")
Back to Journals » International Journal of Nanomedicine » Volume 19
Artesunate-Nanoliposome-TPP, a Novel Drug Delivery System That Targets the Mitochondria, Attenuates Cisplatin-Induced Acute Kidney Injury by Suppressing Oxidative Stress and Inflammatory Effects
Authors Zhang J, Gu L, Jiang Y, Ma Y, Zhang Z, Shen S, Shen S, Peng Q, Xiao W
Received 11 October 2023
Accepted for publication 25 January 2024
Published 12 February 2024 Volume 2024:19 Pages 1385—1408
DOI https://doi.org/10.2147/IJN.S444076
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Misra
Jiaxing Zhang,1,2,* Liwei Gu,3,* Yumao Jiang,4,* Yun Ma,5,* Ziyue Zhang,3 Shengnan Shen,3 Shuo Shen,3 Qing Peng,6 Wei Xiao2,7
1Institute of Digestive Disease, The Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People’s Hospital, Qingyuan, Guang Dong, People’s Republic of China; 2School of Traditional Chinese Medicine, Southern Medical University, Guangzhou, Guang Dong, People’s Republic of China; 3Artemisinin Research Center and Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 4Gannan Medical University, Ganzhou, Jiang Xi, People’s Republic of China; 5Department of Pharmacy, Nanfang Hospital, Southern Medical University, Guangzhou, Guang Dong, People’s Republic of China; 6Institute of Basic Medical Sciences, Xiyuan Hospital, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 7Key Laboratory of Glucolipid Metabolic Disorder, Ministry of Education, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qing Peng; Wei Xiao, Email [email protected]; [email protected]
Background: Acute kidney injury (AKI) is a syndrome, posing a substantial healthcare burden. The pathological basis of AKI is associated with inflammation and oxidative stress which cause additional damage to mitochondria. Artesunate (ATS) is a derivative of artemisinin isolated from Artemisia annua L. that is an effective treatment for malaria and favored for the prevention and treatment of kidney diseases. However, there are still challenges related to its efficacy, including poor water solubility, limited oral bioavailability and short half-life. Liposome-based nanoparticles are used for drug delivery due to their ideal biocompatibility and their ability to improve the bioavailability of specific drugs and enhance drug efficacy.
Methods: In this study, a novel TPP-based natural ATS-nanoliposome, namely T-A-Ls, was applied for the treatment of AKI. ATS was encapsulated with or without triphenylphosphonium (TPP)-modified nanoliposomes. AKI was induced by cisplatin in C57BL/6J mice and a cisplatin-induced injury model was generated in HK-2 cells in vitro. Blood urea nitrogen (BUN), serum creatinine (Scr) measurements and section staining were utilized to assess renal protective effect of T-A-Ls. Inflammatory-related factors and proteins were quantified via Elisa, Immunofluorescence and Western Blot (WB). The anti-mitochondrial oxidative stress effect of T-A-Ls was determined by ROS, JC-1 and oxygen consumption rate (OCR) kits. Immunohistochemistry and WB were conducted to measure associated protein expressions. In vivo biodistribution and the concentration of T-A-Ls in kidney were also explored.
Results: T-A-Ls exhibited good oxidative resistance, preferential renal uptake, mitochondrial targeting, and it ameliorated kidney injury in cisplatin-induced AKI mice. Mitochondrial dysfunction, ATP production and respiratory capacity were improved in damaged HK-2 cells; ROS content decreased while mitochondrial membrane potential recovered. T-A-Ls exerted renal protection by inhibiting inflammation and reducing oxidative stress; these effects were mediated by a downregulation in the expression of RAGE and iNOS and an upregulation in both Nrf2 and HO-1.
Conclusion: T-A-Ls could improve the delivery of ATS to the kidney, offering a promising avenue to treat AKI.
Keywords: artesunate, liposomes, AKI, cisplatin, oxidative stress, inflammation
Graphical Abstract:
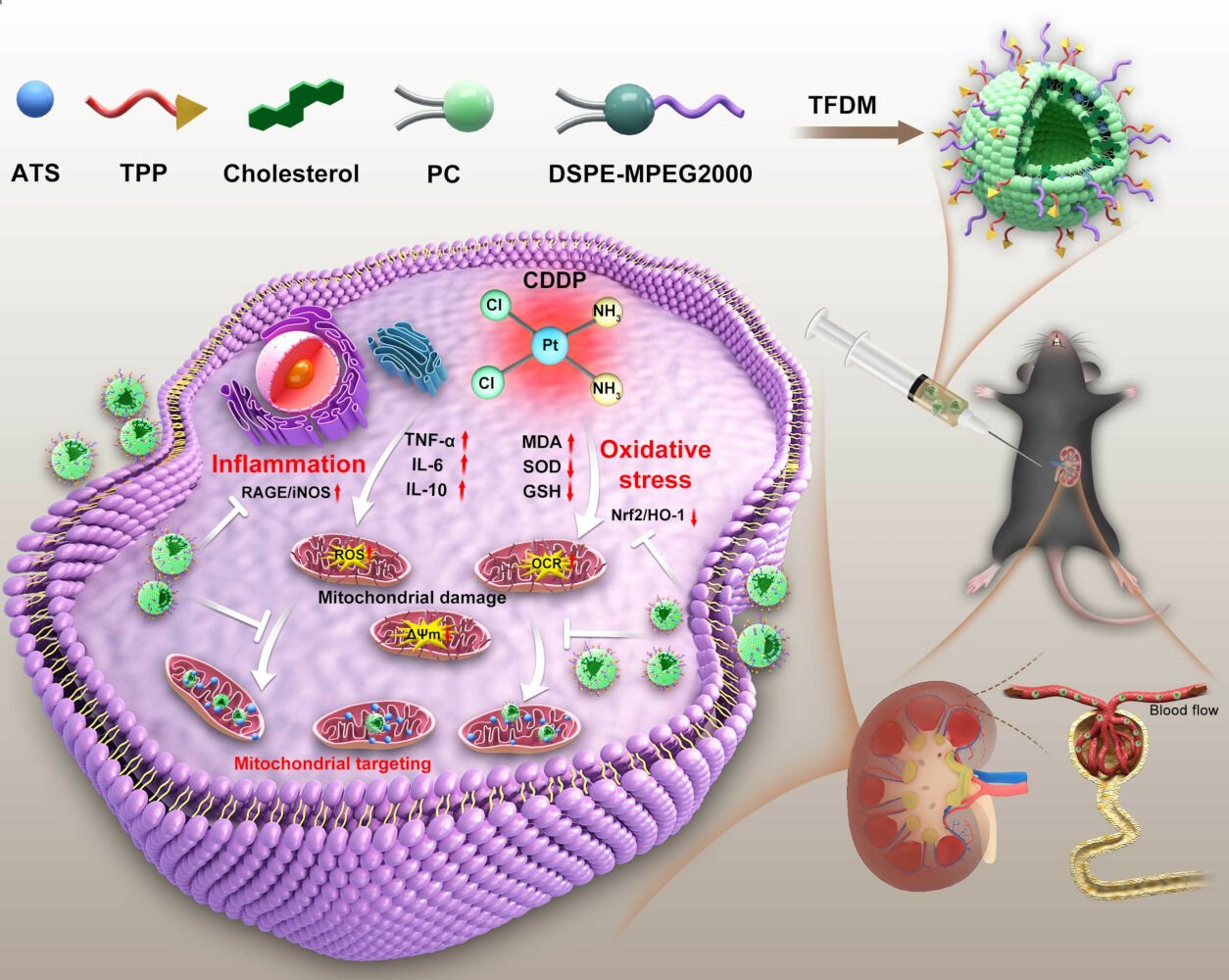
Introduction
Acute kidney injury (AKI)-A clinical syndrome that is characterized by the rapid loss of renal function and can progress into chronic kidney disease (CKD) or even end-stage renal disease (ESRD)-is a major global public health problem.1 AKI accounts for 10–15% of all hospitalized patients,2 while the prevalence among patients in intensive care units (ICUs) sometimes exceeds 50%,3 and AKI affects the lives of approximately 13 million patients every year.4 AKI can be caused by various factors, although the main causes are renal ischemia-reperfusion injury (IRI), nephrotoxic injury, and sepsis infection,5–8 and these conditions pose a serious threat to human health. Drug-induced kidney injury is also a major cause of AKI. The pathophysiology and mechanisms underlying this injury are complex and can vary with different drugs.9–11
Cisplatin (CDDP; cis-diamminedichloroplatinum (II)) was the first inorganic platinum anticancer drug to be approved by the FDA and is one of the most common anticancer drugs available clinically.12 Cisplatin is widely used in the treatment of lung, breast, liver, prostate, and cervical cancers. However, although cisplatin is a powerful drug, it has many side effects and causes significant harm to the human body. The clinical value of this drug is limited by time-and dose-effects and strong nephrotoxicity.13,14 Clinically, the risk of nephrotoxicity in patients taking cisplatin ranges from 20% to 35%,15,16 and different doses of cisplatin can cause different degrees of nephrotoxicity.17 Pharmacokinetic studies have shown that the nephrotoxicity of cisplatin is predominantly due to its wide distribution in kidney and long-term accumulation.18,19 Anticancer treatment with platinum compounds usually aims to damage the integrity of cells by destroying nuclear DNA (nDNA), thus leading to cell death. However, the proportion of cisplatin adducts of mitochondrial DNA was 300–500 times that of nDNA in an analysis of cells containing damaged mitochondria following cisplatin treatment,20 which illustrates the importance of mitochondria in the cisplatin response. Mitochondrial damage is an important early event that occurs prior to renal tubular cell injury.21 However, there is currently no effective renal protective agent for the prevention and treatment of cisplatin-induced mitochondrial damage. Therefore, it is significant value in identifying drugs that have better improved mitochondrial function on cisplatin-induced renal injury.
Artesunate (ATS) is a semi-synthetic derivative of artemisinin that is isolated from Artemisia annua.22 As a drug, ATS is safe, fast, efficient, and does not induce resistance easily, it has become one of the first-line drugs recommended by the WHO for the treatment of malaria.23–26 In addition to the treatment of malaria, ATS and its metabolites are also known to exert anti-inflammatory,27 anti-viral,28 and immune regulation effects29 and can inhibit the occurrence of leukemia and several types of cancer, including colorectal, lung, liver, and breast cancer.30 It has been also reported that ATS can alleviate acute renal injury by inhibiting macrophage mincle-mediated tubular epithelial cell necrosis and inflammation.31 However, ATS has high fat solubility and low water solubility,32 is predominantly transported by simple diffusion in vivo without any form of targeting, and has a half-life in vivo of only 15–30 min,33 these characteristics present significant limitations to the wider application of ATS in the clinic. Liposomes were the first nanoparticle to be successfully applied in clinical treatment and were first described by Bangham and Standish in 1965.34 The bimolecular structure of the liposome membrane is similar to that of the cell membrane and exhibits good biocompatibility, thus, liposomes are more conducive to drug binding and are considered to be the best drug carrier system thus far.35 Furthermore, the combination of nanoscience and bioactive natural products is attracting increasing attention.36–38
In this study, triphenylphosphonium (TPP)-decorated modified liposomes loaded with ATS (T-A-Ls) were used for mitochondrial targeting with the aim of improving cisplatin-induced acute renal injury in mice. TPP is a lipophilic cationic compound that is easily attracted by the negatively charged mitochondrial membrane and can pass through the phospholipid bilayer and accumulate in the mitochondria. It is assumed that the T-A-Ls would be able to deliver ATS selectively and efficiently to the mitochondria to block oxidative damage in the epithelial cells of the renal tubules. The properties of the liposomes including particle size, zeta potential distribution, particle size dispersion coefficient (PDI), encapsulation efficiency (EE), and stability were investigated. It demonstrated that T-A-Ls enhanced the renal protection of ATS in a cisplatin-induced mouse model of AKI. In addition, the mechanism by which T-A-Ls effectively attenuated the nephrotoxicity associated with cisplatin involved activation of the Nrf2/HO-1 signaling pathway and suppression of inflammation. Findings from this study provide a new perspective for the treatment of AKI and the development of new ATS-based liposomes.
Materials and Methods
Chemicals and Reagents
ATS was obtained from Kunyao Group Wulingshan Pharmaceutical Co., Ltd (Chongqing, China). A.V.T. (Shanghai) Pharmaceutical Co., Ltd. supplied the soybean lecithin (PC95), cholesterol, and DSPE-MPEG2000 (Shanghai, China). AAT Bioquest, Inc. supplied DiR iodide (USA). Shanghai TITAN Technology Co., Ltd. supplied dichloromethane and methanol (Shanghai, China). Adamas Reagent Co., Ltd. provided (4-carboxybutyl) triphenylphosphonium bromide (TPP). Thermo Fisher Scientific, Inc. supplied the acetonitrile (HPLC) grade (Massachusetts, USA). A.S. Watson Group Ltd. supplied purified water (Hong Kong, China). Merk Millipore Ltd provided centrifugal ultrafiltration tubes (Darmstadt, Germany). The HPLC chromatographic BEH C18 column (2.1×100 mm, 1.7 μm) was purchased from Waters (USA) and Luna C18(2) column (4.6×100 mm, 3 μm) was purchased from Phenomenex (USA). DMEM/F12 -Dulbecco’s Modified Eagle Medium (DMEM/F12; Corning, USA) and fetal bovine serum (FBS) were obtained from ABW (Animal Blood Ware, China). 0.25% trypsin was purchase from NCM Biotech (Suzhou, China). A Cell Counting Kit-8 (CCK-8) was purchased from DOJINDO (Japan). The primary antibodies used in this study were as follows: iNOS (Cat# 22226-1-AP), HO-1 (Cat# 10701-1-AP) and Nrf2 (Cat# 16396-1-AP) were supplied by Proteintech, Inc. (Proteintech Group, USA), β-actin (Cat# T0022) and GAPDH (Cat# T0004) were obtained from Affinity Biosciences (USA), and RAGE (Cat# bs-4999R) obtained from Bioss (Beijing, China).
Animals and Cell Lines
C57BL/6J wild-type male mice (6–8 weeks-of-age) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Certificate of Quality Number: SCXK (JING) 2016–0006, SCXK (JING) 2021–0011, Beijing, China), were kept under a specific laboratory environment (22–24°C; 55–60% relative humidity; 12 h light/dark cycle). All animal experiments received ethical approval from the Laboratory Animal Ethics Committee of Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences (2022B030). Human proximal tubular epithelial cells (HK-2 cells) were acquired from Procell Life Science & Technology Co., Ltd (Wuhan, China). These cells were cultured in DMEM/F-12 complete medium with 100 U/mL of penicillin and 100 μg/mL of streptomycin at 37°C under 5% CO2.
Preparation of T-A-Ls
T-A-Ls were prepared using a thin-film hydration method.39,40 Briefly, soybean lecithin, ATS, TPP, cholesterol, and DSPE-MPEG2000 were dissolved in dichloromethane and the solution was dried using rotary evaporation. Subsequently, the proper amount of water was added into the bottle for hydration to obtain the liposomes solution. The solution was then treated with ultrasound for 3 min to generate T-A-Ls. Finally, the liposomes were extruded through a 0.22 µm membrane filter.
Preparation of A-Ls and DiR-T-A-Ls
A-Ls were formulated by dissolving soybean lecithin, ATS, cholesterol, and DSPE-MPEG2000 in dichloromethane, then the rest of the method was the same as that of T-A-Ls. DiR-T-A-Ls were prepared by dissolving an alcohol solution of DiR, soybean lecithin, TPP, ATS, cholesterol, and DSPE-MPEG2000 in dichloromethane and then following the process for T-A-Ls.
Characterization of T-A-Ls
Particle Size, Zeta Potential and Polydispersity Index (PDI)
Malvern Zetasizer Nano ZS (Malvern Instruments, UK) was used for determining the particle size, zeta potential and PDI of liposomes at 25°C. Data analysis was based on the average value of three parallel tests.
Encapsulation Efficiency (EE) of Liposomes
The ultrafiltration technique was used to determine the encapsulation efficiency (EE) of T-A-Ls.39,40 The total ATS content (Mtotal drug) of the liposomes was measured by high-performance liquid chromatography (HPLC). Free ATS was separated by ultrafiltration and the free ATS content (Mfree drug) was also measured by HPLC. Centrifuge conditions for the ultrafiltration tubes (Millipore Amicon Ultra 4 mL; 10 kDa) were 5000 rpm at 4°C for 120 min to obtain ultrafiltrates. HPLC conditions were:40 Column: Luna C18(2) column (4.6×100 mm, 3 μm); mobile phase: acetonitrile/0.1% phosphoric acid/water 44:56, mobile phase flow rate: 1.0 mL min−1; column temperature: 30°C; detection wavelength: 216 nm. Mean data were calculated from three parallel experiments. The following equation was used to calculate EE:

Transmission Electron Microscopy (TEM) of T-A-Ls
T-A-Ls were diluted twice with purified water and an appropriate amount of liposomes diluent was dropped onto a copper mesh. After the copper mesh was dried at room temperature, the T-A-Ls were photographed by TEM (HITACHI H7650, Japan).
In vitro Release of T-A-Ls
The T-A-Ls release experiment was performed by dialysis in PBS medium (pH 7.4) containing 20% ethanol. 2mL of liposomes was added and sealed in the dialysis bags (MWCO: 3500 Da) (n = 3), the dialysis bag containing T-A-Ls was placed into 50mL release medium and incubated at 37 °C with a shaking speed of 80 rpm (n = 3). Samples were taken at 1h, 2h, 3h, 4h, 5h, 6h, 8h, 10h and 12h and an equal volume of release medium was immediately supplemented at each sampling point. The HPLC (waters Arc HPLC system, USA) was used to determine the release amount of ATS. Other HPLC conditions were the same as “Encapsulation efficiency (EE) of liposomes”.40
Liposome Stability
The entrapment efficiency, particle size, and PDI of the T-A-Ls were determined at 0, 1, 2, 4, and 6 weeks after sample preparation. These measurements were used to evaluate the stability of the sample stored at 4°C.
Models of CDDP-Induced AKI and Drug Administration
For pharmacological research, male SPF grade mice (weighing approximately 22 g) were randomly divided into five groups (n = 6 animals per group): a normal control, a CDDP group (20 mg/kg), a CDDP + ATS group (30 mg/kg), a CDDP + A-Ls group (30 mg/kg) and a CDDP + T-A-Ls group (30 mg/kg). Different drugs with a dose of 30mg/kg were used to pretreat the mice for 72 h, and CDDP (20 mg/kg) was administered to the mice via a single intraperitoneal injection to induce AKI. Then different drug treatment was continued once daily for additional 72 h. The control group received one dose of physiological saline. Blood was collected from the orbits of each mouse, centrifuged and serum was stored at −20°C. Kidneys were harvested for subsequent experiments.
To further investigate the protective effect of T-A-Ls on CDDP induced AKI, SPF grade C57BL/6J male mice (weighing approximately 22g) were randomly divided into four groups (n = 6 in each group): a normal control group, a CDDP group (20 mg/kg), a CDDP+T-A-Ls group (15 mg/kg), and a CDDP+T-A-Ls group (30 mg/kg). Two different doses were utilized to determine the mechanism by which T-A-Ls alleviate CDDP induced AKI through the inhibition of oxidative stress and inflammation.
Histopathological Analysis
Hematoxylin and Eosin (H&E) Staining
At the end of the experiment, mice were sacrificed by cervical dislocation and the left kidney was removed from each mouse. Then, 4 μm thick sections were cut and stained with hematoxylin and eosin (H&E) kits and photographed with a BX51 optical microscope (Olympus, Japan) to assess kidney damage. The kidney tissues used for H&E staining (x200) were performed as reported,41 and the scoring criteria of the renal tubular interstitial injury index are investigated based on the method described previously.42 There are some parameters, namely renal tubular dilation, renal tubular cell vacuolization, renal tubular atrophy, red blood cell cast, white blood cell cast, protein cast, and interstitial inflammation cell infiltration, for scoring. Each parameter is set to have 4 levels: normal 0 points, mild 1 point, moderate 2 points, severe 3 points. According to the H&E staining evaluations of various groups, renal tubulointerstitial pathological changes are assessed.
Masson’s Trichrome Staining
The level of renal fibrosis in each mouse was assessed using Masson’s trichrome staining, following a method previously described.43 The semi-quantitative renal interstitial fibrosis index was calculated as follows: collagen area < 2%, 0 point; collagen area: 2–10%, 1 point; collagen area: 11–20%, 2 points; collagen area: 21–30%, 3 points; collagen area: > 30%, 4 points.
Blood Physiochemical Assays and Safety Evaluation of T-A-Ls in Healthy Mice
Blood was collected from the abdominal aorta immediately after the mice were sacrificed. The serum levels of urea, nitrogen and creatinine were then determined by biochemical kits (Nanjing Jiancheng, Nanjing, China). To test whether the treatment was safe, T-A-Ls were injected into normal mice for 5 days. Then, we determined a series of standard hematological markers, including white blood cells (WBC), red blood cells (RBC), platelets (PLT), hemoglobin (HGB), hematocrit (HCT), mean platelet volume (MPV), mean corpuscular hemoglobin (MCH), mean corpuscular volume (MCV), platelet distribution width (PDW) and parameters relating to liver and kidney function, including alanine transaminase (ALT), aspartate transaminase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), and serum creatinine (Scr). All these parameters were determined by an automatic biochemical analyzer (Tokyo, Japan). Further, we selected the heart, liver, spleen, lung and kidney of these normal mice for histopathological analysis, and the results were analyzed by H&E staining.
Detection of Inflammatory Factors in the Serum
Blood samples were collected and maintained at room temperature for 2 h. Then, serum was separated from the whole blood by centrifugation at 3500 rpm for 15 min at 25°C. The serum levels of TNF-α, IL-6 and IL-1β were then detected with an ELISA assay kit in accordance with the manufacturer’s protocols.
Measurement of GSH and MDA Levels and the Activity of SOD
To test the antioxidant activity of T-A-Ls, we took kidney tissue samples from each group (0.2–1g) and homogenized each sample on ice with normal saline. Next, the lysate was centrifuged at 2000 rpm for 15 min and the levels of Glutathione (GSH) and Malondialdehyde (MDA), along with the activity of Superoxide Dismutase (SOD), were measured using commercial kits.
Immunohistochemistry (IHC) and Immunofluorescence (IF)
IHC analysis was performed as described previously.44 In brief, primary antibodies, including anti-HO-1 (1:250) and anti-Nrf2 (1:250), were incubated with paraffin-embedded tissue sections in a humidified chamber overnight at 4°C. Next, the sections were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 2 h at room temperature (RT). Brownish yellow regions in the sections were considered to represent positive staining. A light microscope was then used to acquire images and Image J software was used to quantify the intensity of positive expression.
For IF staining, the primary antibodies, including anti-iNOS (1:100), anti-4-HNE (1:50), and anti-RAGE (1:200) were incubated with kidney sections overnight at 4°C in a humidified chamber. The sections incubated with iNOS and RAGE antibodies were then incubated with DyLight 488-labeled secondary antibody (1:200) for 2 h at RT. The slices incubated with 4-HNE antibody (Cat# ab48506, Abcam, USA) were probed by Alexa Fluor 594-labeled secondary antibody (1:200) for 1 h at RT. Hoechst 33258 was used to stain the cell nuclei. Laser scanning confocal microscopy (Laser TCS SP8, Wetzlar, Germany) was used to photograph the images and Image J was used to quantify the intensity of IF.
Western Blotting
Renal tissue was lysed in radio immunoprecipitation assay (RIPA) dissolution buffer and total protein was extracted by centrifugation at 4°C and 13,000 rpm. Next, 10% or 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate the proteins (25 µg/lane), which were then transferred to a PVDF membrane. Membranes were subsequently blocked with skimmed milk for 1 h at RT and then incubated with primary antibodies against RAGE (1:1000), iNOS (1:1000), Nrf2 (1:5000), and HO-1 (1:400) overnight at 4°C. Next, the membranes were incubated with secondary antibodies (1:5000) for 1 h at RT. Finally, ECL substrate (Millipore, MA, USA) was used to detect positive signals and Image J software was used to analyze the intensity of the bands.
Cytotoxicity Analysis, Cellular Uptake and Distribution in vitro
For the cytotoxicity assay, 5×103 HK-2 cells/well were seeded in 96-well plates and incubated for 24 h before being treated with or without drugs at a concentration of 0–200 µM. Subsequently, the cytotoxicity of T-A-Ls to HK-2 cells was determined by using the CCK-8 kit in accordance with the manufacturer’s protocol.
To explore the distribution of T-A-Ls in vitro, HK-2 cells were seeded in a confocal dish at a density of 1×104 for 24 h. FITC-labeled T-A-Ls were prepared spontaneously by self-assembly in ethanol. The cells were then incubated with or without T-A-Ls for 24 h. Following culture, the cells were washed three times in cold PBS and then fixed with 4% formaldehyde. Hoechst 33258 was used as a counterstain for the nucleus.
We used Mito-Tracker Red CMXRos, a mitochondrial red fluorescent probe (#C1049B; Beyotime, China), to label mitochondria, and strictly following the instructions for operation. Finally, the cells were observed by confocal laser scanning microscopy (Zeiss LSM 510 meta, Oberkochen, Germany).
To detect cellular uptake, HK-2 cells were cultured in 24-well plates for 24 h, then 0, 50, and 100 µM of T-A-Ls were added and incubated with the cells for 24 h. Next, the cells were collected, centrifuged, and filtered, and then flow cytometry was used to determine fluorescence intensity.
ROS Detection
Next, we detected the production of Reactive Oxygen Species (ROS) in HK-2 cells to evaluate whether T-A-Ls could effectively reduce the levels of ROS in HK-2 cells after cisplatin injury. First, HK-2 cells were inoculated in confocal dishes for 24 h. Then, cells were treated with T-A-Ls (50 µM, 100 µM) and cultured for 24 h with or without cisplatin (20µM). In this experiment, we used the cell peroxidase reagent 2,7 dichlorofluorescein diacetate (DCFDA). This reagent deacetylates into a non-fluorescent compound by cell esterase which is then oxidized into 2,7 dichlorofluorescein (DCF) by ROS. The intracellular levels of ROS level were determined by detecting the fluorescence intensity of DCF. The cells were fixed with paraformaldehyde and then washed three times with PBS. After blocking with glycerol, semi-quantitative analysis was carried out using images produced by an inverted fluorescence microscope and Image J software.
Determination of Mitochondrial Membrane Potential (MMP, Δψm)
HK-2 cells were cultured in 6‑well plates at a density of 1×105 cells/well for 24 h and then treated with different concentrations of T-A-Ls (0, 50, and 100 µM) for 24 h. Next, the cells were cultured for 24 h with or without cisplatin. The mitochondrial membrane potential was then detected with a JC-1 Kit (Beyotime Biotechnology); first, cells were washed twice in PBS and the medium replaced with serum-free medium. Then, the cells were stained with JC-1 dye at 37°C for 30 min in accordance with the manufacturer’s instructions.
Mitochondrial Respiration Function Measurement
To assess mitochondrial function, the XF96 extracellular flux analyzer (Seahorse Bioscience, Billerica, USA) was used to detect real-time changes in oxygen consumption rate (OCR). All agents were supplied by Agilent Seahorse XF (USA). In brief, HK-2 cells (5×104 cells per well) were seeded into culture plates and cultured overnight. Prior to measurement, cells were treated with various concentrations of T-A-Ls for 24 hours. After that, the cells were washed three times; next, we added 1mM glutamine solution, 1 mM pyruvate and 2 mM glutamine in DMEM base medium for washing and culture. We used the Seahorse XF Cell Mito Stress Test Kit (Seahorse, Billerica, USA) containing 2 µM Oligomycin, 1 µM FCCP, and 0.5 µM Rotenone Antimycin A as final concentrations for OCR analysis. The measurements were conducted in accordance with the manufacturer’s instructions and normalized to the number of cells stained with Hoechst 33258 (Solarbio, Beijing, China) in each well.
Mitochondrial Morphology Observation
A total of 2×106 cells were inoculated into 10 cm culture dishes, and treated with 50 µM and 100µM T-A-Ls, respectively. Cells were washed after 24 hours, scraped off with glutaraldehyde 2.5%, and centrifuged at 1000 rpm for 5 minutes. After clumps of precipitated cells were precipitated, they were fixed with 2.5% glutaraldehyde at room temperature for two hours. Next, the cells were transferred to 4°C, washed with PBS, mounted with 1% osmic acid for 2 hours, dehydrated in ethanol and acetone, and embedded in Epon812. After polymerization, the samples were cut into thin sections with an ultrathin slicing machine (Leica UC7), counterstained with 2% oil and citric lead acetate, and examined under transmission electron microscopy (Hitachi, HT7800).
In vivo Biodistribution and Preliminary the Concentration of T-A-Ls in Kidney
The biodistribution of T-A-Ls in AKI model mice was investigated using fluorescence imaging. First, 8-week-old male AKI mice were injected each day for 3 days with DIR-T-A-Ls at a dose of 30 mg/kg; the same dose was administered to healthy C57BL/6J mice (the normal control group). Five mice were euthanized under anesthesia and their hearts, livers, spleens, lungs, and kidneys were collected. An IVIS Lumia III system (PerkinElmer, USA) was used for in vivo imaging and to observe the fluorescence intensity of T-A-Ls in kidney tissue. Next, a suitable amount of physiologic saline was added to the kidney tissues of each group and the tissues were placed in a water bath of ice to prepare a homogenate with a weight-to-volume ratio of 1:2 (g/mL). Subsequently, 400 μL kidney tissue homogenate had 4 times the amount of ethyl acetate added and was rotated for 2 min, stood for 5 min, and centrifuged at 3500 rpm for 10 min at 4°C. The supernatant was collected, dried with a rotary evaporator at RT, and then 200 μL methanol was added and the solution was centrifuged for 10 min at 4°C and 12,000 rpm. To evaluate the T-A-Ls levels, ultra-high-performance liquid chromatography mass spectrometry (UPLC-MS) was used with the following conditions: mobile phase: 0.1% formic acid aqueous solution (A)-0.1% formic acid acetonitrile (B); elution procedure: equivalent elution, 55% B, 4 min. The flow rate was 0.3 mL/min, the column temperature was 40°C, and the injection volume was 5 μL. Mass spectrometry analysis was performed in the positive ion mode (curtain gas: 30.0 psi; collision gas: 9 psi; ionspray voltage: 5500 V; temperature: 500°C; ion source gas 1:50 psi; ion source gas 2:50 psi).
Statistical Analysis
Data were statistically analyzed with GraphPad Prism 8.0.1 software (San Diego, CA, USA). The Student’s t-test was used to compare data between two groups while one-way analysis of variance (ANOVA) was used to compare multiple groups. A p value <0.05 was considered statistically significant.
Results
Preparation and Characterization of T-A-Ls
T-A-Ls were translucent appearance with a particle size of 81.13±2.18 nm, polydispersity index (PDI) of 0.264±0.008, zeta potential of −17.23±0.64 mV, entrapment efficiency (EE) of 88.07±0.16% and drug-loading rate of 0.30±0.00%, TEM shows that T-A-Ls were spherical in shape (Figures 1A–C and 2). In addition, the particle size, PDI, zeta potential, EE and drug-loading rate of A-Ls were 97.10±0.56 nm, 0.330±0.002 and −26.5±2.67 mV, 88.72±0.22% and 0.31±0.00%, respectively.
 |
Figure 1 Characteristics of T-A-Ls. (A) The appearance of T-A-Ls; (B) The particle size distribution curve of T-A-Ls; (C) TEM of T-A-Ls; (D) Release of ATS from T-A-Ls (n=3); (E and F) The stability of T-A-Ls for 6 weeks (n=3). |
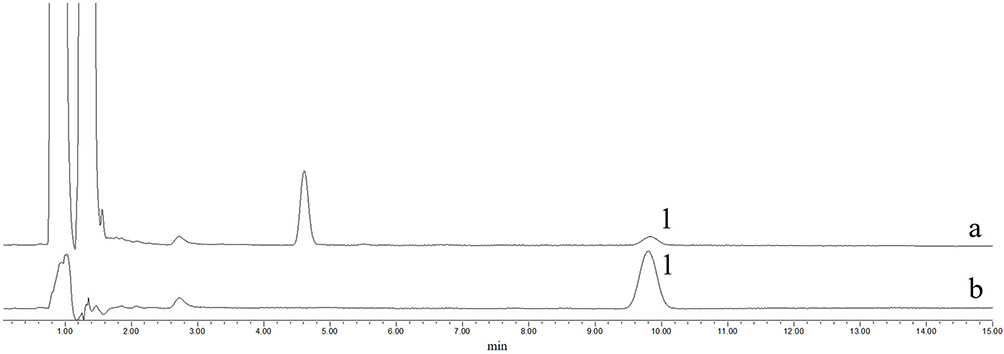 |
Figure 2 The HPLC chromatograms for encapsulation efficiency of T-A-Ls. a. Chromatogram of ultrafiltration solution. b. chromatogram of reference substance. 1. The chromatographic peak of ATS. |
The release experiment (Figure 1D) of T-A-Ls results showed that after 6 hours of drug release experiment, the accumulated release percentage of ATS reached 88.78% and the release rate of the drug rapidly decreased after 6 hours. During the 0–6 hours of the drug release experiment, the drug release was consistent with the law of Higuchi kinetics equation and the release rate of the ATS within 0–6 hours was 2.8366 mg·h−1/2.
The stability study showed that the relative standard deviation (RSD) of particle size, PDI, and EE of T-A-Ls was 1.91%, 1.88%, and 0.39% at different sampling points, respectively. Furthermore, T-A-Ls always presented as a translucent liquid with light blue opalescence during the stability study. These results showed that T-A-Ls had good stability at 4°C for 6 weeks (Figure 1E and F).
T-A-Ls Did Not Exert Toxic Effects on Key Organs
After the administration of T-A-Ls, pathological examinations were performed on the key organs (heart, liver, spleen, lung, and kidneys). The heart, liver, spleen, lung, and kidney tissues of mice treated with the T-A-Ls were similar to those of mice in the normal saline treatment group and no pathological changes were observed (Figure 3A). With regards to the heart, the morphology and structure of cardiomyocytes in the normal control and each administration group were normal, the myocardial fiber cells were arranged in an orderly manner, the transverse lines were clear, the cell mem-brane was complete, and staining was uniform. The appearance of the liver in different administration group was similar to those of the normal control group, which implied that T-A-Ls and A-Ls did not aggravate the liver. For the spleen, the junction between the spleen cortex and medulla was present in all groups, along with a dense distribution of lymphocytes in the white and red pulps of the spleen. With regards to the lungs, there were no signs of edema in the surrounding tissues, and the alveolar structure was complete and clear. For the kidneys, in both the normal control and each administration group, the epithelial cells of the renal tubules were neatly arranged and the renal capsule cavity and glomerular structure were clear and uniform. Collectively, these results demonstrated that the administration of T-A-Ls did not exert toxicity in any of the key organs tested.
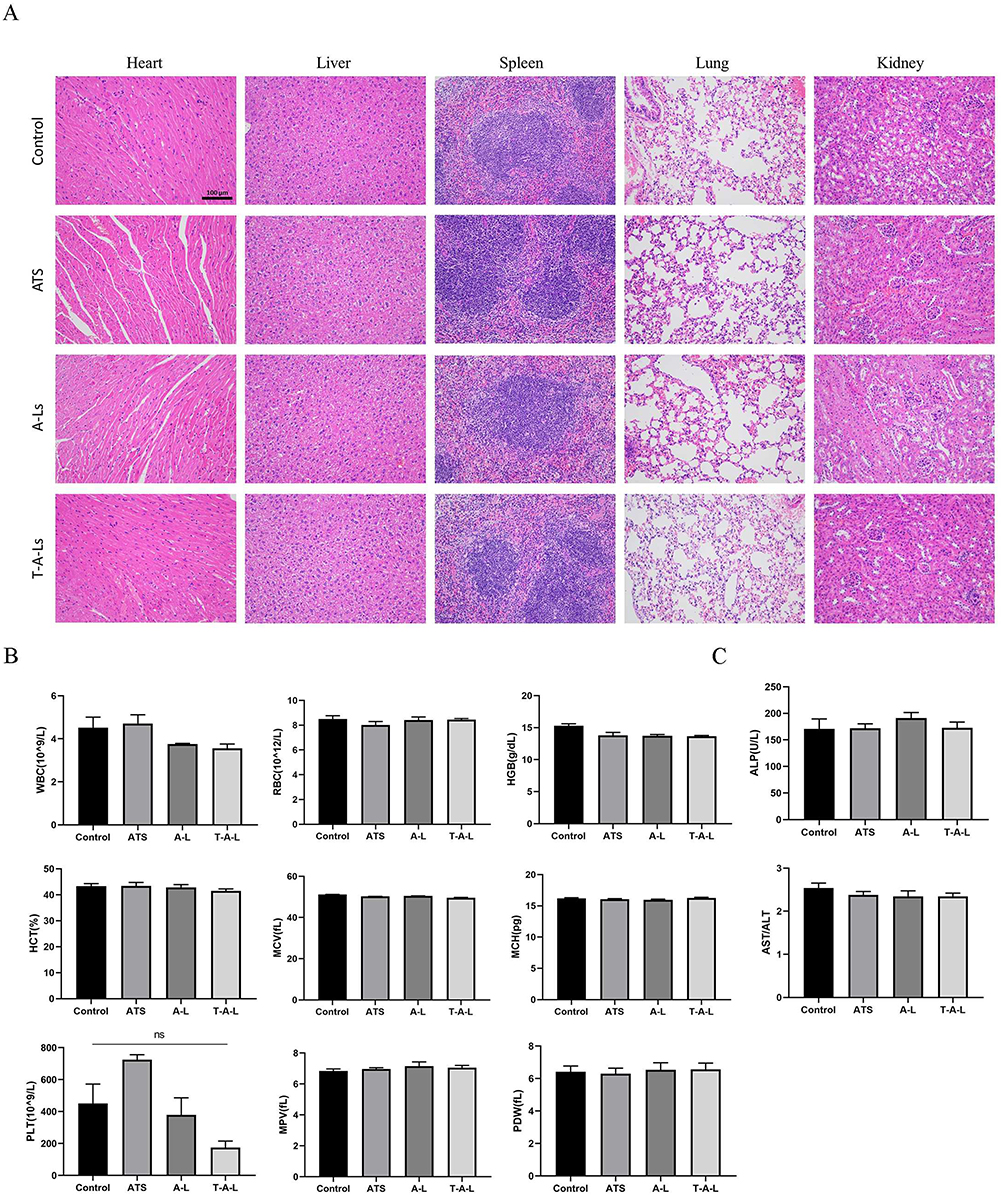 |
Figure 3 In vivo safety evaluation of the T-A-Ls self-assembly system. (A) H&E staining of major organs. Scale bar: 100 μm; (B) Blood routine analysis on day 5 post-injection; (C) Liver function examination. (n = 6). Abbreviations: ns, no significant; WBC, White blood cells; RBC, Red blood cells; HGB, Hemoglobin; HCT, Hematocrit; MCV, Mean corpuscular volume; MCH, Mean corpuscular hemoglobin; PLT, Platelets; MPV, Mean platelet volume; PDW, Platelet distribution width; ALP, Alkaline phosphatase; AST, Aspartate transaminase; ALT, Alanine transaminase. |
Next, the in vivo toxicology and potential side effects of T-A-Ls treatment were systematically investigated using a range of standard hematological markers, including WBC, RBC, PLT, HGB, HCT, MCH, MCV, MPV, and PDW (Figure 3B). There were no significant differences between the normal controls and the treatment groups with respect to any of these parameters (p > 0.05). These results demonstrated that the T-A-Ls treatments did not cause significant injury or inflammation in the experimental mice. Blood biochemical analyses, including AST/ALT and ALP, were also conducted to investigate the liver function in the experimental mice (Figure 3C). There were no significant differences between the normal controls and the treatment groups with respect to any of these parameters (p > 0.05). These results demonstrated that T-A-Ls treatment did not have any significant effects on the blood chemistry of experimental mice and did not induce hepatic toxicity.
T-A-Ls Treatment Abolished CDDP-Evoked AKI
Next, the therapeutic efficacy of T-A-Ls, A-Ls, and ATS in CDDP-induced AKI in five groups of C57BL/6J mice was systemically evaluated; the experimental design is depicted in Figure 4A. Figure 4B shows the relative changes in renal morphology. Compared with the control group (no CDDP), H&E staining and Masson’s trichrome staining of the kidney tissues after the administration of CDDP revealed significant differences with regards to cast formation, tubular necrosis, erosion of the epithelium. These changes were accompanied by significant increases in kidney index (kidney weight to body weight, KI), tubular injury score, renal interstitial fibrosis index, BUN, and Scr (p < 0.01, p < 0.001 or p < 0.0001) (Figure 3C–H). These findings showed that the model of CDDP-induced AKI had been successfully established. After treatment with T-A-Ls, A-Ls, and ATS (30 mg/kg), the pathological condition of the renal tissue was significantly improved (Figure 4B and D), and the KI (Figure 4C), tubular injury score (Figure 4E), renal interstitial fibrosis index (Figure 4F), BUN (Figure 4G), and Scr (Figure 4H) were all downregulated when compared with those of the CDDP group (p < 0.05, p < 0.01, p < 0.001 or p < 0.0001). T-A-Ls exhibited the most significant improvements in these indicators and exhibited better efficacy compared with the other treatments.
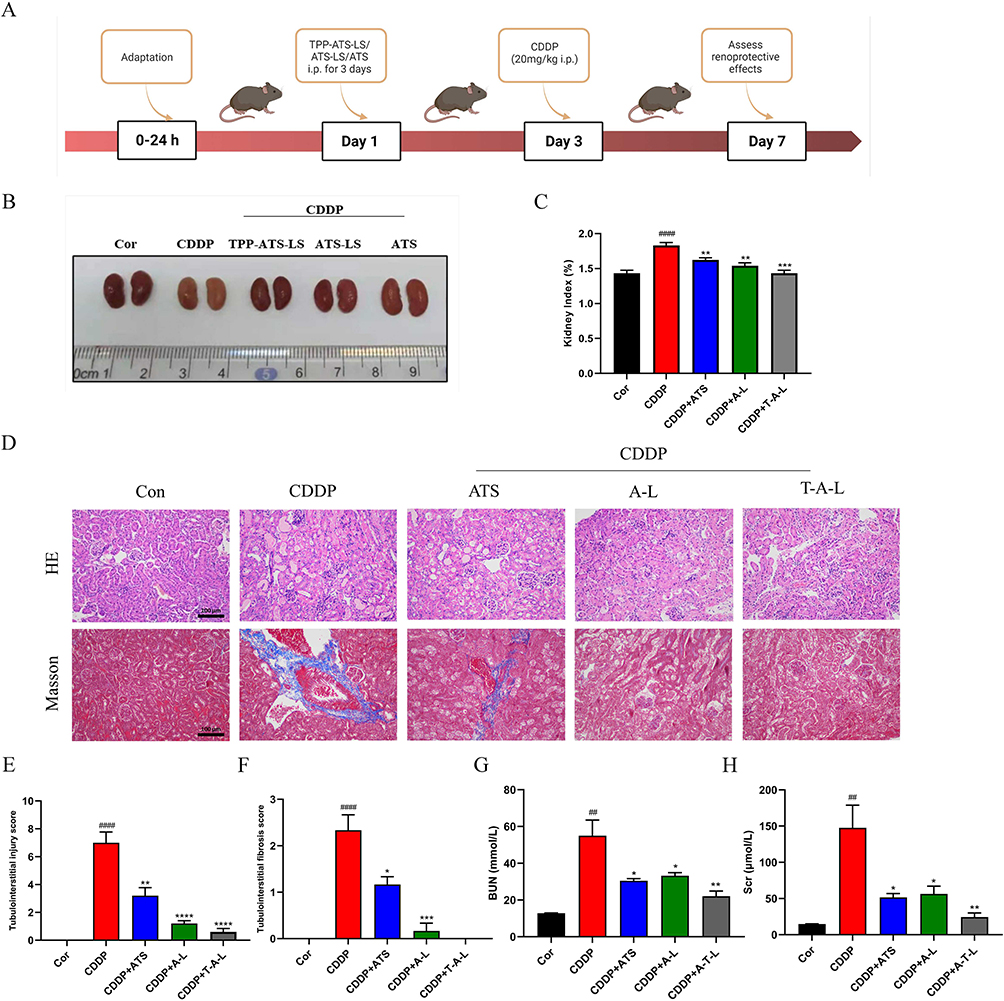 |
Figure 4 Protective effect of T-A-Ls on CDDP-induced AKI. (A) Outline of the experiments; (B) Representative morphology photos; (C) Kidney index; (D) H&E and Masson’s trichrome staining. Scale bar: 100 μm; (E) Tubulointerstitial injury score; (F) Tubulointerstitial fibrosis index; (G) BUN; (H) Scr. Compared with control, ##p < 0.01, ###p < 0.001, ####p < 0.0001; compared with CDDP, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; compared with CDDP+ATS, *p < 0.05, **p < 0.01, ****p < 0.0001; CDDP+A-L vs CDDP+ T-A-L, *p < 0.05, **p < 0.01 (n = 5). Abbreviations: BUN, Blood urea nitrogen; Scr, Serum creatinine. |
T-A-Ls Mitigated the Expression of Inflammatory Cytokines in CDDP-Induced AKI and Modulated the RAGE/iNOS Signaling Pathway
The inflammatory response is instrumental in AKI.45,46 To further verify the mechanism by which T-A-Ls could improve AKI, the expression of key inflammatory factors was examined in the serum of mice in each group. As shown in Figure 5A–C, the expression levels of TNF-α, IL-6, and IL-1β in the serum were significantly upregulated when exposed to CDDP (p < 0.01 or p < 0.001). However, the administration of T-A-Ls inhibited the upregulation of key inflammatory factors induced by CDDP (p < 0.05, p < 0.01 or p < 0.001). IF staining and Western blotting further revealed that T-A-Ls (15 mg/kg, 30 mg/kg) significantly suppressed the levels of RAGE and iNOS that were induced by CDDP exposure (p < 0.05, p < 0.01, or p < 0.0001) (Figure 5D–K); these effects were dose-dependent.
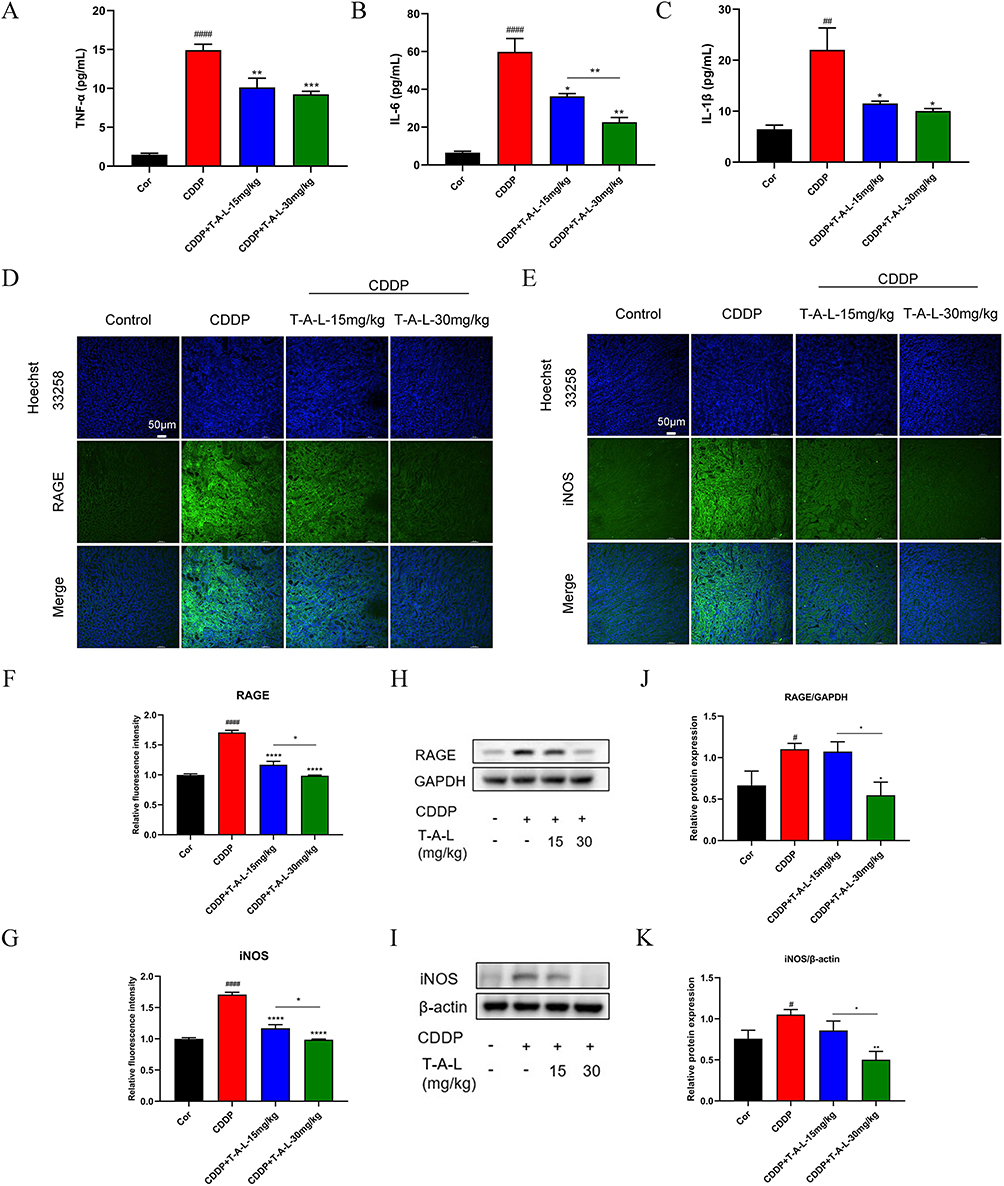 |
Figure 5 T-A-Ls mitigated inflammation in CDDP-induced AKI. (A–C) Detection of inflammatory factors in serum by ELISA; (D and E) IF showing RAGE and iNOS expression in renal tissues. Scale bar: 50 μm; (F and G) relative intensity of IF depicted by column chart (n = 5); (H and I) Western blot images of RAGE and iNOS expression levels; (J and K) quantitative analysis of scanning densitometry of relative protein from Western blot analysis (n = 5). Compared with control, #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001; compared with CDDP, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; CDDP+T-AL-L-15 mg/kg vs CDDP+T-AL-L-30 mg/kg, *p < 0.05, **p < 0.01. |
T-A-Ls Attenuated Oxidative Stress in CDDP-Induced AKI and Activated the Nrf2/HO-1 Signaling Pathway
In recent years, oxidative stress and nitrosate stress have been revealed as critical drivers of cisplatin-induced nephrotoxicity.47 Oxidative stress is inextricably linked with mitochondrial dysfunction and the production of ROS; therefore, markers of these processes were examined in kidney tissues from experimental mice. IF staining showed that lipid peroxidation levels of 4-hydroxynonenal (4-HNE) in tissues from the CDDP group were significantly elevated when compared with those of the normal group (p < 0.01), while T-A-Ls pre-treatment (15 mg/kg, 30 mg/kg) significantly suppressed these changes (p < 0.01 and p < 0.01) (Figure 6A and B). Compared with the normal group, the levels of SOD (Figure 6D) and GSH (Figure 6E) were significantly downregulated, and the levels of MDA (Figure 6C) were significantly upregulated, following exposure to CDDP (20 mg/kg) (p < 0.0001). However, these changes were abolished following T-A-Ls pre-treatment (15 mg/kg, 30 mg/kg).
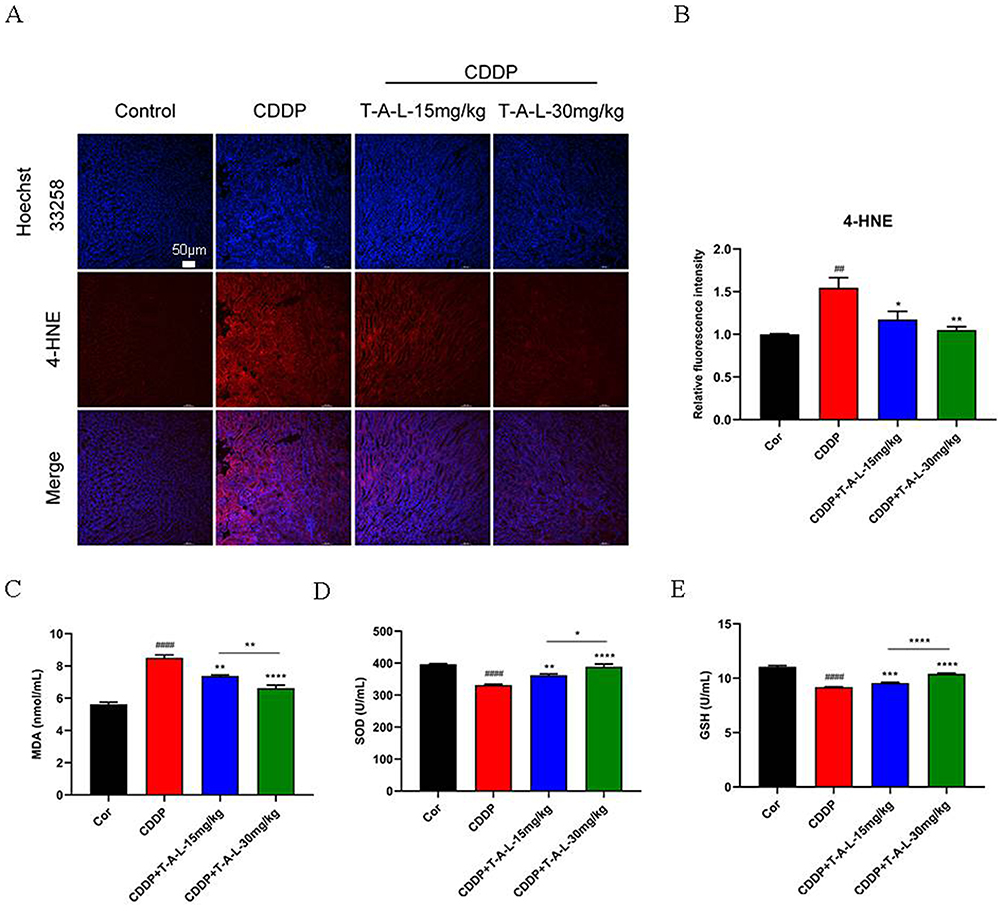 |
Figure 6 T-A-Ls pre-treatment protected against CDDP-induced renal oxidative stress. (A) IF staining of 4-HNE in kidney tissues. Scale bar: 50 μm; (B) relative fluorescence intensity of 4-HNE was analyzed by ImageJ and depicted in a column chart. (C) MDA, (D) SOD, and (E) GSH in renal tissues were detected by ELISA (n = 5). Compared with control, ##p < 0.01, ####p < 0.0001; compared with CDDP, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; CDDP+T-AL-L-15 mg/kg vs CDDP+T-AL-L-30 mg/kg, *p < 0.05, **p < 0.01 and ****p < 0.0001. Abbreviations: MDA, Malondialdehyde; SOD, Superoxide Dismutase; GSH, Glutathione. |
The levels of nuclear factor erythroid 2-related factor 2 (Nrf2) and the Phase II antioxidant enzyme heme oxygenase-1 (HO-1) in the kidney tissues were significantly lower in the CDDP group when compared with those of the normal group. Immunohistochemical (Figure 7A–C) and WB (Figure 7D–G) results both indicate a decrease in the content of these two proteins in CDDP-induced AKI mice (p < 0.05 or p < 0.01 or p < 0.001); T-A-Ls pre-treatment significantly reversed this tendency (p < 0.05 or p < 0.01 or p < 0.001). These results demonstrated that pre-treatment with T-A-Ls effectively inhibited CDDP-induced oxidative stress injury.
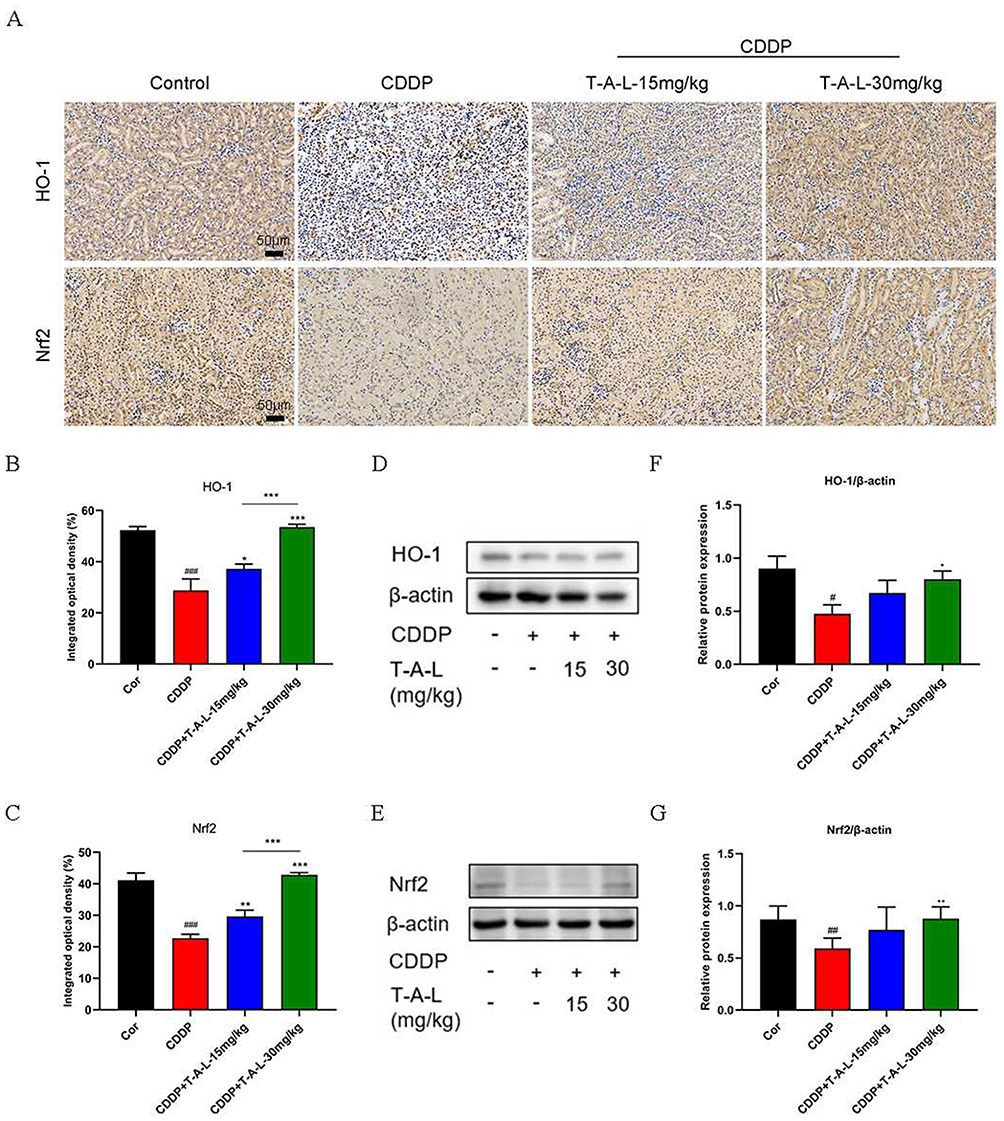 |
Figure 7 T-A-Ls alleviated CDDP-induced renal oxidative stress by regulating the expression of HO-1 and Nrf2 proteins. (A) IHC for expression levels of HO-1 and Nrf2, with (B and C) Statistical results of expression in (A) (n = 3). Scale bar: 50 μm; (D and E) The proteins from kidney tissues were detected by Western blot, with β-actin was used as the loading control (n = 5). (F and G) Quantitative analysis of scanning densitometry of relative protein from Western blot analysis. Compared with control, #p < 0.05, ##p < 0.01, ###p < 0.001; compared with CDDP, *p < 0.05, **p < 0.01, ***p < 0.001; CDDP+T-AL-L-15 mg/kg vs CDDP+T-AL-L-30 mg/kg, ***p < 0.001. |
In vitro Cytotoxicity Assays, Cellular Uptake and Subcellular Localization
Next, the CCK-8 method was used to detect the cytotoxicity of the nanoliposomes in HK-2 cells. After incubation with T-A-Ls for 24 h, there were no signs of toxicity in the HK-2 cells; even the high concentration (200 µM) of T-A-Ls showed good biocompatibility (Figure 8A).
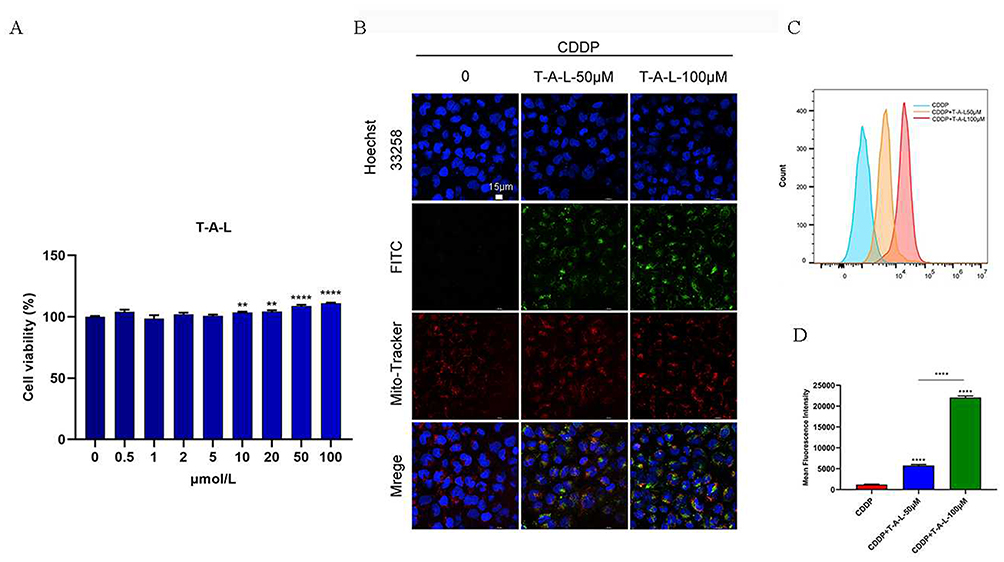 |
Figure 8 In vitro uptake of T-A-Ls by HK-2 cells. (A) CCK-8 was used to detect the cell viability of T-A-Ls on HK-2 cells for 24h. (B). Confocal images show intracellular localization of T-A-Ls with different concentrations after incubation for 24h. Scale bar: 15 μm; (C) Histogram profiles and (D) mean fluorescence intensity analyzed by flow cytometry after incubation with T-A-Ls (50 μM or 100 μM) for 24 h (n = 3). **p < 0.01, ****p < 0.0001. |
The cellar uptake of T-A-Ls in HK-2 cells was then explored. As shown in Figure 8B–D, the uptake of T-A-Ls by HK-2 cells was dose-dependent (Figure 8C and D) and co-localized with the mitochondria in CDDP induced HK-2 cells (Figure 8B). These results suggested that the T-A-Ls system could facilitate the entry of ATS into the mitochondria to improve drug treatment indices.
T-A-Ls Inhibited CDDP-Induced ROS and Elevated Δψm in HK-2 Cells
In this study, a DCFH-DA probe was used to analyze the levels of ROS in HK-2 cells induced by CDDP. Compared with the control group, the levels of ROS in HK-2 cells exposed to CDDP were significantly increased (p < 0.001); however, the administration of T-A-Ls (50 and 100 µM) significantly reduced the levels of ROS (p < 0.001). These results demonstrated that T-A-Ls reduced the generation of ROS induced by cisplatin in HK-2 cells (Figure 9A and B).
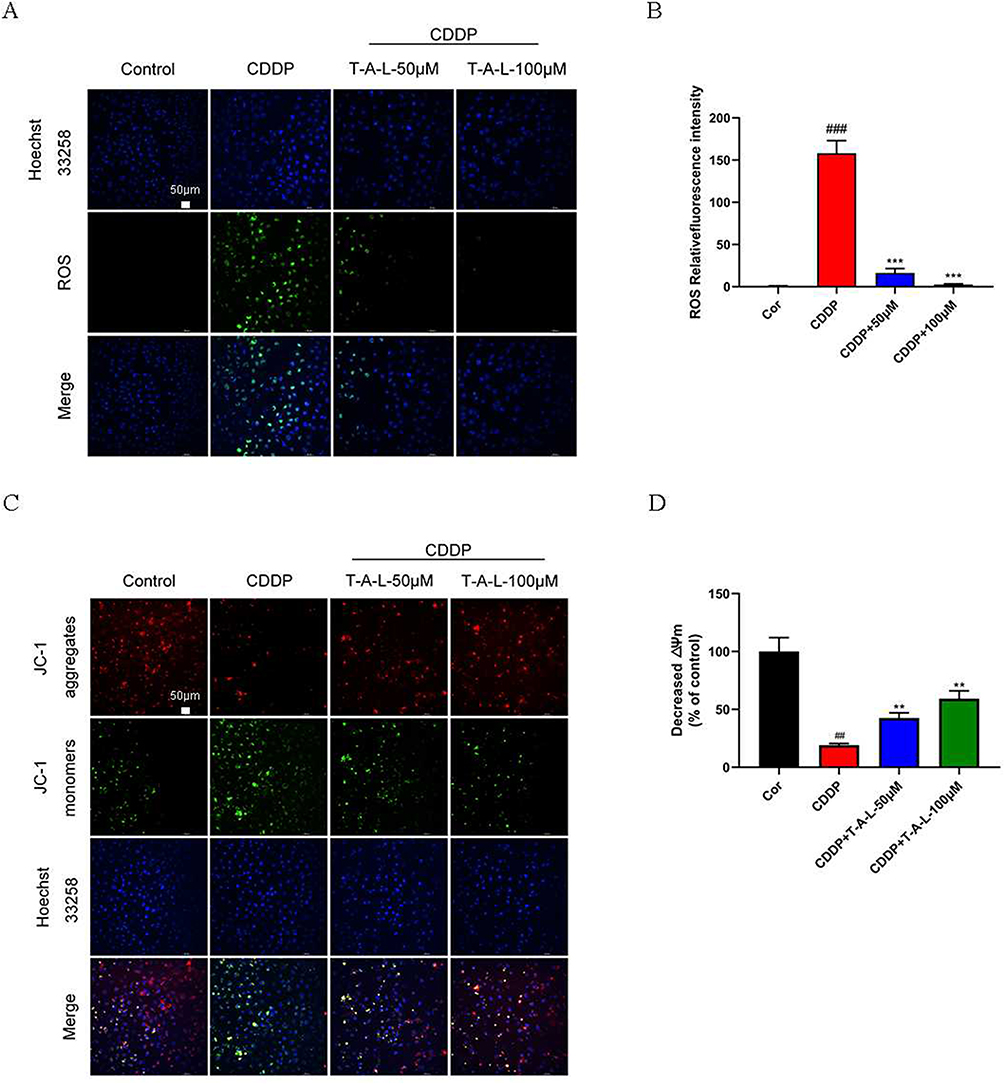 |
Figure 9 T-A-Ls limit the ROS increase induced by CDDP and improve mitochondrial membrane potential in vitro. (A and B) After 24 h of modeling with CDDP, ROS levels in HK-2 cells were measured using a DCFH-DA probe and confocal laser scanning microscopy. Scale bar: 50 μm; (C and D) JC-1 staining (n = 3). Scale bar: 50 μm. Data are shown as means ± SD. Compared with control, ##p < 0.01, ###p < 0.001; compared with CDDP, **p < 0.01, ***p < 0.001. |
During the process of respiratory oxidation, mitochondria can store the energy generated in the inner membrane of mitochondria as electrochemical potential energy, with the protons and other ion concentrations on both sides of the inner membrane forming an asymmetric distribution, thus resulting in a difference in transmembrane potential. This is known as the mitochondrial membrane potential (MMP; ΔΨ m) and reflects the integrity of mitochondrial function and represents a sensitive index with which to evaluate mitochondrial function. JC-1 is a widely used reagent for detecting ΔΨ m in the mitochondrial membrane and acts as a fluorescent probe. JC-1 has two forms (a monomeric form and a J-aggregate form) and accumulates in the mitochondria in a potential-dependent manner. In normal mitochondria, JC-1 accumulates in the mitochondrial matrix to form a polymer and emits strong red fluorescence (ex=585 nm, em=590 nm). When there is a reduction or loss of membrane potential in unhealthy mitochondria, JC-1 diffuses into the cytoplasm and can only exist in that location as a monomeric form that produces green fluorescence (ex=514 nm, em=529 nm). Thus, a reduction in cell membrane potential can be easily detected by the transformation of JC-1 from red fluorescence to green fluorescence. As shown in Figure 9C and D, the ΔΨ m in HK-2 cells with cisplatin-induced damage was lower than that in normal control cells (p < 0.01); the administration of T-A-Ls reversed the ΔΨ m induced by cisplatin in HK-2 cells (p < 0.01).
T-A-Ls Mitigated Mitochondrial Respiration and Mitochondrial Injury in CDDP-Induced AKI
To investigate the specific metabolic activity alterations induced by T-A-Ls, the oxygen consumption rate (OCR) was examined with a Seahorse XF96 extracellular flux analyzer and a Cell Mito Stress Test Kit. As shown in Figure 10A, T-A-Ls ameliorated the abnormal mitochondrial respiration of HK-2 cells caused by CDDP. Furthermore, transmission electron microscopy showed that there was no significant change in the mitochondrial structure of normal control HK-2 cells. However, in the CDDP-induced HK-2 cells, there were significant changes in mitochondrial morphology and structure, including increased membrane density, decreased mitochondrial volume, and decreased or missing mitochondrial cristae. After 24 h of intervention with T-A-Ls, the mitochondrial morphology improved significantly in a dose-dependent manner (Figure 10B). The electron microscopy observations are summarized below.
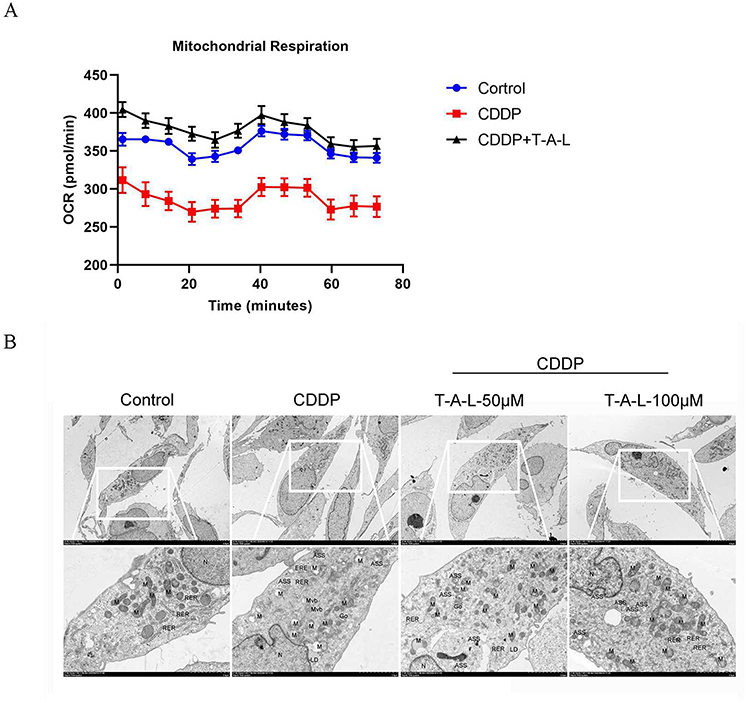 |
Figure 10 T-A-Ls attenuate CDDP‐induced mitochondrial injury and mitochondrial energy metabolism in vitro. (A) OCR levels were quantified in HK-2 cells (n = 6). (B) Micrographs of mitochondria in HK-2 cells taken by TEM. Abbreviations: OCR, Oxygen consumption rate; N, Cell nucleus; M, Mitochondria; RER, Rough endoplasmic reticulum; Go, Golgi apparatus; LD, Lipid droplets; Mvb, Multivesicular bodies; ASS, Autophagic lysosomes. |
Normal group: HK-2 cells had relatively normal morphology, no obvious swelling of mitochondria, normal structure, intact membrane, and parallel cristae.
AKI model group: cellular edema, most cellular organelles were visibly swollen, some appeared vacuolated. The cell nucleus (N) was irregular, with intact nuclear membrane. The mitochondria (M) were abundant and obviously swollen, some were slightly larger, with a reduced or missing cristae structure, and there was vacuolization in some serious cases. The rough endoplasmic reticulum (RER) was significantly shortened and the membrane was damaged. Golgi apparatus (Go) has damaged membrane and incomplete structure. Lipid droplets (LD) existed individually, and there were large number of multivesicular bodies (Mvb). A small amount of autophagic lysosomes (ASS) were present-three were visible in the field shown.
T-A-Ls low-dose group: HK-2 cells showed mild edema, with intact cell membranes and abundant organelles and slightly swollen. The nucleus appears irregular in shape, chromatin was uniform, and the nuclear membrane was intact, the perinuclear space was acceptable. The mitochondria were abundant, slightly swollen, with some cristae rupture. The rough endoplasmic reticulum was locally expanded, and the Golgi apparatus exhibited mild hypertrophy with slight membrane expansion. There was a small amount of lipid droplets and an abundance of autophagy lysosomes.
T-A-Ls high-dose group: The cells show slight edema, intact cell membrane, homogeneous cytoplasm, abundant organelles, and some organelles are mildly swollen. The nucleus has an irregular shape, uniform chromatin, intact nuclear membrane, abundant mitochondria, most of which are structurally intact, with intact membranes and some parallel cristae. Most of the rough endoplasmic reticulum structures were acceptable. The Golgi apparatus does not show obvious hyperplasia or hypertrophy. Autophagosomes are present in large amounts.
In vivo Biodistribution and the Concentration in Kidney
Finally, the distribution of T-A-Ls was observed in vivo using an IVIS Lumia III imaging system. Representative fluorescence images of the heart, liver, spleen, lung, and kidney were obtained in AKI or healthy mice treated with DiR-T-A-Ls (Figure 11A), and the total radiant efficiency was depicted in a column chart (Figure 11B). The content of DiR-T-A-Ls in the kidney tissue of AKI mice was significantly increased compared with that of healthy kidneys 2 h after tail vein injection (p < 0.05) (Figure 11C and D). UPLC-MS analyses showed that the concentration of ATS in the kidney tissue of AKI mice was significantly higher than that in normal mice (p < 0.0001) (Figure 11E and F). Based on these findings, we conclude that T-A-Ls could enhance the absorption of ATS and the accumulation of this drug in AKI kidneys after acute CDDP injury. Interestingly, while the ATS concentration in the kidneys of AKI mice was significantly higher than that in the healthy animals, the concentration of ATS in the heart, liver, spleen, and lung tissue samples of the AKI model mice was not increased compared with the control animals. These results demonstrate that T-A-Ls have good potential for treating CDDP-induced AKI.
 |
Figure 11 In vivo biodistribution of T-A-Ls and the concentration of ATS in the kidneys. (A) In vivo representative fluorescence imaging of T-A-Ls, and (B) histogram statistical analysis. Data are presented as mean ± SD (n = 5). (C) Biodistribution fluorescence images of the kidney of AKI or healthy mice treated with T-A-Ls for 2 h (n = 6), and (D) histogram depicting statistical analysis of the total radiant efficiency in the kidney of AKI or healthy mice. (E) Content of T-A-Ls in the kidney at 2 h, as measured by UPLC-MS, and (F) histogram profiles of the distribution. *p < 0.05, ****p < 0.0001. |
Discussion
Oxidative stress and inflammation are instrumental in the occurrence and development of AKI.48–50 Oxidative stress increases the production of inflammatory cytokines, which then stimulate the production of ROS.51 Previous animal studies have shown that taking various natural antioxidants can reduce the nephrotoxicity of cisplatin.52,53 However, considering the limitations of traditional antioxidants and that mitochondria are the main source of ROS that cause oxidative damage, the development of mitochondrial-targeted antioxidants to selectively block mitochondrial oxidative damage may be a necessary means to inhibit the occurrence of AKI. Recently, it was reported that ATS-A key antimalarial compound purified from Artemisia annua L.-could exert certain renal protective effects in lupus-prone MRL/lpr mice via its immunosuppressive effects.54 In addition, studies have shown that ATS not only has antimalarial and anti-inflammatory effects but also exhibits antioxidant effects that can reduce the damage incurred by renal cells by inhibiting inflammation and reducing ROS.55,56 However, the specific effects of ATS on AKI have yet to be elucidated.
Liposomes are one of the most successful nanoparticles used in the field of medicine.57 The continuous development of biotechnology and nanomaterials over recent years has led to an increase in the number of applications for nanodrugs entering late clinical trials.58 Compared with single prototype drugs, liposomes, as a nano-biomaterial, enhance the water solubility of drugs, prolong the activity of drugs, and increase drug accumulation in target tissues, thus enhancing drug efficacy.59 However, traditional liposomes often show poor targeting effects, which impacts their therapeutic effects.
At present, ATS is predominantly administered as tablets and injections in clinical practice. However, the use of ATS in these forms is limited by low bioavailability, a short half-life, and poor patient compliance, thus placing serious restrictions on efficacy.60 To further improve the renal protective effects of ATS, we prepared ATS anti-AKI liposomes that target damaged HK-2 cells and intracellular mitochondria based on TPP. In this study, we described the synthesis and characterization of these nano-particles, termed T-A-Ls. We also used cisplatin-induced AKI mice as a model for pharmacodynamic evaluation to investigate the therapeutic effects of T-A-Ls on AKI and identify the mechanisms involved.
The mean size of the ATS liposomes prepared by the thin-film dispersion method was 81.13±2.18 nm. Previous research showed that liposomes of 50–100 nm exhibited enhanced enrichment and penetration at abnormal tissue sites owing to their enhanced permeability and the retention effect of the abnormal tissue.61,62 The zeta potential of the ATS liposomes was 9.86±0.76 mV and the PDI was 0.264±0.008; these characteristics indicated that the new ATS liposomes had a uniform particle size and a good dispersion performance. The stability of liposomes can be evaluated by analyzing temperature changes during phase transition. In vitro stability tests showed that our T-A-Ls could be stored at 4°C for 6 weeks and that the encapsulation efficiency, particle size, PDI, and zeta characterization of the liposomes were consistent with previous test results, thus indicating that the T-A-Ls had good stability.
To investigate how T-A-Ls play a protective role in the kidneys, a cisplatin-induced mouse model of AKI was established for in vivo analyses, and cisplatin was also used to cause damage in renal tubule epithelial cells in vitro. The effects of T-A-Ls on cisplatin-induced nephrotoxicity were studied from two aspects: inflammation and oxidative stress. The acute renal injury induced by cisplatin is closely related to the occurrence of inflammatory reactions. In mice experiencing cisplatin-induced renal injury, numerous studies have identified significant increases in the serum levels of TNF-α, IL-1β, IL-6, and other proinflammatory factors.63–66 Previous studies demonstrated that inhibition or knockout of TNF-α significantly reduced the renal injury caused by cisplatin.67 Congruent with these studies, our research showed that IL-1β, IL-6, and TNF-α were significantly increased in AKI when induced by cisplatin. However, following the administration of T-A-Ls, these indices all decreased in a dose-dependent manner. Advanced glycation end-products (AGEs) are irreversible products of the amino non-enzymatic reduction of sugars and proteins or peptides, which are metabolized and excreted by the kidneys. However, if not metabolized, AGEs can be deposited in the kidneys and bind to the AGE receptor (RAGE), thus causing pathological changes, including inflammation and oxidative stress.68 RAGE over-expression increases the levels of iNOS,69 thus promoting the formation of NO–, which, in turn, reacts with O2– to produce ONOO–, a molecule with strong oxidation and nitrification characteristics that can cause further cellular damage. Here, IF and Western blotting were used to detect the activation of RAGE and iNOS in the cisplatin-induced AKI model. T-A-Ls significantly inhibited the expression of RAGE and iNOS proteins in the kidney tissue of cisplatin-induced AKI mice, inhibited the inflammatory response, and reduced renal injury. In addition, after cisplatin modeling, the content of MDA in the renal tissue of mice increased significantly while the levels of GSH and SOD activity decreased significantly; these changes were consistent with those reported in the literature.70,71 Following the administration of T-A-Ls, MDA decreased in a dose-dependent manner, and the levels of GSH and SOD activity began to increase. 4-HNE, as a reliable biomarker for the degree of lipid peroxidation, can reflect the status of free radical reactions.72 In this study, IF assays revealed that 4-HNE levels were higher in the CDDP group compared with the levels in the normal group; however, this increase was significantly mitigated when the mice were treated with T-A-Ls. To further verify the effect of T-A-Ls on proteins related to oxidative stress in cisplatin-induced AKI, the expression of Nrf2 and HO-1 in the kidneys of mice in each group were detected by IHC and Western blotting.
Nrf2 is a transcription factor that plays a key role in the regulation of antioxidant processes and controls the transcription of antioxidant genes through the antioxidant response element (ARE) sequence, Keap1. Therefore, Nrf2 activation is regarded as a critical mechanism for antioxidants to exert their cytoprotective effects.73 HO-1 is a cytoprotective protein that is activated by Nrf2 and can prevent apoptosis and oxidative stress under various pathological conditions. Numerous studies have shown that increased expression of HO-1 can reduce the damage caused by AKI.74,75 In this study, Nrf2 and HO-1 protein levels decreased significantly in cisplatin-induced AKI mice; however, following T-A-Ls intervention, the expression levels of Nrf2 and HO-1 were significantly increased in a dose-dependent manner. Finally, investigation of ROS, MMP, and mitochondrial energy metabolism in vitro showed that the T-A-Ls effectively reduced the levels of ROS in HK-2 cells induced by cisplatin, increased the MMP, and restored the abnormal mitochondrial energy metabolism. Furthermore, electron microscopy revealed that the morphology of the mitochondria in HK-2 cells in the model group was almost normal after 24 h of T-A-Ls treatment, because the kidney is a highly metabolic organ, there are large number of ATP and mitochondria in the kidney, and mitochondria play an important role in the process of kidney injury.76 These results were consistent with our concept of ATS liposomes based on TPP mitochondrial targeting.
Based on our research, intervention against mitochondrial damage and oxidative stress has great potential for the targeted therapy of AKI in the future. The findings of this study provide a potential new treatment for cisplatin-induced kidney damage in mitochondria.
Conclusion
We successfully constructed a mitochondrial-targeted ATS liposome system that named T-A-Ls. ATS alone is inefficient for the treatment of AKI owing to the lack of a specific targeting ability, hence the need to develop a targeted delivery system. The T-A-Ls significantly enhanced the inhibitory effect of ATS on the development of AKI, thus indicating their therapeutic potential for CDDP-induced renal injury. Moreover, the T-A-Ls had excellent targeting characteristics for the mitochondria and damaged kidneys. Our study verified the renal protective effects of T-A-Ls on AKI, both in vivo and in vitro. Furthermore, the T-A-Ls had very few side effects with regards to hematology, biochemistry, and histology. Preliminary investigations of the anti-AKI mechanism showed that the T-A-Ls resisted renal injury by inhibiting oxidative stress and inflammation via upregulation of Nrf2 and HO-1 and downregulation of RAGE and iNOS. Future in-depth studies of renal protection will explore and improve the efficacy of ATS. The findings from this study provide a new research strategy for the development of new renal protective drugs based on ATS.
Ethics Approval and Informed Consent
All animal experiments received ethical approval from the Laboratory Animal Ethics Committee of Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences (2022B030). The experiments were conducted in strict accordance with the Guidelines for Ethical Review of Laboratory Animal Welfare in China (GB/ T35892-2018).
Acknowledgments
The authors thank Ms. Guihua Yu in the Institute of Chinese Materia Medica, China Academy of Chinese Medical Sciences for her technical support. The work was supported by National Natural Science Foundation of China (Grant No. 81873332); Guang Dong Basic and Applied Basic Research Foundation (Grant No. 2021A1515110381); Fundamental Research Funds for the Central public welfare research institutes (Grant No. ZZ13-YQ-103; ZZ16-ND-10-11).
Author Contributions
All authors made significant contributions to the design and conception of the study, as well as the acquisition, execution, analysis, and interpretation of the data. Additionally, they participated in drafting, revising, or critically reviewing the manuscript and gave final approval for the version to be published. They also have agreed on the journal to which the article has been submitted, and agreed on all versions of the article before submission, during revision, the final version accepted for publication. All authors reached an agreement to take responsibility for all aspects of the article, ensuring that any questions regarding the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Disclosure
None of the authors have any conflicts of interest to disclose.
References
1. Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380(9843):756–766. doi:10.1016/s0140-6736(11)61454-2
2. Al-Jaghbeer M, Dealmeida D, Bilderback A, Ambrosino R, Kellum JA. Clinical decision support for in-hospital AKI. J Am Soc Nephrol. 2018;29(2):654–660. doi:10.1681/asn.2017070765
3. Hoste EA, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423. doi:10.1007/s00134-015-3934-7
4. Li HD, Meng XM, Huang C, Zhang L, Lv XW, Li J. Application of herbal traditional Chinese medicine in the treatment of acute kidney injury. Front Pharmacol. 2019;10:376. doi:10.3389/fphar.2019.00376
5. Kellum JA, Chawla LS, Keener C, et al. The effects of alternative resuscitation strategies on acute kidney injury in patients with septic shock. Am J Respir Crit Care Med. 2016;193(3):281–287. doi:10.1164/rccm.201505-0995OC
6. Linkermann A. Nonapoptotic cell death in acute kidney injury and transplantation. Kidney Int. 2016;89(1):46–57. doi:10.1016/j.kint.2015.10.008
7. Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. 2008;3(3):844–861. doi:10.2215/cjn.05191107
8. Wang W, Chen J, Hu D, et al. SARS-CoV-2 N protein induces acute kidney injury via Smad3-dependent G1 cell cycle arrest mechanism. Adv Sci. 2022;9(3):e2103248. doi:10.1002/advs.202103248
9. Aloy B, Janus N, Isnard-Bagnis C, Deray G, Launay-Vacher V. Renal toxicity of anticancer drugs. Nephrol Ther. 2021;17(7):553–563. doi:10.1016/j.nephro.2021.09.001
10. Liu S, Zhao J, Wang F. Acute kidney injury in cancer patients. Clin Exp Nephrol. 2022;26(2):103–112. doi:10.1007/s10157-021-02131-7
11. Rosner MH, Perazella MA, Ingelfinger JR. Acute kidney injury in patients with cancer. N Engl J Med. 2017;376(18):1770–1781. doi:10.1056/NEJMra1613984
12. Ghosh S. Cisplatin: the first metal based anticancer drug. Bioorg Chem. 2019;88:102925. doi:10.1016/j.bioorg.2019.102925
13. Ojha S, Venkataraman B, Kurdi A, Mahgoub E, Sadek B, Rajesh M. Plant-derived agents for counteracting cisplatin-induced nephrotoxicity. Oxid Med Cell Longev. 2016;2016:4320374. doi:10.1155/2016/4320374
14. Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 2008;73(9):994–1007. doi:10.1038/sj.ki.5002786
15. Fang CY, Lou DY, Zhou LQ . Natural products: potential treatments for cisplatin-induced nephrotoxicity. Acta Pharmacol Sin. 2021;42(12):1951–1969. doi:10.1038/s41401-021-00620-9
16. Li J, Wu Y, Chen C, Zhang, W, Yue, L, Liu, T, et al. A systematic review for prevention of cisplatin-induced nephrotoxicity using different hydration protocols and meta-analysis for magnesium hydrate supplementation. Clin Exp Nephrol. 2024;28(1):1–12. doi:10.1007/s10157-023-02386-2
17. Fukushima K, Futatsugi A, Maekawa M, Naito S, Okada A, Sugioka N. Comparison of cisplatin-induced nephrotoxicity between single-dose and split-dose administration to rats. Biomed Pharmacother. 2022;147:112619. doi:10.1016/j.biopha.2022.112619
18. Darwish MA, Abo-Youssef AM, Khalaf MM, Abo-Saif AA, Saleh IG, Abdelghany TM. Resveratrol influences platinum pharmacokinetics: a novel mechanism in protection against cisplatin-induced nephrotoxicity. Toxicol Lett. 2018;290:73–82. doi:10.1016/j.toxlet.2018.03.023
19. Nakamura T, Yonezawa A, Hashimoto S, Katsura T, Inui K. Disruption of multidrug and toxin extrusion MATE1 potentiates cisplatin-induced nephrotoxicity. Biochem Pharmacol. 2010;80(11):1762–1767. doi:10.1016/j.bcp.2010.08.019
20. Kleih M, Böpple K, Dong M, et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. 2019;10(11):851. doi:10.1038/s41419-019-2081-4
21. Brooks C, Wei Q, Cho SG, Dong Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J Clin Invest. 2009;119(5):1275–1285. doi:10.1172/jci37829
22. Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. Science. 1985;228(4703):1049–1055. doi:10.1126/science.3887571
23. Dentinger CM, Rakotomanga TA, Rakotondrandriana A, et al. Efficacy of artesunate-amodiaquine and artemether-lumefantrine for uncomplicated Plasmodium falciparum malaria in Madagascar, 2018. Malar J. 2021;20(1):432. doi:10.1186/s12936-021-03935-4
24. Visser MT, Zonneveld R, Peto TJ, van Vugt M, Dondorp AM, van der Pluijm RW. Are national treatment guidelines for falciparum malaria in line with WHO recommendations and is antimalarial resistance taken into consideration? - A review of guidelines in non-endemic countries. Trop Med Int Health. 2022;27(2):129–136. doi:10.1111/tmi.13715
25. Hess KM, Goad JA, Arguin PM. Intravenous artesunate for the treatment of severe malaria. Ann Pharmacother. 2010;44(7–8):1250–1258. doi:10.1345/aph.1M732
26. Shah PJ, Koshy J, Everett N, Attia E. Severe Plasmodium falciparum malaria treated with investigational artesunate in the United States. J Pharm Pract. 2020;33(1):108–112. doi:10.1177/0897190018782367
27. Li B, Zhang Z, Fu Y. Anti-inflammatory effects of artesunate on atherosclerosis via miR-16-5p and TXNIP regulation of the NLRP3 inflammasome. Ann Transl Med. 2021;9(20):1558. doi:10.21037/atm-21-4939
28. Ghosh AK, Miller H, Knox K, Kundu M, Henrickson KJ, Arav-Boger R. Inhibition of human coronaviruses by antimalarial peroxides. ACS Infect Dis. 2021;7(7):1985–1995. doi:10.1021/acsinfecdis.1c00053
29. Shakir L, Hussain M, Javeed A, Ashraf M, Riaz A. Artemisinins and immune system. Eur J Pharmacol. 2011;668(1–2):6–14. doi:10.1016/j.ejphar.2011.06.044
30. Efferth T, Dunstan H, Sauerbrey A, Miyachi H, Chitambar CR. The anti-malarial artesunate is also active against cancer. Int J Oncol. 2001;18(4):767–773. doi:10.3892/ijo.18.4.767
31. Lei XY, Tan RZ, Jia J, et al. Artesunate relieves acute kidney injury through inhibiting macrophagic mincle-mediated necroptosis and inflammation to tubular epithelial cell. J Cell Mol Med. 2021;25(18):8775–8788. doi:10.1111/jcmm.16833
32. Shen S, Liu SZ, Du MB, Ge KY, Song LH, Ye ZG. Determination of equilibrium solubility and apparent oil /water partition coefficient of artesunate. Chin J Exp Traditional Med Formulae. 2013;19(19):9–12.
33. Fanello C, Hoglund RM, Lee SJ, et al. Pharmacokinetic study of rectal artesunate in children with severe malaria in Africa. Antimicrob Agents Chemother. 2021;65(4). doi:10.1128/aac.02223-20
34. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi:10.1016/j.addr.2012.09.037
35. Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomed. 2015;10:975–999. doi:10.2147/ijn.S68861
36. Amekyeh H, Alkhader E, Sabra R, Billa N. Prospects of curcumin nanoformulations in cancer management. Molecules. 2022;27(2):361. doi:10.3390/molecules27020361
37. Khan S, Hussain A, Attar F, et al. A review of the berberine natural polysaccharide nanostructures as potential anticancer and antibacterial agents. Biomed Pharmacother. 2022;146:112531. doi:10.1016/j.biopha.2021.112531
38. Quijia CR, Chorilli M. Piperine for treating breast cancer: a review of molecular mechanisms, combination with anticancer drugs, and nanosystems. Phytother Res. 2022;36(1):147–163. doi:10.1002/ptr.7291
39. Shen S, Du M, Liu Q, et al. Development of GLUT1-targeting alkyl glucoside-modified dihydroartemisinin liposomes for cancer therapy. Nanoscale. 2020;12(42):21901–21912. doi:10.1039/d0nr05138a
40. Gu L, Zhang J, Liu D, et al. Development of artesunate intelligent prodrug liposomes based on mitochondrial targeting strategy. J Nanobiotechnology. 2022;20(1):376. doi:10.1186/s12951-022-01569-5
41. Deng F, Zhang H, Zhou W, et al. TRPA1 promotes cisplatin-induced acute kidney injury via regulating the endoplasmic reticulum stress-mitochondrial damage. J Transl Med. 2023;21(1):695. doi:10.1186/s12967-023-04351-9
42. Radford MG, Jr., Donadio JV, Jr., Bergstralh EJ, Grande, JP, et al. Predicting renal outcome in IgA nephropathy. J Am Soc Nephrol. 1997;8(2):199–207. doi:10.1681/asn.V82199
43. Xie SB, Wang WM, Chen N. Progression of renal tubulointerstitial fibrosis and expression of α-SMA,TGF-β_1 and VDR in rat UUO models. J Shanghai Jiaotong Univ. 2010;30(7):1.
44. Li W, Xu Q, He YF, et al. Anti-tumor effect of steamed codonopsis lanceolata in H22 tumor-bearing mice and its possible mechanism. Nutrients. 2015;7(10):8294–8307. doi:10.3390/nu7105395
45. Lingaraju MC, Pathak NN, Begum J, et al. Betulinic acid attenuates renal oxidative stress and inflammation in experimental model of murine polymicrobial sepsis. Eur J Pharm Sci. 2015;70:12–21. doi:10.1016/j.ejps.2015.01.001
46. Ozkok A, Ravichandran K, Wang Q, Ljubanovic D, Edelstein CL. NF-κB transcriptional inhibition ameliorates cisplatin-induced acute kidney injury (AKI). Toxicol Lett. 2016;240(1):105–113. doi:10.1016/j.toxlet.2015.10.028
47. Mukhopadhyay P, Rajesh M, Pan H, et al. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med. 2010;48(3 457–467). doi:10.1016/j.freeradbiomed.2009.11.022
48. Hagar H, Medany AE, Salam R, Medany GE, Nayal OA. Betaine supplementation mitigates cisplatin-induced nephrotoxicity by abrogation of oxidative/nitrosative stress and suppression of inflammation and apoptosis in rats. Exp Toxicol Pathol. 2015;67(2):133–141. doi:10.1016/j.etp.2014.11.001
49. Gu H, Gwon MG, Kim JH, Leem J, Lee SJ, Ahmed M. Oridonin attenuates cisplatin-induced acute kidney injury via inhibiting oxidative stress, apoptosis, and inflammation in mice. Biomed Res Int. 2022;2022:3002962. doi:10.1155/2022/3002962
50. Zaaba NE, Beegam S, Elzaki O, et al. The nephroprotective effects of α-bisabolol in cisplatin-induced acute kidney injury in mice. Biomedicines. 2022;10(4):842. doi:10.3390/biomedicines10040842
51. Yan X, Tan XY, Li YX, et al. A stepwise targeting curcumin derivative, Ser@TPP@CUR, for acute kidney injury. ACS Med Chem Lett. 2022;13(4):554–559. doi:10.1021/acsmedchemlett.1c00585
52. Hao Q, Xiao X, Zhen J, et al. Resveratrol attenuates acute kidney injury by inhibiting death receptor‑mediated apoptotic pathways in a cisplatin‑induced rat model. Mol Med Rep. 2016;14(4):3683–3689. doi:10.3892/mmr.2016.5714
53. Zhou L, Yu P, Wang TT, et al. Polydatin attenuates cisplatin-induced acute kidney injury by inhibiting ferroptosis. Oxid Med Cell Longev. 2022;2022:9947191. doi:10.1155/2022/9947191
54. Dang WZ, Li H, Jiang B, et al. Therapeutic effects of artesunate on lupus-prone MRL/lpr mice are dependent on T follicular helper cell differentiation and activation of JAK2-STAT3 signaling pathway. Phytomedicine. 2019;62:152965. doi:10.1016/j.phymed.2019.152965
55. Chen Q, Wang Z, Lv J, et al. Efficacy and safety of artesunate for patients with IgA nephropathy: a study protocol for a multicenter, double-blind, randomized, placebo-controlled trial. Trials. 2022;23(1):444. doi:10.1186/s13063-022-06336-3
56. Sun Z, Ma Y, Chen F, Wang S, Chen B, Shi J. Artesunate ameliorates high glucose-induced rat glomerular mesangial cell injury by suppressing the TLR4/NF-κB/NLRP3 inflammasome pathway. Chem Biol Interact. 2018;293:11–19. doi:10.1016/j.cbi.2018.07.011
57. Patra JK, Das G, Fraceto LF, et al. Nano based drug delivery systems: recent developments and future prospects. J Nanobiotechnology. 2018;16(1):71. doi:10.1186/s12951-018-0392-8
58. Lammers T, Ferrari M. The success of nanomedicine. Nano Today. 2020;31. doi:10.1016/j.nantod.2020.100853
59. Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomed. 2012;7:49–60. doi:10.2147/ijn.S26766
60. Hu C, Liang K, An R, Wang XH, You LS. Tissue distribution of TPGS modified artesunate liposome and its metabolites in rats. Zhongguo Zhong Yao Za Zhi. 2018;43(2):325–331. doi:10.19540/j.cnki.cjcmm.20171023.006
61. Karim N, Shishir MRI, Rashwan AK, Ke H, Chen W. Suppression of palmitic acid-induced hepatic oxidative injury by neohesperidin-loaded pectin-chitosan decorated nanoliposomes. Int J Biol Macromol. 2021;183:908–917. doi:10.1016/j.ijbiomac.2021.05.010
62. Li Y, Xu F, Li X, et al. Development of curcumin-loaded composite phospholipid ethosomes for enhanced skin permeability and vesicle stability. Int J Pharm. 2021;592:119936. doi:10.1016/j.ijpharm.2020.119936
63. Ramesh G, Reeves WB. TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest. 2002;110(6):835–842. doi:10.1172/jci15606
64. Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci. 2019;20(12):3011. doi:10.3390/ijms20123011
65. Kim DU, Kim DG, Choi JW, et al. Loganin attenuates the severity of acute kidney injury induced by cisplatin through the inhibition of ERK activation in mice. Int J Mol Sci. 2021;22(3). doi:10.3390/ijms22031421
66. Faubel S, Lewis EC, Reznikov L, et al. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther. 2007;322(1):8–15. doi:10.1124/jpet.107.119792
67. Ramesh G, Reeves WB. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int. 2004;65(2):490–499. doi:10.1111/j.1523-1755.2004.00413.x
68. Zeng L, Lin L, Xiao W, Li Y. L-theanine protects rat kidney from D-galactose-induced injury via inhibition of the AGEs/RAGE signaling pathway. Eur J Pharmacol. 2022;927:175072. doi:10.1016/j.ejphar.2022.175072
69. Shen C, Ma Y, Zeng Z, Yin Q, Hong Y, Hou X and Liu X. (2017). RAGE-Specific Inhibitor FPS-ZM1 Attenuates AGEs-Induced Neuroinflammation and Oxidative Stress in Rat Primary Microglia. Neurochem Res, 42(10), 2902–2911. 10.1007/s11064-017-2321-x
70. Giridharan VV, Thandavarayan RA, Bhilwade HN. Schisandrin B, attenuates cisplatin-induced oxidative stress, genotoxicity and neurotoxicity through modulating NF-κB pathway in mice. Free Radic Res. 2012;46(1):50–60. doi:10.3109/10715762.2011.638291
71. Gao X, Wang J, Wang Y . Fucoidan-ferulic acid nanoparticles alleviate cisplatin-induced acute kidney injury by inhibiting the cGAS-STING pathway. Int J Biol Macromol. 2022;223(Pt A):1083–1093. doi:10.1016/j.ijbiomac.2022.11.062
72. Peiro G, Alary J, Cravedi JP, Rathahao E, Steghens JP, Guéraud F. Dihydroxynonene mercapturic acid, a urinary metabolite of 4-hydroxynonenal, as a biomarker of lipid peroxidation. Biofactors. 2005;24(1–4):89–96. doi:10.1002/biof.5520240110
73. Ansari MA. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed Pharmacother. 2017;93:646–653. doi:10.1016/j.biopha.2017.06.085
74. Zhao W, Li J, Li Y, Chen Y, Jin H. Preventive effect of collagen peptides from acaudina molpadioides on acute kidney injury through attenuation of oxidative stress and inflammation. Oxid Med Cell Longev. 2022;2022:8186838. doi:10.1155/2022/8186838
75. Diao C, Chen Z, Qiu T, et al. Inhibition of PRMT5 attenuates oxidative stress-induced pyroptosis via activation of the Nrf2/HO-1 signal pathway in a mouse model of renal ischemia-reperfusion injury. Oxid Med Cell Longev. 2019;2019:2345658. doi:10.1155/2019/2345658
76. Tran MT, Zsengeller ZK, Berg AH, et al. PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature. 2016;531(7595):528–532. doi:10.1038/nature17184
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.