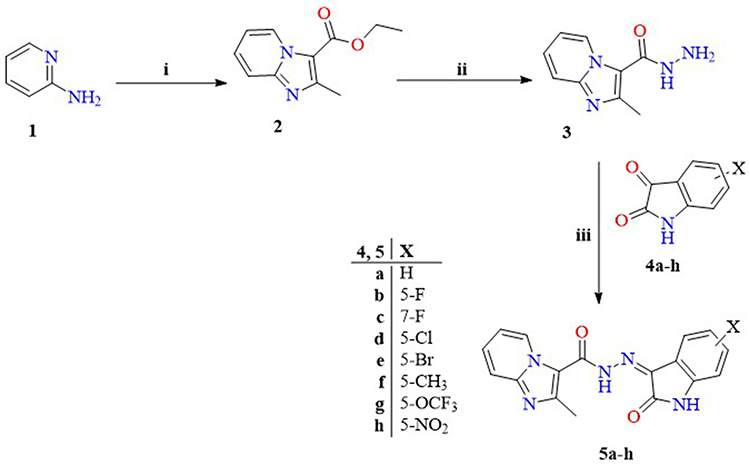 |
Scheme 1 Synthesis of compounds 5a-h. Reagents and conditions: (i) Ethyl 2-chloroacetoacetate, abs. EtOH, reflux, 6 h; (ii) Hydrazine hydrate, abs. EtOH, reflux, 6 h; (iii) abs. EtOH, drops of gl. AcOH as a catalyst, reflux, 10 h. |
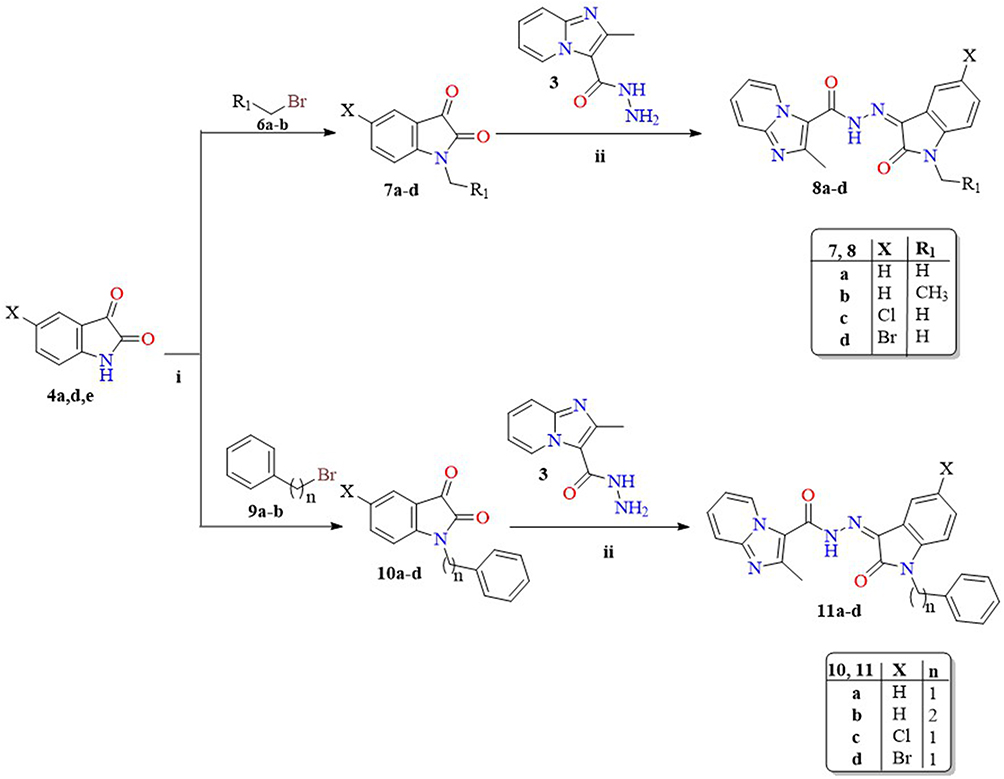 |
Scheme 2 Synthesis of compounds 8a-d and 11a-d. Reagents and conditions: (i) Anh. K2CO3, dry DMF, reflux, 6 h; (ii) EtOH, gl. AcOH, reflux, 10 h. |
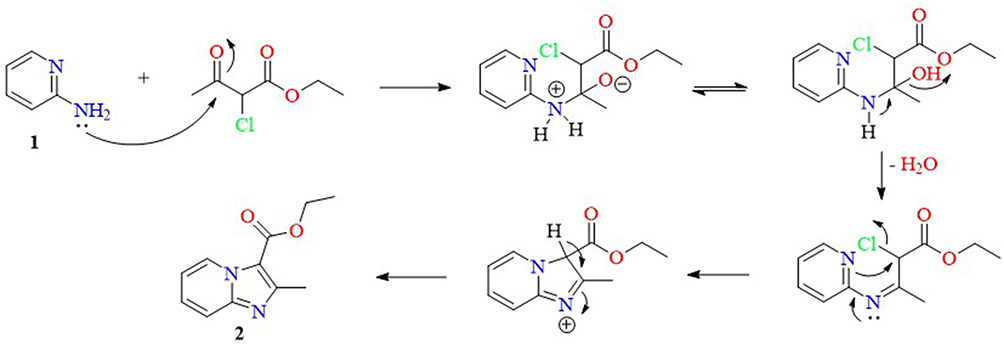 |
Figure 3 Plausible cyclization mechanism and formation of intermediate 2. |
In Scheme 2, selected isatin derivatives 4a,d,e was dissolved in dry DMF in presence of anhydrous potassium carbonate as a base16,52–55 and subjected to alkylation with methyl bromide 6a, ethyl bromide 6b, phenylmethyl bromide 9a and 2-phenylethyl bromide 9b to get N-alkyl isatin derivatives 7a-d and 10a-d, which condensed with the previously prepared key intermediate 3 in glacial AcOH affording the target compounds 8a-d and 11a-d, respectively.
The structures of all synthesized compounds were unequivocally confirmed through comprehensive spectral data, such as NMR (Supplementary Figures S1–S15) and HRMS (Figures S16–S31), and microanalysis, which aligned perfectly with the proposed structural frameworks.
By using 1H NMR analysis, the structural integrity of the target compounds 5a-h, 8a-d, and 11a-d was clarified. The spectra showed that the two NH2 protons of the hydrazide 3 completely vanished at 4.56 ppm,56 and that the newly created aromatic systems were responsible for an increase in aromatic proton signals. A notable downfield shift of one NH proton in intermediate 3, from 9.18 ppm56 to 13.24–13.44 ppm, significantly supported the successful synthesis of the Schiff base moiety. The suggested structures were supported by complementary 13C NMR spectroscopy data, which showed a noticeable upfield shift of the ketonic carbonyl carbon signal of isatin derivatives, shifting from about 180 ppm to ~140 ppm across all produced compounds.
Theoretically, the produced compounds might form both single and mixed (E and Z) stereoisomers. However, a single stereoisomer (either E or Z) was detected in the target derivatives’ 1H NMR spectra. The discovery of a single distinctive NH signal of the hydrazide moiety, which appeared around 13.24–13.44 ppm, served as proof of this. According to earlier reports, the creation of an intramolecular hydrogen bond with the carbonyl group of the indolinone ring is responsible for the significant downfield shift of this NH proton, resulting in a pseudo–six-membered ring structure.57,58 The Z-configuration, which is thought to be the thermodynamically preferred form under the given reaction circumstances, is strongly supported by this spectrum discovery. Factors like reaction temperature, starting material concentration, and the product’s intrinsic stability can all be used to explain why this isomer forms preferentially.59
The target compounds, including both chlorinated and brominated derivatives, showed molecular ion peaks and their distinctive isotopic patterns in high-resolution mass spectrometric analysis. Additionally, the estimated values for the suggested chemical formulae and the microanalysis results showed excellent agreement, with differences limited to ±0.4%.
Biological Evaluations
In vitro Anti-Leishmanial Activities
Using miltefosine as the reference medication, the anti-leishmanial potential of sixteen newly synthesized imidazopyridine–isatin hybrids (5a-h, 8a-d, and 11a-d) against both promastigote and amastigote forms of Leishmania major was assessed. Table 1 summarizes the IC50 values, shown in Figures 4 and 5, which are the micromolar concentrations required to prevent 50% of parasite growth. The IC50 dose–response curve for the representative compound 5b is presented in Supplementary Figure S32. In contrast to miltefosine (IC50 = 7.83 μM), all examined drugs (IC50 = 0.84–4.84 μM) showed remarkable effectiveness against the promastigote stage of L. major. Also, all synthesized compounds (IC50 = 1.28 to 7.18 μM) showed a higher anti-leishmanial activity than the reference (IC50 = 8.07 μM) except for 8d (IC50 = 8.18 μM) against amastigotes. Specifically, three compounds (5b, 5c, and 5h) showed IC50 values of 1.28 μM or less, indicating that they were 6 to 9 times more effective against promastigotes than miltefosine. They also showed notable anti-amastigote activity, approximately 4 to 6 times that of miltefosine. Also, promastigote forms were more susceptible to most of the newly synthesized compounds compared to the amastigote forms of the L. major parasite. While both L. major forms showed similar susceptibility to 5a, 11c, and 11d (IC50 = 2.33 to 3.84 μM).
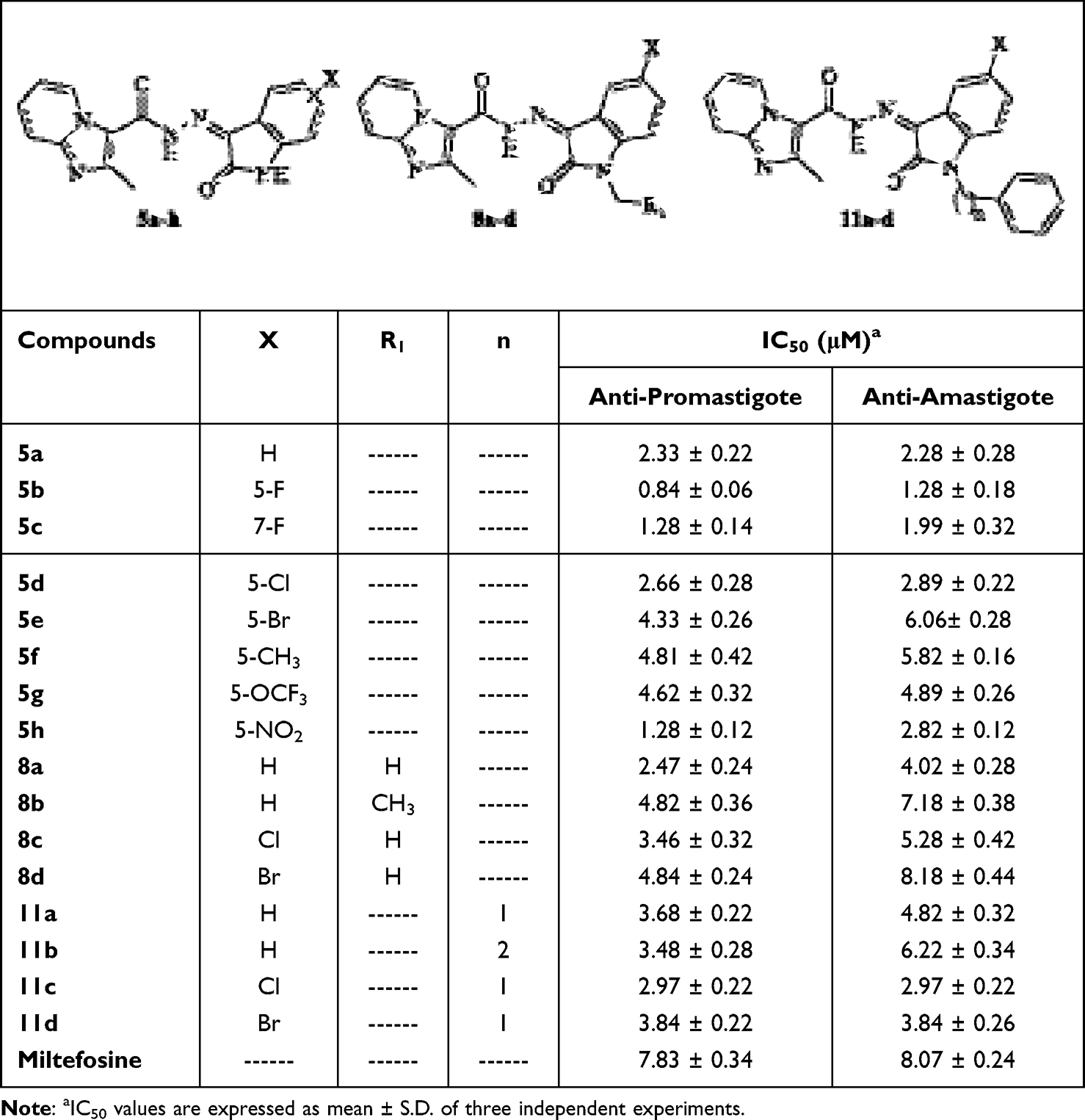 |
Table 1 Comparative in vitro Anti-Leishmanial Activity (IC50, μM) of Miltefosine and the Synthesized Compounds Against Leishmania major Promastigote Forms |
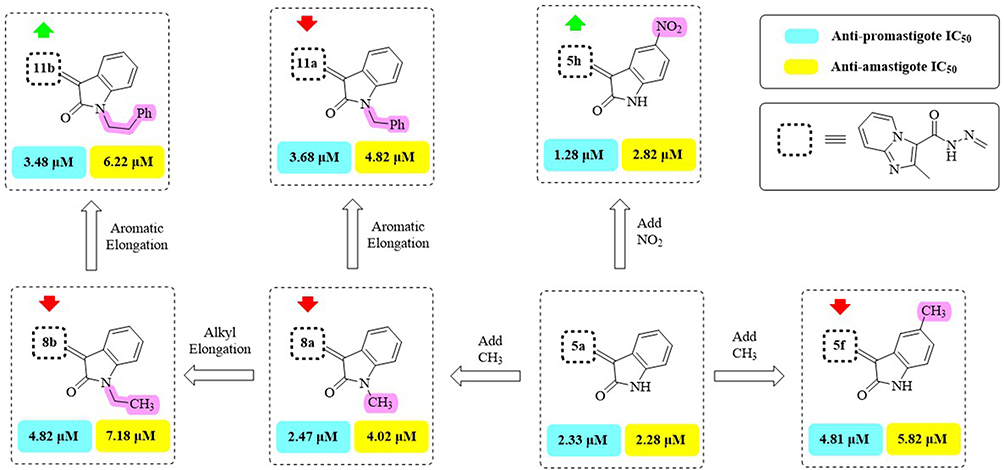 |
Figure 4 Effect of alkylated and non-halogenated isatins on both forms of L. major. The green arrow indicates increased activity, and the red arrow indicates decreased activity. |
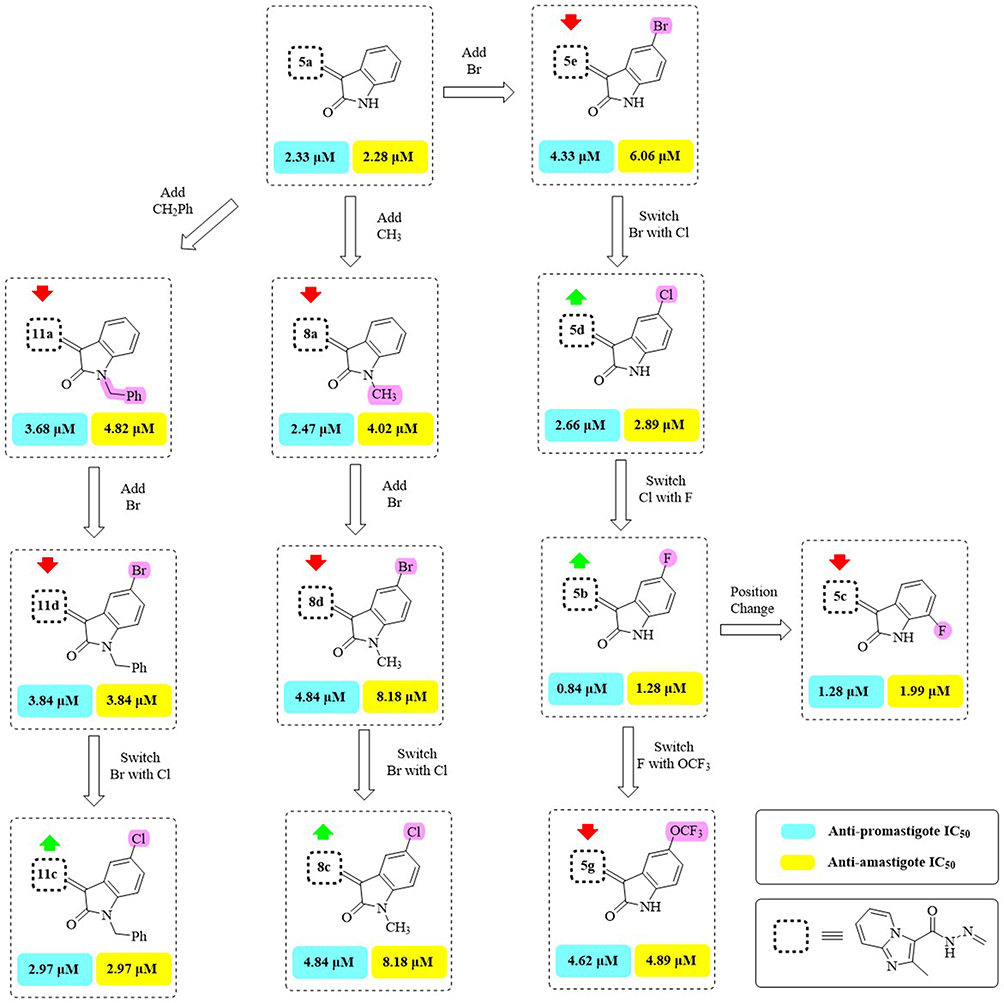 |
Figure 5 Effect of halogenated isatins on both forms of L. major. The green arrow indicates increased activity, and the red arrow indicates decreased activity. |
Based on alterations at either C5 or C7 of the isatin ring, the SAR of series 5a-h was assessed. As a result of fluorine’s electronegativity and lipophilicity, the C5-fluoro derivative 5b demonstrated the strongest anti-leishmanial activity against promastigotes (IC50 = 0.84 ± 0.06 μM) and amastigotes (IC50 = 1.28 ± 0.18 μM), followed by the C7-fluoro 5c. On the other hand, activity was reduced by bigger halogens like chlorine (5d) or bromine (5e) at C5, with 5e being the least active in this series. These results show that the anti-leishmanial potency is strongly influenced by the size and electronegativity of the halogen at C5.
Activity was affected by substitutions with strongly electronegative groups at C5. In comparison to the un-substituted 5a, the OCF3 derivative 5g exhibited lower activity against promastigotes (IC50 = 4.62 ± 0.32 μM) and amastigotes (IC50 = 4.89 ± 0.26 μM), whereas the NO2 derivative 5h showed higher activity against promastigotes (IC50 = 1.28 ± 0.12 μM) but lower activity against amastigotes (IC50 = 2.82 ± 0.12 μM). These effects most likely result from the substituents’ steric and electronic impacts on target interactions.
Introducing an electron-donating methyl group at C5 (5f) resulted in the lowest activity in the series (IC50 = 4.81 μM for promastigotes; 5.82 μM for amastigotes). Overall, the activity of series 5a-h depended on the electronic nature and position of the substituent on the isatin ring. Electron-withdrawing and small electronegative groups, such as F (5b) and NO2 (5h), generally enhanced activity, whereas larger or electron-donating groups (Cl, Br, OCF3, CH3) decreased it. Activity at C5 was superior to C7, as demonstrated by 5b (5-F) versus 5c (7-F).
N-Alkylation and C5 substitution of the isatin ring affected activity in the second series (8a-d). The most active N-methyl unsubstituted derivative was 8a (IC50 = 2.47 ± 0.24 μM for promastigotes; 4.02 ± 0.28 μM for amastigotes), while activity was generally reduced by increasing the N-alkyl size to ethyl (8b) or adding electronegative substituents at C5 (8c: Cl; 8d: Br). Interestingly, 8d exhibited the least amount of activity against both parasite types in this series. These findings suggest that anti-leishmanial efficacy is negatively impacted by bulky N-alkyl groups or by combining N-alkylation with C5-electronegative substitutions.
The third series (11a-d) showed moderate to good activity when lipophilic aralkyl groups were added at the isatin ring’s N1. In this series, the N-benzyl derivative with a C5-chlorine substitution (11c) exhibited the maximum potency (IC50 = 2.97 ± 0.22 μM), whereas the unsubstituted 11a and the C5-bromo derivative 11d were less active. These results imply that anti-leishmanial action is increased when N-aralkylation is combined with a moderately electronegative C5 substituent.
However, the same compound was the least active of the third series (IC50 = 3.84 ± 0.22 μM) against promastigote forms, assuming that the presence of the large-sized electronegative group with lipophilic aralkyl side chain decreased the availability and penetration against this form.
The inverse was noticed for the N-phenethyl derivative 11b (IC50 = 6.22 ± 0.34 μM) that showed a visible decrease in activity compared to the N-benzyl derivative 11a (IC50 = 4.82 ± 0.32 μM) against amastigote forms. In particular, the activity of this series seemed to be dependent on both the size of the halogen and its electronegativity at C5 of the isatin moiety and on the size of the aralkyl group on N1. Hence, the order of the decrease in the activity for amastigote forms was: Cl with N-benzyl > Br with N-benzyl > H with N-benzyl > H with N-phenethyl, and the order of the decrease in the activity for the promastigote forms was: Cl with N-benzyl > H with N-phenethyl > H with N-benzyl > Br with N-benzyl.
Reversal of Anti-Leishmanial Activity of the Most Potent Compound 5b by Folic Acid and Folinic Acid
Using the methodology described by Mendoza-Martínez et al,60 the anti-folate mechanism of the most active molecule, 5b, was examined. The test compound’s impact on L. major promastigotes’ survival was assessed in this assay both with and without folic acid/folinic acid at doses higher than their IC50 values. Trimethoprim decreased parasite survival to 72% as a positive control; supplementing with folic acid improved this to over 99%. Without folic acid, parasite survival for compound 5b dropped to 22%. This technique depends on folic acid’s capacity to compete for DHFR and PTR1 active sites and its function in DNA synthesis without the need for previous activation. The anti-leishmanial effect of 5b was reversed by the addition of folinic acid, resulting in survival rates ranging from 82% to 87% (Table 2). According to these findings, compound 5b’s anti-leishmanial action is likely due to an anti-folate mechanism that involves dual inhibition of DHFR and PTR1.
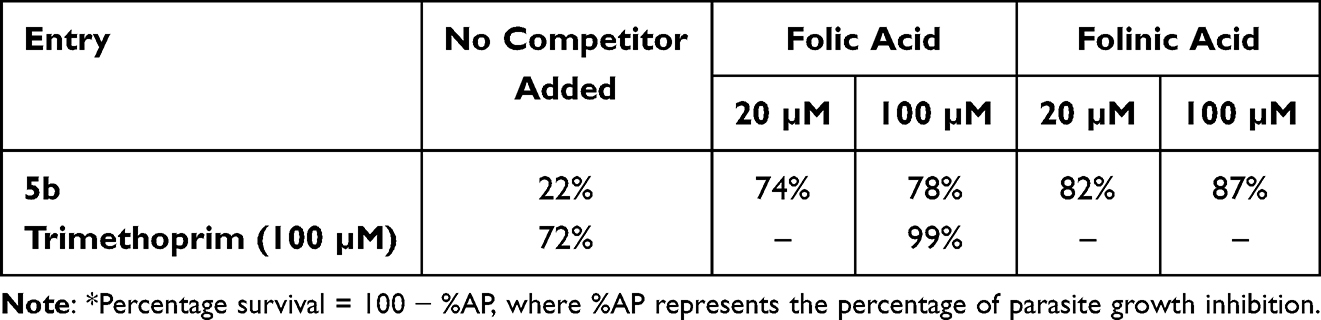 |
Table 2 In vitro Evaluation of Folate Pathway Inhibition Expressed as Percentage Survival* |
In vitro Cytotoxic Activity
Potential anti-leishmanial drugs must have strong anti-amastigote action with little cytotoxic effects on host cells to be deemed successful.61 To evaluate its safety profile, the most active molecule, 5b, was put through a cytotoxicity test against African green monkey kidney (VERO) cells, as previously reported.62 In summary, the 50% cytotoxic concentration (CC50), which is the quantity needed to kill 50% of fibroblast cells, was found after VERO cells were exposed to different doses of the substance for 72 hours (Table 3). According to Table 3, the selectivity index (SI) was determined by dividing the compound’s IC50 value against the amastigote form (μM) by its CC50 value (μM). One important metric for determining a proposed drug’s therapeutic window is the SI. High selectivity and safety for mammalian cells were indicated by the results, which showed that compound 5b’s CC50 against the parasite was significantly higher than its IC50. Interestingly, 5b’s SI value was almost 35 times higher than miltefosine’s.
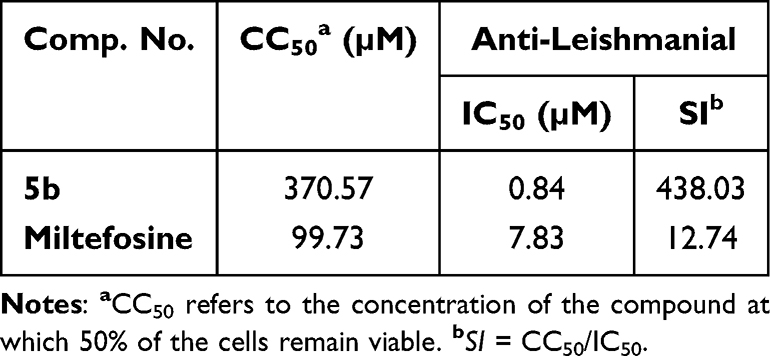 |
Table 3 Compound 5b Exhibits Cytotoxic Activity (CC50) Against Normal VERO Cells and Computed Selectivity Indices (SI) Against Promastigote Forms of Leishmania major |
Computational Studies
Predicted ADMET Studies
Good biochemical activity and advantageous ADMET characteristics (which include absorption, distribution, metabolism, excretion, and toxicity) are essential for a potential therapeutic candidate. Many online resources and computational techniques have been created recently to forecast the ADMET profiles of possible anti-cancer medication candidates.63–65 Notably, researchers have embraced ADMETlab 3.0, a completely rebuilt and currently commercially available web service, for the in silico assessment of drug-like characteristics.66–68 Using ADMETlab 3.0, the ADMET properties of compound 5b and miltefosine were predicted for the current investigation. Miltefosine met all of the ADMETlab 3.0 requirements, with the exception of the number of rotatable bonds (nRot), as shown in Figure 6. Compound 5b, in contrast, satisfied every assessed criterion, indicating a better ADMET profile and emphasizing its room for improvement.
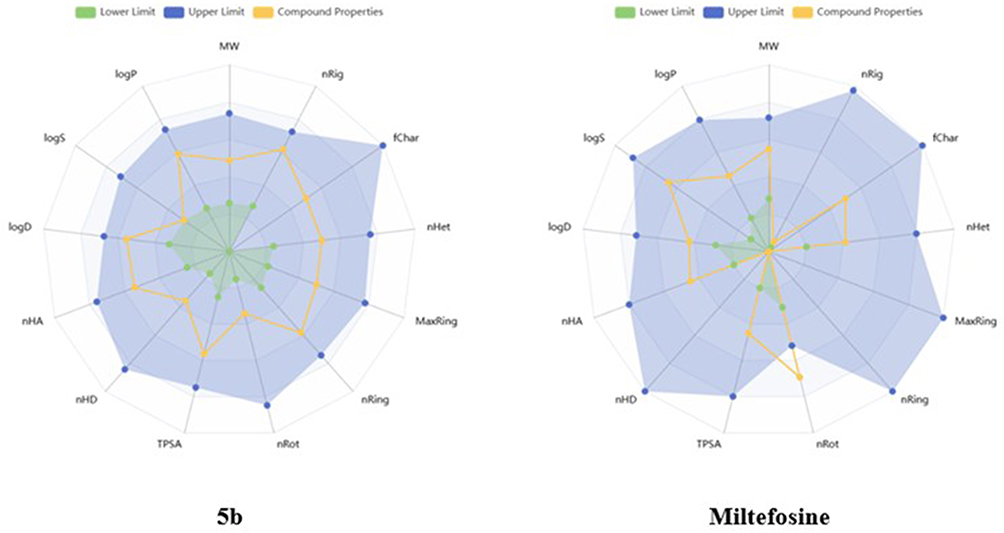 |
Figure 6 A) Pharmacokinetic mapping of compound 5b and miltefosine obtained from ADMETlab 3.0. Orangish lines indicate the compound parameters, while bluish regions define the upper limit, and greenish regions define the lower limit. |
Molecular Docking
Leishmanial DHFR-TS
To indicate the mechanism of the anti-leishmanial activity of the most active compounds via the anti-folate pathway, our consideration was mainly focused on the protein structure of dihydrofolate reductase-thymidylate synthase (DHFR-TS). The most active compound 5b showed an excellent and superior docking score (−16.5 Kcal/Mol) compared to the reference inhibitor (−13.4 Kcal/Mol). This anticipates favorable binding towards DHFR-TS and hence confirms the anti-folate mechanism validated by the in vitro experiment (Section: Reversal of the anti-leishmanial activity via folic and folinic acid). Perceiving the intermolecular interactions, compound 5b showed a strong binding affinity compared to the reference, consistent with the obtained in vitro results, where the docking pose of 5b showed several hydrogen bonds with Asn108, Ser111, and Leu164. Moreover, the methyl imidazo[1,2-a]pyridine ring was oriented into the hydrophobic pocket, forming several hydrophobic interactions with Met55, Phe58, Ile112, Phe116, and Leu164. In addition, the 5-fluoro-isatin moieties interacted with residues Leu40, Val45, and Leu46 through hydrophobic interactions Figure 7.
 |
Figure 7 The 2D interaction between 5b and DHFR-TS PDB ID: (1j3k). |
Leishmanial PTR1
Additionally, we conducted a docking experiment of the most active molecule 5b against the Leishmania major PTR1 (Lm-PTR1) enzyme in comparison to the reference Lm-PTR1 to elucidate the potential molecular mechanism underlying the anti-malarial activity. The docking study result of 5b was well matched with the in vitro enzymatic inhibition assay result. The tested compound 5b was able to properly dock into the enzyme binding site, achieving a docking score of −13.9 Kcal/Mol, similar to the reference inhibitor, having a docking score of −14.1 Kcal/Mol. Clearly, achieving a comparable binding mode for the reference inhibitor and establishing comparable key interactions with the amino acid residues inside the binding sites determined the enzymatic activity of the tested compound 5b. For the imidazo[1,2-a]pyridine moiety, the core ring interacted with Ser111 and NADP301 through hydrophobic interactions. In addition, the 2-CH3 group formed a hydrophobic interaction with the Met179 amino acid residue. For the carbohydrazine linker, the C=O and the imino N=C groups contributed to the hydrogen bonding interactions with Ser111, Tyr194, Lys198, and NADP301. Moreover, the 5-fluoro-isatin moiety was involved in several hydrophobic interactions with Phe113 and Asp181 residues (Figure 8).
 |
Figure 8 The 2D interaction between 5b and Leishmania major PTR1 (Lm-PTR1) ID: (6rxc). |
Conclusions
In this work, a novel series of imidazo[1,2-a]pyridine–isatin hybrids (5a-h, 8a-d, and 11a-d) was designed and synthesized using a molecular hybridization technique as possible anti-leishmanial agents. Benchmarked against the reference medication miltefosine, the developed framework incorporates an imidazo[1,2-a]pyridine core that is connected to C5/C7- and/or N1-substituted isatin derivatives at the C3 position via a hydrazide spacer. All compounds showed greater activity against Leishmania major promastigotes (IC50 = 0.84–4.84 μM vs. 7.83 μM) than miltefosine, according to in vitro evaluation. Most compounds were also better against amastigotes (IC50 = 1.28–7.18 μM vs. 8.07 μM), with the exception of 8d (IC50 = 8.18 μM). Using trimethoprim as a positive control, the most potent molecule, 5b, showed sub-micromolar potency and an anti-folate mechanism of action; in the presence of folic or folinic acid, its anti-leishmanial activity was eliminated. Compound 5b demonstrated exceptional safety with a high selectivity index (SI = 438.03) against normal VERO cells. The parasite selectivity of 5b was highlighted by molecular docking studies that showed substantial binding to the active sites of T. cruzi DHFR-TS and L. major pteridine reductase (PTR1), with no interaction in human DHFR. Additionally, positive physicochemical and pharmacokinetic characteristics were revealed by in silico research. This study is limited by the absence of in vivo assessment of the antileishmanial activity. However, this gap outlines a clear direction for future work, in which a thorough evaluation of efficacy, pharmacokinetics, and potential acute and chronic toxicities is needed to fully validate the therapeutic potential of the compounds. Overall, the obtained results position compound 5b as a strong antileishmanial candidate with promising biocompatibility and selectivity, warranting further investigation given the current scarcity and toxicity of available leishmaniasis therapies.
Highlights
- A new set of sixteen hybrids was developed.
- The biological efficacy of the compounds against the intracellular amastigote and promastigote forms of Leishmania major was assessed.
- With IC50 values ranging from 0.84 to 4.84 μM (promastigotes) to 1.28 to 8.18 µM (amastigotes), several hybrids performed better than miltefosine.
- In addition to having low cytotoxicity and good selectivity indices, the lead compound (5b) demonstrated strong dual-stage inhibition.
Acknowledgment
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through Small Research Project under grant number (RGP1/212/46).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Antinori S, Cascio A, Parravicini C, Bianchi R, Corbellino M. Leishmaniasis among organ transplant recipients. Lancet Infect Dis. 2008;8:191–20.
2. Desjeux P. Focus: leishmaniasis. Nat Rev Microbiol. 2004;2(9):692. doi:10.1038/nrmicro981
3. Zulfiqar B, Shelper TB, Avery VM. Leishmaniasis drug discovery: recent progress and challenges in assay development. Drug Discovery Today. 2017;22(10):1516–1531. doi:10.1016/j.drudis.2017.06.004
4. Hansen C, Paintsil E. Infectious diseases of poverty in children: a tale of two worlds. Pediatr Clin N Am. 2016;63(1):37. doi:10.1016/j.pcl.2015.08.002
5. Vickers TJ, Beverley SM, Docampo R. Folate metabolic pathways in Leishmania. Essays Biochem. 2011;51:63–80. doi:10.1042/bse0510063
6. Nare B, Hardy LW, Beverley SM. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem. 1997;272(21):13883–13891. doi:10.1074/jbc.272.21.13883
7. Leite FHA, Froes TQ, da Silva SG, et al. An integrated approach towards the discovery of novel non-nucleoside Leishmania major pteridine reductase 1 inhibitors. Eur J Med Chem. 2017;132:322–332. doi:10.1016/j.ejmech.2017.03.043
8. Dube D, Periwal V, Kumar M, Sharma S, Singh TP, Kaur P. 3D-QSAR based pharmacophore modeling and virtual screening for identification of novel pteridine reductase inhibitors. J Mol Model. 2012;18(5):1701–1711. doi:10.1007/s00894-011-1187-0
9. Roatt BM, de Oliveira Cardoso JM, De Brito RCF, Coura-Vital W, de Oliveira Aguiar-Soares RD, Reis AB. Recent advances and new strategies on leishmaniasis treatment. Appl Microbiol Biotechnol. 2020;104(21):8965–8977. doi:10.1007/s00253-020-10856-w
10. Adam R, Bilbao-Ramos P, López-Molina S, et al. Triazolopyridyl ketones as a novel class of antileishmanial agents. DNA binding and BSA interaction, Bioorg. Med Chem. 2014;22:4018–4027.
11. Eldehna WM, Almahli H, Ibrahim TM, et al. Synthesis, in vitro biological evaluation and in silico studies of certain arylnicotinic acids conjugated with aryl (thio)semicarbazides as a novel class of anti-leishmanial agents. Eur J Med Chem. 2019;179:335–346. doi:10.1016/j.ejmech.2019.06.051
12. Surur AS, Chan CF, Bartz F-M, et al. Fexinidazole optimization: enhancing anti-leishmanial profile, metabolic stability and hERG safety. RSC Med Chem. 2024;15(11):3837–3852. doi:10.1039/D4MD00426D
13. Khatun S, Singh A, Bader GN, Sofi FA. Imidazopyridine, a promising scaffold with potential medicinal applications and structural activity relationship (SAR): recent advances. J Biomol Struct Dyn. 2022;40(24):14279–14302. doi:10.1080/07391102.2021.1997818
14. Devi N, Singh D, Rawal RK, Bariwal J, Singh V. Medicinal attributes of imidazo [1, 2-a] pyridine derivatives: an update. Curr Top Med Chem. 2016;16(26):2963–2994. doi:10.2174/1568026616666160506145539
15. Marhadour S, Marchand P, Pagniez F, et al. Synthesis and biological evaluation of 2,3-diarylimidazo[1,2-a]pyridines as antileishmanial agents. Eur J Med Chem. 2012;58:543–556. doi:10.1016/j.ejmech.2012.10.048
16. Eldehna WM, El-Hamaky AA, Giovannuzzi S, et al. Development of isatin-functionalized benzenesulfonamides as novel carbonic anhydrase II and VII inhibitors with antiepileptic potential. Eur J Med Chem. 2025;292:117706. doi:10.1016/j.ejmech.2025.117706
17. Elsebaie HA, Abdulla M-H, Elsayed ZM, et al. Unveiling the potential of isatin-grafted phenyl-1, 2, 3-triazole derivatives as dual VEGFR-2/STAT-3 inhibitors: design, synthesis and biological assessments. Bioorg Chem. 2024;151:107626. doi:10.1016/j.bioorg.2024.107626
18. Eldehna WM, Nocentini A, Al-Rashood ST, et al. Tumor-associated carbonic anhydrase isoform IX and XII inhibitory properties of certain isatin-bearing sulfonamides endowed with in vitro antitumor activity towards colon cancer. Bioorg Chem. 2018;81:425–432. doi:10.1016/j.bioorg.2018.09.007
19. Elkaeed EB, Taghour MS, Mahdy HA, et al. New quinoline and isatin derivatives as apoptotic VEGFR-2 inhibitors: design, synthesis, anti-proliferative activity, docking, ADMET, toxicity, and MD simulation studies. J Enzyme Inhib Med Chem. 2022;37(1):2191–2205. doi:10.1080/14756366.2022.2110869
20. Saied S, Shaldam M, Elbadawi MM, et al. Discovery of indolinone-bearing benzenesulfonamides as new dual carbonic anhydrase and VEGFR-2 inhibitors possessing anticancer and pro-apoptotic properties. Eur J Med Chem. 2023;259:115707. doi:10.1016/j.ejmech.2023.115707
21. Shaldam MA, Almahli H, Angeli A, et al. Discovery of sulfonamide-tethered isatin derivatives as novel anticancer agents and VEGFR-2 inhibitors. J Enzyme Inhib Med Chem. 2023;38(1):2203389. doi:10.1080/14756366.2023.2203389
22. Fares M, Eldehna WM, Abou-Seri SM, Abdel-Aziz HA, Aly MH, Tolba MF. Design, Synthesis and In Vitro Antiproliferative Activity of Novel Isatin-Quinazoline Hybrids, Arch. Pharm. 2015;348:144–154.
23. Eldehna WM, Mahmoud ST, Elshnawey ER, et al. Novel indolinone-tethered benzothiophenes as anti-tubercular agents against MDR/XDR M. tuberculosis: design, synthesis, biological evaluation and in vivo pharmacokinetic study. Bioorg Chem. 2024;143:107009. doi:10.1016/j.bioorg.2023.107009
24. Elsayed ZM, Eldehna WM, Abdel-Aziz MM, et al. Development of novel isatin–nicotinohydrazide hybrids with potent activity against susceptible/resistant Mycobacterium tuberculosis and bronchitis causing–bacteria. J Enzyme Inhib Med Chem. 2021;36(1):384–392. doi:10.1080/14756366.2020.1868450
25. Al-Warhi T, Rashad NM, Almahli H, et al. Design and synthesis of benzo[b]thiophene-based hybrids as novel antitubercular agents against MDR/XDR Mycobacterium tuberculosis. Arch Pharm. 2024;357(2):2300529. doi:10.1002/ardp.202300529
26. Khatoon S, Aroosh A, Islam A, et al. Novel coumarin-isatin hybrids as potent antileishmanial agents: synthesis, in silico and in vitro evaluations. Bioorg Chem. 2021;110:104816. doi:10.1016/j.bioorg.2021.104816
27. Sabt A, Eldehna WM, Ibrahim TM, Bekhit AA, Batran RZ. New antileishmanial quinoline linked isatin derivatives targeting DHFR-TS and PTR1: design, synthesis, and molecular modeling studies. Eur J Med Chem. 2023;246:114959. doi:10.1016/j.ejmech.2022.114959
28. Khatoon S, Asif R, Kalsoom S, et al. Novel thiosemicarbazones of coumarin incorporated isatins: synthesis, structural characterization and antileishmanial activity. J Biomol Struct Dyn. 2025;1–13. doi:10.1080/07391102.2025.2498072
29. Barros Freitas LA, Caroline da Silva Santos A, de Cássia Silva G, et al. Structural improvement of new thiazolyl-isatin derivatives produces potent and selective trypanocidal and leishmanicidal compounds. Chem Biol Interact. 2021;345:109561. doi:10.1016/j.cbi.2021.109561
30. Yousuf M, Mukherjee D, Dey S, et al. Synthesis and biological evaluation of polyhydroxylated oxindole derivatives as potential antileishmanial agent. Bioorg Med Chem Lett. 2018;28(6):1056–1062. doi:10.1016/j.bmcl.2018.02.023
31. Hassan NW, Sabt A, El-Attar MAZ, et al. Modulating leishmanial pteridine metabolism machinery via some new coumarin-1,2,3-triazoles: design, synthesis and computational studies. Eur J Med Chem. 2023;253:115333. doi:10.1016/j.ejmech.2023.115333
32. Samala G, Nallangi R, Devi PB, et al. Identification and development of 2-methylimidazo[1,2-a]pyridine-3-carboxamides as Mycobacterium tuberculosis pantothenate synthetase inhibitors. Bioorg Med Chem. 2014;22(15):4223–4232. doi:10.1016/j.bmc.2014.05.038
33. Abo-Ashour MF, Eldehna WM, Nocentini A, et al. Novel hydrazido benzenesulfonamides-isatin conjugates: synthesis, carbonic anhydrase inhibitory activity and molecular modeling studies. Eur J Med Chem. 2018;157:28–36. doi:10.1016/j.ejmech.2018.07.054
34. Eldehna WM, El Hassab MA, Abo-Ashour MF, et al. Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: synthesis, biological and molecular dynamics investigations. Bioorg Chem. 2021;110:104748. doi:10.1016/j.bioorg.2021.104748
35. Ibrahim TM, Abada G, Dammann M, et al. Tetrahydrobenzo[h]quinoline derivatives as a novel chemotype for dual antileishmanial-antimalarial activity graced with antitubercular activity: design, synthesis and biological evaluation. Eur J Med Chem. 2023;257:115534. doi:10.1016/j.ejmech.2023.115534
36. Batran RZ, Ebaid MS, Nasralla SN, et al. Synthesis and mechanistic insights of Coumarinyl-Indolinone hybrids as potent inhibitors of Leishmania major. Eur J Med Chem. 2025;288:117392. doi:10.1016/j.ejmech.2025.117392
37. Bekhit AA, Lodebo ET, Hymete A, et al. New pyrazolylpyrazoline derivatives as dual acting antimalarial-antileishamanial agents: synthesis, biological evaluation and molecular modelling simulations. J Enzyme Inhib Med Chem. 2022;37(1):2320–2333. doi:10.1080/14756366.2022.2117316
38. Bekhit AA, Saudi MN, Hassan AM, et al. Synthesis, molecular modeling and biological screening of some pyrazole derivatives as antileishmanial agents. Future Med Chem. 2018;10(19):2325–2344. doi:10.4155/fmc-2018-0058
39. Temraz MG, Elzahhar PA, Bekhit AE-DA, Bekhit AA, Labib HF, Belal AS. Anti-leishmanial click modifiable thiosemicarbazones: design, synthesis, biological evaluation and in silico studies. Eur J Med Chem. 2018;151:585–600. doi:10.1016/j.ejmech.2018.04.003
40. Teixeira M, de Jesus Santos R, Sampaio R, Pontes-de-carvalho L, dos-Santos WL. A simple and reproducible method to obtain large numbers of axenic amastigotes of different Leishmania species. Parasitol Res. 2002;88(11):963–968. doi:10.1007/s00436-002-0695-3
41. Osorio Y, Travi BL, Renslo AR, Peniche AG, Melby PC, Geary TG. Identification of small molecule lead compounds for visceral leishmaniasis using a novel ex vivo splenic explant model system. PLoS Negl Trop Dis. 2011;5(2):e962. doi:10.1371/journal.pntd.0000962
42. Ahsan MJ, Samy JG, Khalilullah H, et al. Molecular properties prediction and synthesis of novel 1,3,4-oxadiazole analogues as potent antimicrobial and antitubercular agents. Bioorg Med Chem Lett. 2011;21(24):7246–7250. doi:10.1016/j.bmcl.2011.10.057
43. Desai NC, Kotadiya GM, Jadeja KA, et al. Synthesis, antitubercular, antimicrobial activities and molecular docking study of quinoline bearing dihydropyrimidines. Bioorg Chem. 2021;115:1–14. doi:10.1016/j.bioorg.2021.105173
44. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
45. El Hassab MA, Eldehna WM, Hassan GS, Abou-Seri SM. Multi-stage structure-based virtual screening approach combining 3D pharmacophore, docking and molecular dynamic simulation towards the identification of potential selective PARP-1 inhibitors. BMC Chem. 2025;19(1):30. doi:10.1186/s13065-025-01389-2
46. Elsebaie HA, El-Moselhy TF, El-Bastawissy EA, et al. Development of new thieno[2,3-d]pyrimidines as dual EGFR and STAT3 inhibitors endowed with anticancer and pro-apoptotic activities. Bioorg Chem. 2024;143:107101. doi:10.1016/j.bioorg.2024.107101
47. Mohamady S, Khalil AF, Naguib BH, Nafie MS, Tawfik HO, Shaldam MA. Tailored horseshoe-shaped nicotinonitrile scaffold as dual promising c-Met and Pim-1 inhibitors: design, synthesis, SAR and in silico study. Bioorg Chem. 2024;143:106988. doi:10.1016/j.bioorg.2023.106988
48. Eldehna WM, Alkabbani MA, Elsayed ZM, et al. Identification of novel triazole–pyrazole conjugates as potential anticonvulsant agents: synthesis and biological evaluations. ACS Chem Neurosci. 2025;16(15):3038–3050. doi:10.1021/acschemneuro.5c00392
49. Nafie MS, Youssef MI, El-Hamaky AA, et al. Triazole-functionalized benzofuran and benzothiophene semicarbazides as novel VEGFR-2-targeted anti-cancer agents. Bioorg Chem. 2025;163:108702. doi:10.1016/j.bioorg.2025.108702
50. Zeidan MA, Alkabbani MA, Giovannuzzi S, et al. Shooting an arrow against convulsion: novel triazole-grafted benzenesulfonamide derivatives as carbonic anhydrase II and VII inhibitors. J Med Chem. 2025;68(8):8873–8893. doi:10.1021/acs.jmedchem.5c00526
51. Eldehna WM, Roshdy E, Abdulla M-H, et al. Discovery of 1-phenyl-1,2,3-triazole ureas as dual VEGFR-2/JNK-1 type II kinase inhibitors targeting pancreatic cancer. Int J Biol Macromol. 2025;308:142372. doi:10.1016/j.ijbiomac.2025.142372
52. Eldehna WM, Tawfik HO, Nafie MS, et al. Novel benzofuran-conjugated indolin-2-ones as anticancer agents; design, synthesis, biological assessments, and molecular modeling insights. Bioorg Chem. 2025;160:108494. doi:10.1016/j.bioorg.2025.108494
53. Eldehna WM, Salem R, Elsayed ZM, et al. Development of novel benzofuran-isatin conjugates as potential antiproliferative agents with apoptosis inducing mechanism in Colon cancer. J Enzyme Inhib Med Chem. 2021;36(1):1423–1434. doi:10.1080/14756366.2021.1944127
54. Eldehna WM, Habib YA, Mahmoud AE, et al. Design, synthesis, and in silico insights of novel N’-(2-oxoindolin-3-ylidene)piperidine-4-carbohydrazide derivatives as VEGFR-2 inhibitors. Bioorg Chem. 2024;153:107829. doi:10.1016/j.bioorg.2024.107829
55. Khaleel EF, Sabt A, Korycka-Machala M, et al. Identification of new anti-mycobacterial agents based on quinoline-isatin hybrids targeting enoyl acyl carrier protein reductase (InhA). Bioorg Chem. 2024;144:107138. doi:10.1016/j.bioorg.2024.107138
56. Pereira TM, Vitório F, Amaral RC, Zanoni KPS, Iha NYM, Kümmerle AE. Microwave-assisted synthesis and photophysical studies of novel fluorescent N-acylhydrazone and semicarbazone-7-OH-coumarin dyes. New J Chem. 2016;40(10):8846–8854. doi:10.1039/C6NJ01532H
57. Emami S, Valipour M, Kazemi Komishani F, et al. Synthesis, in silico, in vitro and in vivo evaluations of isatin aroylhydrazones as highly potent anticonvulsant agents. Bioorg Chem. 2021;112:104943. doi:10.1016/j.bioorg.2021.104943
58. Sharma PK, Balwani S, Mathur D, et al. Synthesis and anti-inflammatory activity evaluation of novel triazolyl-isatin hybrids. J Enzyme Inhib Med Chem. 2016;31(6):1520–1526. doi:10.3109/14756366.2016.1151015
59. Jakusová K, Gáplovský M, Donovalová J, et al. Effect of reactants’ concentration on the ratio and yield of E, Z isomers of isatin-3-(4-phenyl) semicarbazone and N-methylisatin-3-(4-phenyl) semicarbazone. Chem Papers. 2013;67(1):117–126. doi:10.2478/s11696-012-0248-x
60. Mendoza-Martínez C, Galindo-Sevilla N, Correa-Basurto J, et al. Antileishmanial activity of quinazoline derivatives: synthesis, docking screens, molecular dynamic simulations and electrochemical studies. Eur J Med Chem. 2015;92:314–331. doi:10.1016/j.ejmech.2014.12.051
61. Tonelli M, Gabriele E, Piazza F, et al. Benzimidazole derivatives endowed with potent antileishmanial activity. J Enzyme Inhib Med Chem. 2018;33(1):210–226. doi:10.1080/14756366.2017.1410480
62. Pessotti J, Do Valle TZ, Corte-Real S, Da Costa SG. Interaction of Leishmania (L.) chagasi with the Vero cell line. Parasite. 2004;11(1):9–102. doi:10.1051/parasite/200411199
63. Tawfik HO, El-Hamaky AA, El-Bastawissy EA, et al. New genetic bomb trigger: design, synthesis, molecular dynamics simulation, and biological evaluation of novel BIBR1532-related analogs targeting telomerase against non-small cell lung cancer. Pharmaceuticals. 2022;15(4):1–28. doi:10.3390/ph15040481
64. Elsebaie HA, El-Bastawissy EA, Elberembally KM, et al. Novel 4-(2-arylidenehydrazineyl)thienopyrimidine derivatives as anticancer EGFR inhibitors: design, synthesis, biological evaluation, kinome selectivity and in silico insights. Bioorg Chem. 2023;140:1–18. doi:10.1016/j.bioorg.2023.106799
65. Hefny SM, El-Moselhy TF, El-Din N, et al. A new framework for novel analogues of pazopanib as potent and selective human carbonic anhydrase inhibitors: design, repurposing rational, synthesis, crystallographic, in vivo and in vitro biological assessments. Eur J Med Chem. 2024;274:116527. doi:10.1016/j.ejmech.2024.116527
66. Ding Y, Zhang Y, Yan L, Liu L. New pyrimidinothiophene derivatives: synthesis, spectroscopic analysis, X-ray, DFT calculation, biological activity studies and ADMET prediction. J Mol Struct. 2023;1290:1–10. doi:10.1016/j.molstruc.2023.135952
67. Xiong G, Wu Z, Yi J, et al. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1):1–10. doi:10.1093/nar/gkab255
68. Al-Karmalawy AA, Nafie MS, Shaldam MA, et al. Ligand-based design on the dog-bone-shaped BIBR1532 pharmacophoric features and synthesis of novel analogues as promising telomerase inhibitors with in vitro and in vivo evaluations. J Med Chem. 2023;66(1):777–792. doi:10.1021/acs.jmedchem.2c01668
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.