Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Aripiprazole: from pharmacological profile to clinical use
Authors Di Sciascio G, Riva MA
Received 7 May 2015
Accepted for publication 11 August 2015
Published 13 October 2015 Volume 2015:11 Pages 2635—2647
DOI https://doi.org/10.2147/NDT.S88117
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Roger Pinder
Guido Di Sciascio,1 Marco Andrea Riva2
1Department of Psychiatry, University Hospital “Policlinico”, Bari, Italy; 2Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
Abstract: Clinical experience with aripiprazole has confirmed the effectiveness and the safety of this novel antipsychotic drug in patients with schizophrenia as well as for the treatment of mania in type I bipolar disorder. However the generalization of the results from clinical trials requires further effort in order to address some issues and to overcome incorrect and partial interpretation of the clinical evidence. This article provides some straightforward guidance that may help clinical psychiatrists to translate the mechanism of action of aripiprazole into clinical setting, thus improving the appropriate use of the drug through rational application of its pharmacological profile. Examples of paradigmatic clinical situations are presented and discussed, suggesting possible intervention strategies, which may contribute to achieving the most appropriate use of the pharmacological properties of aripiprazole in real life settings.
Keywords: partial agonist, dopamine receptor, antipsychotic switch, neuronal plasticity
Introduction
Aripiprazole, defined as a third-generation antipsychotic drug (APD) due to its unique pharmacological profile, represents an innovative therapeutic tool for major psychiatric disorders. Its modulatory properties on different neurotransmitter systems (see section: Overview of the pharmacological profile) highlight a drug with a broad pharmacological profile and clinical effectiveness in the acute and long-term treatment of different psychopathological conditions, as demonstrated in clinical trials.
Aripiprazole was shown to be effective and well tolerated in short- (4–8 weeks) and intermediate- to long-term (26 and 52 weeks) clinical trials for the treatment of schizophrenia and schizoaffective disorder. The data on aripiprazole efficacy and tolerability in schizophrenia have been recently reviewed,1 and support the overall effectiveness of this antipsychotic for the short- and long-term treatment of schizophrenia.
Second-generation antipsychotics have been shown to be equally, or more, effective than classic antimanic agents, such as lithium and valproate, in the treatment of mania associated with bipolar disorder.2,3
Aripiprazole was also shown to be effective and well tolerated in the manic and maintenance phases of bipolar disorder, in monotherapy as well as in combination with mood stabilizers, such as lithium or valproate.4,5
Guidelines for the treatment of mania, edited in 2009 by the World Federation of Societies of Biological Psychiatry and the update of Canadian Network for Mood and Anxiety Treatment guidelines for the management of bipolar disorder, edited in 2013 by Canadian Network for Mood and Anxiety Treatment and International Society for Bipolar Disorder, include aripiprazole among those treatments with the highest level of evidence with regard to efficacy and tolerability profiles.2,3
While a recent meta-analysis did not give aripiprazole a high ranking for efficacy and tolerability in the treatment of acute mania,6 it must be borne in mind that, clinical efficacy of a given drug should not only be viewed as the result of short-term treatment (3 weeks) but primarily with respect to the long-term, which represents the final goal for patient treatment and for functional recovery.7
Complete data regarding aripiprazole efficacy and tolerability for short- and long-term treatment of bipolar disorder8–14 are summarized in Table 1 and they have also been reviewed elsewhere.4,5
 | Table 1 Summary of aripiprazole trials in bipolar patients |
In both clinical conditions (schizophrenia and bipolar disorder) the treatment of psychomotor agitation represents one of the most challenging tasks. It is currently agreed that best outcomes and compliance to treatment are obtained when the remission of agitation symptoms is induced without sedation. With aripiprazole, compliance increased both in the acute phase and during follow-up. Remission of agitation 2 hours after administration was achieved with intramuscular aripiprazole 9.75 mg, with no sedative effects on patients; on the contrary, sedation was present after treatment with intramuscular lorazepam 2 mg.15
All these evidences suggest aripiprazole represents one of the most effective therapeutic options in both the short- and long-term treatment of schizophrenia and bipolar disorder.
Critical issues in the use of aripiprazole in real life settings
Five years of clinical experience with aripiprazole have confirmed the effectiveness and safety of aripiprazole in patients with both schizophrenia and type I bipolar disorder, although physicians sometimes feel that not all clinical trial results could be generalized into real life settings. In particular, the evidence retrieved from clinical trials may not be immediately reflected in clinical practice: in most cases, the psychiatrist may perceive the results of clinical trials as coming from an “ideal world”, quite remote from “real life”. This aspect may be particularly relevant for aripiprazole, whose novel mechanism of action requires a better knowledge of the molecule for different medical settings in order to achieve the most appropriate use in real life settings. On this basis, we will specifically refer to some criticisms that have emerged since its marketing:
- aripiprazole is usually considered more effective on negative symptoms of schizophrenia than on positive symptoms, the latter being particularly prominent in patients with concomitant acute psychosis;
- aripiprazole is not often considered a first choice treatment for psychomotor agitation, even in the intramuscular formulation;
- the correct dosage for acute conditions, both in schizophrenia and mania, is sometimes an issue;
- psychiatrists often feel uncomfortable facing the need to switch from other APDs to aripiprazole;
- the use of aripiprazole as add-on therapy in partial responsers.
In light of this scenario, the aim of this article is to provide some useful practical hints, based on our long-standing clinical experience, which may help clinicians to generalize data from clinical trials. The unique pharmacological properties of aripiprazole are reviewed, with particular emphasis on how they may relate to the clinical activity of the drug. Furthermore, in order to “translate” these properties into real clinical settings, we present five frequently observed clinical situations that address some of the specific scenarios discussed earlier, and provide guidance on appropriate clinical intervention through an increased understanding of the pharmacological rationale of aripiprazole.
Aripiprazole: overview of the pharmacological profile
Neurotransmitter receptors represent the primary synaptic target of APDs. The importance of receptor mechanisms is at least threefold: first, it is thought that, by binding to specific receptors, APDs can normalize altered activity of selected neurotransmitters, which contribute to the functional abnormalities that are present in schizophrenic and bipolar patients. Second, receptor interaction initiates a series of intracellular events that will eventually lead to changes in gene expression and to more profound structural modifications that are critical for long-term improvement and patient stabilization. Last, receptor profiles are often responsible for the occurrence of unwanted side effects that may hinder patient compliance and treatment adherence.
When examining the receptor profile of aripiprazole, one should consider similarities and differences not only with respect to first-generation APDs (FGAs) but also with respect to second-generation APDs (SGAs). Indeed it has become apparent that SGAs display rather heterogeneous pharmacodynamic profiles that may lead to different synaptic and post-synaptic effects.16,17
A common feature of APD activity is represented by their inhibitory effects on dopamine neurotransmission, particularly at dopamine D2/D3 receptors. The interaction of APDs with dopamine D2/D3 receptors is essential for antipsychotic activity,18 and may normalize the excessive dopaminergic function in subcortical regions. However, this effect can be achieved through different mechanisms. Firstly, there is a clear distinction between FGAs and SGAs: while FGAs are potent antagonists of dopamine D2/D3 receptors and determine prominent receptor blockade, SGAs have a lower affinity for dopamine D2 receptors which, in some cases, can be due to the more rapid dissociation rate from the receptor.19
It has also been shown, mainly through in vitro experiments, that most APDs may act as inverse agonists, which implies that they may reduce the basal constitutive activity at D2/D3 receptors.20
The key feature of aripiprazole is represented by its activity as a partial agonist at dopamine D2 receptors, which differentiates this drug from FGA and SGA.21
The definition of partial agonist implies the ability to bind and activate a given receptor, but with partial efficacy relative to the full agonist, which means that the activity of the partial agonist stands between a normal agonist and an antagonist. This suggests that the drug may effectively “substitute” the endogenous neurotransmitter at a given receptor site, the consequences of this being dependent upon the functional state of the neurotransmitter. Indeed, in general terms, if there is an over-activation of the neurotransmitter at a given receptor, the partial agonist will interact with the receptor and will reduce the functional response. However, under conditions of defective transmission, partial agonists will be able to restore, at least in part, the physiological function of the neurotransmitter, taking advantage of its ability to activate the given receptor. How can this contribute to the clinical activity of aripiprazole in schizophrenia? We know that psychotic symptoms are primarily associated with enhanced dopamine neurotransmission in mesolimbic areas and excessive activation of dopamine D2 receptors.22,23
FGAs, being potent antagonists at these receptors, can effectively prevent the activation of dopamine D2 receptors and reduce psychotic symptoms. However, it is known that if more than 80% of dopamine D2 receptors are blocked by APDs, amelioration of psychotic symptoms is associated with an increased incidence of motor side effects, as well as elevated circulating levels of prolactin. This occurs to a high degree with FGAs, but less so with SGAs that, through different mechanisms, may determine a less pronounced blockade of dopamine D2 receptors. Conversely, aripiprazole, by virtue of its high affinity for these receptors, results in high occupancy of dopamine D2 receptors, with the minimal effective dose of 10 mg yielding >80% receptor occupancy, reaching 90%–94% at higher doses (30 mg).24
The high occupancy of D2 receptors with aripiprazole occurs without inducing extrapyramidal symptoms in most subjects, due to the fact that its partial agonism produces a much lower level of functional antagonism of D2 receptor-mediated neurotransmission than is seen with full antagonists.20,21,24,25
The term “functional antagonism” means to separate the receptor occupancy of a given drug from the functional effects produced by drug-receptor interaction. Indeed, for classical receptor antagonists (FGAs and SGAs), since receptor blockade is equal to receptor occupancy the functional effects will depend upon the extent of occupancy. Conversely, in the case of partial agonists, such as aripiprazole, the extent of receptor blockade is due to occupancy minus its intrinsic activity, indicating that the functional effect produced by the drug is not proportionate to the occupancy of the receptor. Thus, in the case of aripiprazole, this will ultimately contribute to blunt excessive pathological activation of dopamine D2 receptors, while preserving physiological neurotransmission.
Studies carried out in rodents have provided support for the separation between dopamine D2 receptor occupancy and functional receptor antagonism with APDs. In fact, while haloperidol and risperidone induce catalepsy (considered a surrogate of motor side effects) at doses producing >80% dopamine D2 receptor occupancy, aripiprazole, despite higher receptor occupancy (>90%), induced no catalepsy. The higher receptor occupancy with aripiprazole was necessary for attaining behavioral effects in the conditioned avoidance response, a surrogate for antipsychotic activity.26
Evidence for the pharmacological profile of aripiprazole on dopamine D2 receptors is not limited to the lack of motor side effects but comes from an analysis of the subjective well-being scale of patients treated with aripiprazole. Indeed, extensive blockade of dopamine D2 receptors is associated with dysphoric experiences, which may also involve extra-striatal brain regions. Indeed, Mizrahi et al have demonstrated that patients switched from antagonist antipsychotics (risperidone, olanzapine) to aripiprazole showed improvement in their subjective well-being, despite the very high levels of dopamine D2 receptor occupancy (82%–99%), an effect that was sustained for 6 months.27
Some of the mechanisms described here are also relevant for the activity of aripiprazole in bipolar disorder, particularly for the control of manic symptoms.8
Indeed, although manic episodes are probably associated with multiple dysfunctions, affecting neurotransmitters as well as intracellular signaling pathways, dopamine hyperactivity has long been indicated as a causative factor for acute mania.28
Hence, similar to FGAs and SGAs, the antimanic effects of aripiprazole are presumably due to its ability to reduce the hyperactivity of dopamine D2 receptors in key brain regions. In summary, the potential of aripiprazole as a partial agonist will be to reduce the hyperactivity of the dopaminergic system to the level that is set by its intrinsic activity (depending on the concentration of the drug) as well as the function of systems downstream from dopamine receptors. Furthermore, as a partial agonist, aripiprazole is unlikely to produce the up-regulation of dopamine D2 receptors, which represents a general consequence of prolonged treatment with dopamine D2 receptor antagonists. Indeed chronic treatment with aripiprazole prevents the development of dopamine supersensitivity and potentially supersensitivity psychosis, suggesting that its stabilizing properties may be helpful for patients with treatment-resistant schizophrenia.29
The potential benefits associated with the partial agonism of aripiprazole at dopamine D2 receptors are not limited to the hyperdopaminergic state associated with psychotic symptoms, but may also be relevant for the reduced activity of the prefrontal cortex, which has important implications for cognitive function.30,31
Although cognitive impairment in schizophrenic subjects is not only due to dopaminergic dysfunction but involves other neurotransmitters through a complex array of mechanisms, it is believed that reduced dopaminergic activity in the prefrontal cortex may contribute to clinical alterations.32
On this basis, an excessive blockade of dopamine D2 receptors, occurring in subjects treated with FGAs, results in further impairment and may worsen the clinical outcome. Since it is expected that the dopamine system in the prefrontal cortex is hypoactive in schizophrenics, the partial agonism of aripiprazole may restore, at least in part, such defective neurotransmission, thus leading to a stabilization of the dopaminergic signal.33
There is also evidence that the unique profile of aripiprazole is not only due to its activity as a partial agonist at dopamine D2 receptors, but may be the consequence of its functional selectivity at dopamine D2 receptors, a concept that broadens the impact of aripiprazole on dopaminergic transmission.34
Indeed, some of the effects of aripiprazole cannot be explained only on the basis of its partial agonism. For example, aripiprazole shows different potency in modulating intracellular signaling pathways coupled to D2 receptors. In fact, it is known that stimulation of D2-like receptors not only leads to inhibition of cAMP accumulation, but modulates other pathways, including phospholipases, ion channels (major effects are due to the regulation of K+ channels via Gbγ subunits), and MAPK.35,36
Aripiprazole has low potency for the activation of MAPK, compared to its effects on arachidonic acid release or inhibition of cAMP accumulation.37
This suggests that the ability of aripiprazole to regulate dopaminergic function will not only depend on the activity of the system (dopaminergic hyperactivity for psychosis and dopaminergic hypoactivity for cognitive dysfunction) but also on the cellular milieu where dopamine D2 receptors are expressed.34–37
Similar to SGAs, aripiprazole can also interact with other neurotransmitter receptors, some of which may contribute to its therapeutic activity. With regard to the serotonergic system, aripiprazole has significant affinity (5–30 nM) for several serotonin (5-HT) receptors (5-HT1A, 5-HT2A, 5-HT2B, and 5-HT7).21,38
The activity at 5-HT2a receptors is similar to that reported for most SGAs that effectively block these receptors, a mechanism that may also modulate the function of dopaminergic neurons at cortical and subcortical level.16,39
In particular, blockade of 5-HT2A receptors may increase dopamine release at striatal level (thus reducing the incidence of motor side effects) and in the prefrontal cortex where, as mentioned above, decreased dopaminergic transmission contributes to cognitive dysfunction. Moreover, in human subjects, aripiprazole exhibits higher occupancy at D2 receptors (87%) than at 5-HT2A receptors (52%), suggesting that, different to SGAs, its “atypical” profile is not related to a high 5-HT2A/D2 binding profile.39
Aripiprazole is also a partial agonist at 5-HT1A receptors, a property shared by other antipsychotics, such as quetiapine, clozapine, ziprasidone, asenapine, and lurasidone.40,41
Drugs characterized by “balanced” 5-HT1A receptor agonism and D2 antagonism (or partial agonism) may be effective in treating a broader range of symptoms of schizophrenia, including depressive as well as cognitive symptoms, and be better tolerated than existing antipsychotics.16,40,42
However, it must be borne in mind that human studies found that aripiprazole displays low in vivo occupancy of 5-HT1A receptors,24 a result shared by recent rodent studies, casting some doubts on the role of this receptor for the clinical activity of aripiprazole. Interestingly, preclinical studies have demonstrated that behavioral abnormalities in the phencyclidine model of schizophrenia can be restored by aripiprazole also through 5-HT1A receptor mechanisms. In particular, social isolation deficits as well as impairment of recognition memory produced by the glutamate N-methyl-d-aspartate receptor antagonist, phencyclidine, can be ameliorated by aripiprazole; an effect that is prevented by 5-HT1A receptor antagonists.43,44
Aripiprazole displays negligible affinity for muscarinic receptors21 that, combined with its partial agonist activity for D2 and 5-HT1A receptors, could explain the reported improvements in neurocognitive functions of schizophrenic patients treated with aripiprazole.45
In summary, similar to other SGAs, aripiprazole has good affinity for several monoamine receptors that, in combination with the activity at dopamine D2 receptors, may provide an add-on value for its therapeutic efficacy in schizophrenia as well as in bipolar disorder.
Receptor profiles are also important for the incidence of side effects associated with antipsychotic therapy. While a general improvement of motor side effects and hyperprolactinemia has been observed with aripiprazole (partial agonist) as well as with other SGAs (due to their different impact on dopamine D2 receptors),46,47 attention has been devoted to other important side effects, such as weight gain, dyslipidemia, diabetes, and cardiometabolic risk.48,49
Although these effects are probably the result of complex mechanisms, there might be a predisposition of schizophrenic and bipolar patients to develop such disorders, which can be exacerbated by antipsychotic therapy.50
The greatest single correlate for weight gain associated with antipsychotic therapy is considered to be the antagonism at H1-histamine receptors,51,52 probably in conjunction with the antagonism for serotonin 5-HT2c receptors. The moderate affinity of aripiprazole for H1-histamine receptors may therefore explain its minimal propensity to induce weight gain.51
Sedation is also due, although not exclusively, to the antagonism of H1 receptors. While sedation may be required, and is necessary during acute psychotic or manic episodes, it represents a negative factor for long-term treatment and for the functional recovery of schizophrenic and bipolar patients. The initial advantage associated with the use of APDs characterized by high affinity at histamine H1 receptors may then become a drawback when sedative effects are no longer required. Aripiprazole has low sedative properties due to its modest antagonism at H1 receptors. For this reason, during an initial phase of treatment it might be useful in association with a benzodiazepine, which can be withdrawn when the acute episode is over (see section: Managing a partial response).
As it will be discussed in detail later on, knowledge of aripiprazole receptor profiles is important not only for a better understanding of its clinical activity, but also for the appropriate management of switch procedures from other antipsychotics that may or may not share similar pharmacodynamic profiles.
If receptor mechanisms represent primary synaptic events, the overall impact on synaptic function relies also on secondary mechanisms due to the modulation of neurotransmitter release. In this regard, and similar to what has been reported for other SGAs, aripiprazole can increase cortical dopamine efflux, an effect that may persist after long-term administration.16,53 Regardless of the mechanisms that sustain such effect, it may be inferred that the ability to increase the synaptic levels of dopamine, or of other neurotransmitters, in key brain regions represents an add-on value for antipsychotic activity and may also contribute to secondary changes set in motion following repeated administration of APDs.
While synaptic mechanisms are responsible for rapid drug effects leading to the control of acute symptomatology, patient stabilization and functional improvement are probably related to different mechanisms that are set in motion after long-term treatment. With this respect, neuroadaptive events that are thought to improve neuronal plasticity and resiliency, represent an important component of long-term drug exposure that will eventually improve the ability to respond and adapt to environmental conditions, such as stress, that may trigger or precipitate psychiatric disorders.16,54–56
Different studies have demonstrated the ability of SGAs to increase the expression of neurotrophic factors and to enhance neurogenesis, whereas FGAs have a negative impact on these mechanisms or, at most, are devoid of any activity.16,17
A number of preclinical studies have been carried out on the investigation of long-term changes produced by aripiprazole administration on gene expression and neuroplastic proteins. Using microarray-based gene expression profiling technology, it has been shown that prolonged (4 weeks) treatment with aripiprazole (10 mg/kg) can induce, within the frontal cortex, differential expression of genes involved in transcriptional regulation and chromatin remodeling, and of genes implicated in the pathogenesis of psychosis.57
Aripiprazole, but not haloperidol, can increase the activity of the BDNF promoter, the levels of the neurotrophin protein as well as related signaling mechanisms.58
In line with these results, Park et al have shown that chronic administration of aripiprazole increases the hippocampal expression of BDNF in normal animals and, more importantly, counteracts the reduction of BDNF levels produced by chronic stress exposure.59
Moreover, the protective and neurotrophic effects of aripiprazole are shared by olanzapine but not by the classical antipsychotic haloperidol.59–61
We have recently demonstrated that chronic aripiprazole treatment may facilitate BDNF transcription under acute challenging conditions, an effect that is particularly evident in the prefrontal cortex.62
This effect may represent an important mechanism that will promote resilience under adverse stressful conditions, an ability that is defective in patients with mental illness. Interestingly Yoshimura et al have shown that subchronic treatment of first-episode schizophrenia patients with aripiprazole increases plasma BDNF levels.63
All in all, these results suggest the potential impact of aripiprazole on “neurotrophic” mechanisms that may be critical for the functional recovery of patients, a property that may be shared by other SGAs.16,17,60,64
Pharmacological mechanisms in specific clinical situations
Managing acute psychosis: why does aripiprazole 30 mg/day monotherapy seem to be more effective than 15 mg/day in the acute setting?
European prescribing information for aripiprazole reports a starting dose of 10 or 15 mg/day and a maintenance dose of 15 mg/day, with a maximum dose of 30 mg/day. Evidence reported from clinical practice suggests the preferential use of aripiprazole at the highest dose (30 mg once daily) in this pathological condition. Indeed aripiprazole monotherapy at a dose of 30 mg/day, administered in the morning, is commonly used in practice for patients with an acute episode mainly with positive symptoms (hallucinations, delusions) due to discontinuation of previous treatment. Based on our clinical experience, remission of symptoms can be obtained earlier and more efficiently with daily aripiprazole 30 mg than with 15 mg in patients with the features highlighted in the above example.
Since psychotic emergency and agitation are characterized by excessive function of the dopaminergic system, there may be some concern among doctors with respect to the intrinsic activity aripiprazole in a situation characterized by overt dopamine hyperactivity. However a careful examination of the pharmacological features of aripiprazole may provide an explanation for the effectiveness of high vs low doses of aripiprazole in controlling psychotic symptoms and agitation in an emergency.
There are at least two important issues that must be considered. First, the intrinsic activity of aripiprazole allows the maintenance of “physiological” activity of the dopaminergic system, without blunting dopamine transmission. Second, a high dose of the drug is more effective in overcoming dopamine hyperactivity with respect to lower doses. This is illustrated in Figure 1, which simulates an example of this situation depicting a dose-response curve of a full receptor agonist (dopamine) in the presence of increasing concentrations (doses) of the partial receptor agonist aripiprazole. Under physiological conditions, dopamine may produce a range of effects that depend upon the brain region involved, as well as on the functional activity of the system (gray shaded area). We can hypothesize that in an “emergency” situation the concentration of dopamine raises in the synaptic cleft thus producing a maximal “supra-physiological” response (as indicated by the dotted line). The presence of a partial agonist, as shown in Figure 1, will shift this dose-response curve to the right and such a “shift” depends upon the concentration of the partial agonist (aripiprazole). If we consider the acute “emergency” situation (dotted line) we can calculate the percentage of maximal effect for each curve that, in our case, represents the response to dopamine. As one can see, in the absence of aripiprazole, dopamine may produce a maximal activation of D2 receptors (estimated around 95%), which is only marginally reduced (to 89%) with a low aripiprazole concentration. Only in the presence of higher concentrations of the partial agonist (aripiprazole) does the response to the endogenous agonist (dopamine) become significantly reduced (<50%). However, the dampening properties of aripiprazole on dopamine hyperactivity will never lead to a complete “blockade” of the system, which is illustrated by the fact that at each concentration of partial agonist the percentage response will never go below the threshold (30%) that is determined by its intrinsic activity.
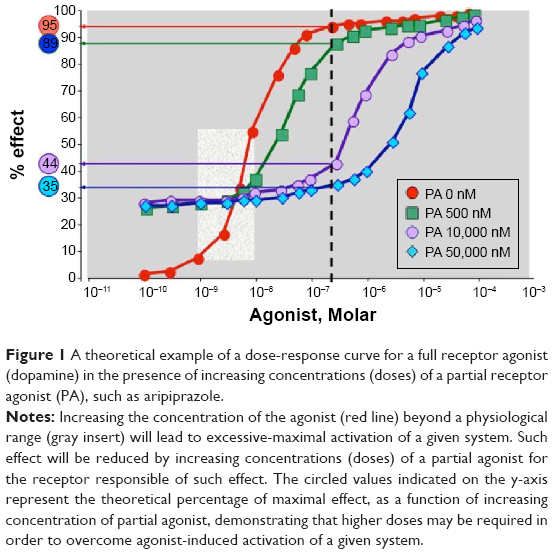 | Figure 1 A theoretical example of a dose-response curve for a full receptor agonist (dopamine) in the presence of increasing concentrations (doses) of a partial receptor agonist (PA), such as aripiprazole. |
When translated to the clinical setting, this example provides some clue as to why a single aripiprazole dose of 30 mg may be more effective than a dose of 15 mg in a psychotic emergency. As described above, doubling the dose (from 15 to 30 mg) could lead to a significant shift from a condition of excessive dopamine activity to a condition of “physiological” dopamine transmission. For the same reason, the administration of two 15 mg doses apart from each other will not provide the same effect as a single 30 mg dose. These considerations are not in contrast with the conclusions drawn by Sparshatt et al in a recent review on the correlation between aripiprazole dose, plasma concentration, receptor occupancy, and response.65
Indeed, the authors show that a strong correlation exists between aripiprazole dose and plasma levels. They state that dopamine receptor occupancy appears to reach a plateau at doses above 10 mg and suggest that, from dose-response studies, the optimal dose of aripiprazole may be 10 mg/day. However, this refers to prolonged treatment with the drug, whereas the point we make here relates to the use of aripiprazole in an acute setting in association with symptom exacerbation, a condition that may be sustained by an overt activation of the dopaminergic system. Hence, as illustrated above, a higher dose of aripiprazole will be necessary to compete with dopamine and blunt receptor activation. For stable patients on oral aripiprazole who would prefer, or are likely to benefit from, a long-acting formulation, long-acting injectable (LAI) aripiprazole may be of particular interest – a new treatment option for adult patients with schizophrenia.
It has been shown that LAI antipsychotics might reduce both unintentional and intentional non-adherence. LAI aripiprazole (Abilify Maintena) was approved for the treatment of schizophrenia by the US Food and Drug Administration on February 28, 2013 and the efficacy as a maintenance treatment for schizophrenia has been demonstrated. Aripiprazole LAI is more effective than placebo, and non-inferior to oral aripiprazole, in delaying relapse and in reducing relapse rates in schizophrenia.66,67
Managing a manic episode in bipolar disorder: what precautions need to be observed when valproate or other mood stabilizers are added to aripiprazole for the treatment of bipolar disorder?
Manic episodes are one of the most frequent clinical issues in hospitals and in first aid. The use of mood stabilizers and APDs with stabilizing properties is currently recommended for the treatment of patients with bipolar disorder with prevalent manic symptoms. Guidelines for the treatment of mania recommend monotherapy although mood stabilizers are frequently combined with second-generation antipsychotics for the treatment of mania, particularly when psychotic symptoms occur with the manic presentation. Combination of mood stabilizers with second-generation antipsychotics improves the effect of “flight of ideas” and restlessness. Aripiprazole is effective and safe in these situations, both as monotherapy and it is often combined with a mood stabilizer, particularly with valproate or lithium, due to the absence of interactions and adverse events.2,3
The improved efficacy of antipsychotics and mood stabilizers, compared with monotherapy using mood stabilizers alone, has been firmly established for bipolar mania. This type of combination may take advantage of the different mechanisms of action that characterize APDs with respect to mood stabilizers. Indeed, as mentioned above, the antimanic action of antipsychotic agents is primarily due to their interaction with dopamine D2 receptors, whereas mood stabilizers act through different mechanisms, including the modulation of ion channels as well as the regulation of intracellular signaling pathways.68,69
Furthermore, the combination of an antipsychotic agent (mostly second-generation drugs) and mood stabilizers could also lead to synergistic effects on the modulation of neuroadaptive mechanisms that are thought to improve neuronal plasticity, and may be particularly relevant for long-term patient stabilization.70,71
These concepts also hold true for aripiprazole. Moreover, no adjustment in the dose of aripiprazole is required when it is combined with lithium or valproate, since no clinically significant effects on the steady-state pharmacokinetics of aripiprazole have been reported.72
Since aripiprazole is primarily metabolized by CYP-2D6 and CYP-3A4, combination therapy with carbamazepine, a CYP-3A4 inducer, leads to a significant decrease in aripiprazole mean peak plasma concentrations and the area under the plasma concentration-time curve of aripiprazole (by 66% and 71%, respectively). On this basis, the combination of carbamazepine and aripiprazole will require a significant adjustment of aripiprazole dosage, approximately double.73 Aripiprazole is also a potential treatment candidate for different types of dependence in individuals diagnosed with comorbid psychoses, such as schizophrenia or bipolar disorder.74
It is known that dopaminergic mechanisms are involved in motivation, reward, and reinforcement of substance abuse.
Acting on the dopaminergic/serotonergic systems, the efficacy of aripiprazole in decreasing alcohol consumption, craving, and psychiatric symptom intensity has been shown. Moreover there is good evidence for the treatment of substance abuse such as cocaine, amphetamine, and tobacco use disorders and as an agent for the prevention of relapse in abstinent cocaine users.75
Voronin et al76 suggested that aripiprazole improves impulse control by enhancing the function of frontal cortex in patients with alcohol dependence and suggest that dopamine release induced by aripiprazole might be associated with increased activation of anterior cingulate which may control cravings for alcohol and substances. In fact it has been shown that patients treated with aripiprazole remained abstinent from any amount of alcohol for a longer time with respect to those treated with naltrexone.77
Managing a partial response: is there a rationale for combining different antipsychotics in a patient who achieves only a partial response to a single drug?
Schizophrenia with prevalent negative symptoms is often resistant to treatment and may require the combination of different antipsychotics. Partial response is one of the most critical problems in clinical practice since it causes dissatisfaction for both the patient and physician.13
Although clozapine remains the choice of treatment for schizophrenic patients resistant to conventional treatments, one-third to two-thirds of these patients fail or partially respond to clozapine.78
Furthermore, clozapine is associated with different adverse effects, including significant weight gain and metabolic disturbances.79
One key question arising from the issue of partial response relates to the choice of combination strategies that may be more effective. However, although combination therapy with different APDs is rather common in clinical practice, it is difficult, based on pharmacodynamic profiles, to provide a scientific rationale for such combinations. The major reason for this is that APDs are often characterized by complex and heterogeneous receptor profiles since, as described above, they interact with several neurotransmitter receptors at clinically relevant doses.38
On this basis, it is somewhat empirical to decide which combination might provide a real benefit at clinical level. Despite the fact that we suggest the contribution of several neurotransmitter receptors in the action of SGAs, there are probably only two undoubtedly efficacy-related mechanisms: the reduced blockade of dopamine D2 receptors (either due to reduced affinity as an antagonist, for most SGAs, or due to partial agonism, as for aripiprazole) and the higher affinity as 5-HT2a receptor antagonists. Most, if not all, SGAs share these properties, suggesting that it will be difficult to expect further benefit from their combination. There is, however, evidence that when aripiprazole is added to patients on haloperidol who are experiencing marked elevations in prolactin levels, there is a significant decrease over time in prolactin levels, with respect to patients receiving placebo in addition to haloperidol.80
Furthermore, the switch from risperidone or olanzapine to aripiprazole 30 mg/day produced a significant decrease in prolactin levels at week 1 and throughout the 8-week study period.81
This effect is probably due to the intrinsic activity of aripiprazole on dopamine D2 receptors. Interestingly, there was no observed clinical deterioration or worsening of symptoms with adjunctive aripiprazole treatment, suggesting that rebound mechanisms due to excessive stimulation of D2 receptors under this scenario should be limited.
However, since SGAs can also bind to neurotransmitter receptors that are responsible for, or may contribute to, the occurrence of side effects, one might expect that the combination of two or more APDs may worsen tolerability due to an increased incidence of side effects. Moreover, sub-threshold blockade of a given receptor subtype by two different drugs might become “at risk” if those drugs are used in combination. The combination clozapine-aripiprazole deserves a separate mention. There is evidence that aripiprazole in combination with clozapine improves therapy-resistant positive and negative symptoms as well as clozapine side effects.82
In a recent randomized, double-blind, placebo-controlled trial carried out in schizophrenia patients treated with clozapine, aripiprazole combined with clozapine resulted in significant improvements in terms of body weight and lipid parameters.83
While on the basis of their complex receptor profiles it is difficult to provide a strong rationale for such a combination, there may be beneficial effects for two major reasons. On one hand, combining aripiprazole with clozapine will result in a wider coverage of receptor systems compared with either drug alone, with the possibility that these mechanisms may synergize at the intracellular level leading to novel changes particularly relevant for treatment-resistant patients. On the other hand, the dose of clozapine may be reduced if the drug is combined with aripiprazole. This may eventually lead to a significant improvement in the side effect profile of clozapine (as some adverse events are dose-dependent) and, consequently, patient compliance may increase.
As previously mentioned, the association of aripiprazole with benzodiazepine is well established and takes advantage of the sedative properties of GABA modulation that may be used “on demand” in the presence of psychotic and manic episodes.
Aripiprazole can also be combined with antidepressant drugs acting on serotonin or noradrenaline transporters (selective serotonin reuptake inhibitors, serotonin and norepinephrine reuptake inhibitors). In this case, although detailed studies are still lacking, the potential advantage of such combinations may be due to the synergic effects of the two drugs (aripiprazole and selective serotonin reuptake inhibitors, for example) that, acting on different molecular targets, may produce more significant changes on brain function.84
This type of association, as demonstrated for the combination fluoxetine-olanzapine,85 may be useful in bipolar disorders as well as in treatment-resistant depression.
Managing the switch to aripiprazole: which pharmacological profiles have to be considered for a safety approach?
Tolerability and lack of efficacy represent the two main reasons for changing a pharmacological therapy. Switching to a different antipsychotic is a complex task for the psychiatrist. An evaluation of the risk–benefit ratio is important for a successful outcome.
While different strategies can be used during the switch, they should take into consideration several factors, which are primarily related to the pharmacological profile of the ongoing treatment as well as that of the new drug. While a detailed description of switching strategies is beyond the scope of the present article,86 we want to highlight some pharmacodynamic mechanisms that may be critical for choosing the proper switch strategy. When considering switching during antipsychotic therapy, the primary issue that has to be taken into account usually relates to the fact that there is a change from a drug with potent antagonism at a specific receptor to a drug with reduced impact on that receptor. Since chronic antagonism at a given receptor leads to an up-regulation of the receptor itself, the risk associated with pharmacological switch is a rebound effect due to excessive stimulation of the receptor by the endogenous neurotransmitter.
Probably the major concern with APDs is represented by supersensitivity psychosis. In fact, chronic APD treatment, especially with FGAs, may lead to an up-regulation of dopamine D2 receptors that, if no longer blocked by the drug, will be available for the endogenous agonist, dopamine. The consequence of this is an “over-responsiveness” of the system that may lead to the occurrence of psychosis. The potential for psychosis rebound may be higher when switching to drugs with lower dopamine D2 affinity (quetiapine, clozapine) or drugs, such as aripiprazole, that are partial agonists at D2 receptors. Under these conditions, it is necessary to perform a slow cross-tapering between the two drugs, or to reach the plateau dose for the incoming drug (aripiprazole) before starting to taper off the first drug.87
One potential advantage in switching to aripiprazole from APDs with D2 dopamine receptor antagonism, is the reduction of side effects, such as extrapyramidal symptoms and hyperprolactinemia, that are associated with excessive blockade of dopamine receptors. Indeed, as previously mentioned, adjunctive treatment with aripiprazole reduces hyperprolactinemia associated with haloperidol, possibly due to the fact that aripiprazole may bind to dopamine D2 receptors more robustly than haloperidol and, taking advantage of its intrinsic activity, can normalize prolactin levels.78
Another potential concern in switching to aripiprazole from different APDs is represented by the rebound mechanisms that are associated with receptors that represent clinically relevant targets of different APDs, including histamine H1 or muscarinic acetylcholine receptors, toward which aripiprazole shows very low affinity. For example, as mentioned above, different APDs are potent antagonists at histamine H1 receptors that have been associated with weight gain and sedation. Two different consequences can be anticipated when switching to aripiprazole from these antipsychotics. With regard to weight gain and metabolic effects, it is expected that side effects would improve after the switch. This has been demonstrated in a 4-month study with patients switched to aripiprazole from olanzapine, with a significant decrease in weight and lipids in overweight patients.88
Recent data have confirmed that switching to aripiprazole can significantly improve the metabolic profile of patients under treatment with other APDs.11,89
If amelioration of APD-associated weight gain and metabolic profile is observed when switching to aripiprazole, insomnia and agitation can be observed due to similar rebound mechanisms as previously mentioned. In this case, the occurrence of these effects may be reduced, or avoided, using slow cross-titration and/or through the addition of anti-histaminergic drugs, or benzodiazepine, if needed.90
Conclusion
In summary, based on the clinical experience accumulated in recent years, aripiprazole can be considered as an innovative APD, with a unique mechanism of action, which translates into an excellent efficacy and tolerability profile. Aripiprazole can be considered a first choice drug both for the treatment of schizophrenia and for the therapy of several phases of bipolar disorder. The intramuscular formulation provides a very efficient tool for rapid control of agitation. Furthermore an extended-release suspension that is an intramuscular depot formulation of aripiprazole has been recently added to the existing formulations. It is licensed for maintenance treatment of schizophrenia in adults whose condition has been stabilized with oral aripiprazole. Treatment with aripiprazole prolonged-release injection was associated with statistically significantly lower impending relapse rates compared with placebo (10% compared with 39.6%).91,92
Depot or LAI antipsychotics may be used to mitigate non-adherence, possibly reducing the risk of relapse, which represents a key issue for long-term functional improvement. Aripiprazole prolonged-release injection may therefore represent an important treatment option for patients who may show limited adherence to treatment.
Acknowledgment
Funding of this work was partly supported by an unrestricted grant from Otsuka Italia. The design, implications, and conclusions of the manuscript are solely the responsibility of the authors, and the funding source had no role in the conception, design, or content of the manuscript.
Author contributions
Guido Di Sciascio and Marco A Riva contributed to the writing and revision of the manuscript. Both authors contributed toward data analysis, drafting, and revising the manuscript and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest that are directly relevant to the content of this review. In the past 5 years, Dr Guido Di Sciascio has received honoraria or research support from Angelini, AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Janssen, Otsuka, and Polifarma, whereas Dr Marco A Riva has received honoraria or research support from Bristol-Myers Squibb, Sumitomo Dainippon Pharma, Eli Lilly, Innova Pharma, Lundbeck, Servier, Sunovion, and Takeda.
References
Stip E, Tourjman V. Aripiprazole in schizophrenia and schizoaffective disorder: A review. Clin Ther. 2010;32 Suppl 1:S3–S20. | ||
Grunze H, Vieta E, Goodwin GM, et al. The World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for the biological treatment of bipolar disorders: update 2009 on the treatment of acute mania. World J Biol Psychiatry. 2009;10(2):85–116. | ||
Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1–44. | ||
Aitchison KJ, Bienroth M, Cookson J, et al. A UK consensus on the administration of aripiprazole for the treatment of mania. J Psychopharmacol. 2009;23(3):231–240. | ||
Mcintyre RS. Aripiprazole for the maintenance treatment of bipolar I disorder: A review. Clin Ther. 2010;32 Suppl 1:S32–S38. | ||
Cipriani A, Barbui C, Salanti G, et al. Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple-treatments meta-analysis. Lancet. 2011;378(9799):1306–1315. | ||
Berk M, Malhi GS. Should antipsychotics take pole position in mania treatment? Lancet. 2011;378(9799):1279–1281. | ||
Keck PE Jr, Marcus R, Tourkodimitris S, et al. A placebo-controlled, double-blind study of the efficacy and safety of aripiprazole in patients with acute bipolar mania. Am J Psychiatry. 2003;160(9):1651–1658. | ||
Keck PE, Calabrese JR, Mcintyre RS, et al. Aripiprazole monotherapy for maintenance therapy in bipolar I disorder: a 100-week, double-blind study versus placebo. J Clin Psychiatry. 2007;68(10):1480–1491. | ||
Keck PE, Orsulak PJ, Cutler AJ, et al. Aripiprazole monotherapy in the treatment of acute bipolar I mania: a randomized, double-blind, placebo- and lithium-controlled study. J Affect Disord. 2009;112(1–3):36–49. | ||
Marcus R, Khan A, Rollin L, et al. Efficacy of aripiprazole adjunctive to lithium or valproate in the long-term treatment of patients with bipolar I disorder with an inadequate response to lithium or valproate monotherapy: a multicenter, double-blind, randomized study. Bipolar Disord. 2011;13(2):133–144. | ||
Vieta E, Bourin M, Sanchez R, et al. Effectiveness of aripiprazole v. haloperidol in acute bipolar mania: double-blind, randomised, comparative 12-week trial. Br J Psychiatry. 2005;187:235–242. | ||
Vieta E, T’joen C, Mcquade RD, et al. Efficacy of adjunctive aripiprazole to either valproate or lithium in bipolar mania patients partially nonresponsive to valproate/lithium monotherapy: a placebo-controlled study. Am J Psychiatry. 2008;165(10):1316–1325. | ||
Vieta E, Owen R, Baudelet C, Mcquade RD, Sanchez R, Marcus RN. Assessment of safety, tolerability and effectiveness of adjunctive aripiprazole to lithium/valproate in bipolar mania: a 46-week, open-label extension following a 6-week double-blind study. Curr Med Res Opin. 2010;26(6):1485–1496. | ||
Sanford M, Scott LJ. Intramuscular aripiprazole : a review of its use in the management of agitation in schizophrenia and bipolar I disorder. CNS Drugs. 2008;22(4):335–352. | ||
Lieberman JA, Bymaster FP, Meltzer HY, et al. Antipsychotic drugs: comparison in animal models of efficacy, neurotransmitter regulation, and neuroprotection. Pharmacol Rev. 2008;60(3):358–403. | ||
Molteni R, Calabrese F, Racagni G, Fumagalli F, Riva MA. Antipsychotic drug actions on gene modulation and signaling mechanisms. Pharmacol Ther. 2009;124(1):74–85. | ||
Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(7):1081–1090. | ||
Miyamoto S, Duncan GE, Marx CE, Lieberman JA. Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry. 2005;10(1):79–104. | ||
Strange PG. Antipsychotic drug action: antagonism, inverse agonism or partial agonism. Trends Pharmacol Sci. 2008;29(6):314–321. | ||
Shapiro DA, Renock S, Arrington E, et al. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28(8):1400–1411. | ||
Laruelle M. The role of endogenous sensitization in the pathophysiology of schizophrenia: implications from recent brain imaging studies. Brain Res Brain Res Rev. 2000;31(2–3):371–384. | ||
Laruelle M. The second revision of the dopamine theory of schizophrenia: implications for treatment and drug development. Biol Psychiatry. 2013;74(2):80–81. | ||
Mamo D, Graff A, Mizrahi R, Shammi CM, Romeyer F, Kapur S. Differential effects of aripiprazole on D(2), 5-HT(2), and 5-HT(1A) receptor occupancy in patients with schizophrenia: a triple tracer PET study. Am J Psychiatry. 2007;164(9):1411–1417. | ||
Kegeles LS, Slifstein M, Frankle WG, et al. Dose-occupancy study of striatal and extrastriatal dopamine D2 receptors by aripiprazole in schizophrenia with PET and [18F]fallypride. Neuropsychopharmacology. 2008;33(13):3111–3125. | ||
Natesan S, Reckless GE, Nobrega JN, Fletcher PJ, Kapur S. Dissociation between in vivo occupancy and functional antagonism of dopamine D2 receptors: comparing aripiprazole to other antipsychotics in animal models. Neuropsychopharmacology. 2006;31(9):1854–1863. | ||
Mizrahi R, Mamo D, Rusjan P, Graff A, Houle S, Kapur, S. The relationship between subjective well-being and dopamine D2 receptors in patients treated with a dopamine partial agonist and full antagonist antipsychotics. Int J Neuropsychopharmacol. 2009;12(5):715–721. | ||
Gerner RH, Post RM, Bunney WE Jr. A dopaminergic mechanism in mania. Am J Psychiatry. 1976;133(10):1177–1180. | ||
Tadokoro S, Okamura N, Sekine Y, Kanahara N, Hashimoto K, Iyo M. Chronic treatment with aripiprazole prevents development of dopamine supersensitivity and potentially supersensitivity psychosis. Schizophr Bull. 2012;38(5):1012–1020. | ||
Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III – the final common pathway. Schizophr Bull. 2009;35(3):549–562. | ||
Willner P. The dopamine hypothesis of schizophrenia: current status, future prospects. Int Clin Psychopharmacol. 1997;12(6):297–308. | ||
Carlsson A, Waters N, Holm-Waters S, Tedroff J, Nilsson M, Carlsson ML. Interactions between monoamines, glutamate, and GABA in schizophrenia: new evidence. Annu Rev Pharmacol Toxicol. 2001;41:237–260. | ||
Grunder G, Hippius H, Carlsson A. The ‘atypicality’ of antipsychotics: a concept re-examined and re-defined. Nat Rev Drug Discov. 2009;8(3):197–202. | ||
Mailman RB, Murthy V. Third Generation Antipsychotic Drugs: Partial Agonism or Receptor Functional Selectivity? Curr Pharm Des. 2010;16(5):488–501. | ||
Huff RM, Chio CL, Lajiness ME, Goodman LV. Signal transduction pathways modulated by D2-like dopamine receptors. Adv Pharmacol. 1998;42:454–457. | ||
Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol Sci. 2007;28(8):390–696. | ||
Urban JD, Vargas GA, Von Zastrow M, Mailman RB. Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology. 2007;32(1):67–77. | ||
Wong EH, Tarazi FI, Shahid M. The effectiveness of multi-target agents in schizophrenia and mood disorders: Relevance of receptor signature to clinical action. Pharmacol Ther. 2010;126(2):173–185. | ||
Kuroki T, Meltzer HY, Ichikawa J. Effects of antipsychotic drugs on extracellular dopamine levels in rat medial prefrontal cortex and nucleus accumbens. J Pharmacol Exp Ther. 1999;288(2):774–781. | ||
Newman-Tancredi A. The importance of 5-HT1A receptor agonism in antipsychotic drug action: rationale and perspectives. Curr Opin Investig Drugs. 2010;11(7):802–812. | ||
Tarazi FI, Riva MA. The preclinical profile of lurasidone: clinical relevance for the treatment of schizophrenia. Expert Opin Drug Discov. 2013;8(10):1297–1307. | ||
Meltzer HY, Sumiyoshi T. Does stimulation of 5-HT(1A) receptors improve cognition in schizophrenia? Behav Brain Res. 2008;195(1):98–102. | ||
Bruins Slot LA, Kleven MS, Newman-Tancredi A. Effects of novel antipsychotics with mixed D(2) antagonist/5-HT(1A) agonist properties on PCP-induced social interaction deficits in the rat. Neuropharmacology. 2005;49(7):996–1006. | ||
Nagai T, Murai R, Matsui K, et al. Aripiprazole ameliorates phencyclidine-induced impairment of recognition memory through dopamine D1 and serotonin 5-HT1A receptors. Psychopharmacology (Berl). 2009;202(1–3):315–328. | ||
Kern RS, Green MF, Cornblatt BA, et al. The neurocognitive effects of aripiprazole: an open-label comparison with olanzapine. Psychopharmacology (Berl). 2006;187(3):312–320. | ||
De Oliveira IR, Juruena MF. Treatment of psychosis: 30 years of progress. J Clin Pharm Ther. 2006;31(6):523–534. | ||
Henderson DC, Doraiswamy PM. Prolactin-related and metabolic adverse effects of atypical antipsychotic agents. J Clin Psychiatry. 2008;69 Suppl 1:32–44. | ||
Stahl SM, Mignon L, Meyer JM. Which comes first: atypical antipsychotic treatment or cardiometabolic risk? Acta Psychiatr Scand. 2009;119(3):171–179. | ||
Young SL, Taylor M, Lawrie SM. “First do no harm.” A systematic review of the prevalence and management of antipsychotic adverse effects. J Psychopharmacol. 2015;29(4):353–362. | ||
Newcomer JW. Medical risk in patients with bipolar disorder and schizophrenia. J Clin Psychiatry. 2006;67 Suppl 9:25–30, discussion 36–42. | ||
Kroeze WK, Hufeisen SJ, Popadak BA, et al. H1-histamine receptor affinity predicts short-term weight gain for typical and atypical antipsychotic drugs. Neuropsychopharmacology. 2003;28(3):519–526. | ||
Wirshing DA, Wirshing WC, Kysar L, et al. Novel antipsychotics: comparison of weight gain liabilities. J Clin Psychiatry. 1999;60(6):358–363. | ||
Li Z, Ichikawa J, Dai J, Meltzer HY. Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur J Pharmacol. 2004;493(1–2):75–83. | ||
Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology. 2009;34 Suppl 1:S208–S216. | ||
Calabrese F, Molteni R, Riva MA. Antistress properties of antidepressant drugs and their clinical implications. Pharmacol Ther. 2011;132(1):39–56. | ||
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455(7215):894–902. | ||
Cheng MC, Liao DL, Hsiung CA, Chen CY, Liao YC, Chen CH. Chronic treatment with aripiprazole induces differential gene expression in the rat frontal cortex. Int J Neuropsychopharmacol. 2008;11(2):207–216. | ||
Park SW, Lee JG, Ha EK, et al. Differential effects of aripiprazole and haloperidol on BDNF-mediated signal changes in SH-SY5Y cells. Eur Neuropsychopharmacol. 2009;19(5):356–362. | ||
Park SW, Phuong VT, Lee CH, et al. Effects of antipsychotic drugs on BDNF, GSK-3beta, and beta-catenin expression in rats subjected to immobilization stress. Neurosci Res. 2011;71(4):335–340. | ||
Fumagalli F, Molteni R, Roceri M, et al. Effect of antipsychotic drugs on brain-derived neurotrophic factor expression under reduced N-methyl-D-aspartate receptor activity. J Neurosci Res. 2003;72(5):622–628. | ||
Park SW, Lee SK, Kim JM, Yoon JS, Kim YH. Effects of quetiapine on the brain-derived neurotrophic factor expression in the hippocampus and neocortex of rats. Neurosci Lett. 2006;402(1–2):25–29. | ||
Luoni A, Fumagalli F, Racagni G, Riva MA. Repeated aripiprazole treatment regulates Bdnf, Arc and Npas4 expression under basal condition as well as after an acute swim stress in the rat brain. Pharmacol Res. 2014;80:1–8. | ||
Yoshimura R, Hori H, Ikenouchi-Sugita A, et al. Aripiprazole altered plasma levels of brain-derived neurotrophic factor and catecholamine metabolites in first-episode untreated Japanese schizophrenia patients. Hum Psychopharmacol. 2012;27(1):33–38. | ||
Luoni A, Hulsken S, Cazzaniga G, Racagni G, Homberg JR, Riva MA. Behavioural and neuroplastic properties of chronic lurasidone treatment in serotonin transporter knockout rats. Int J Neuropsychopharmacol. 2013;16(6):1319–1330. | ||
Sparshatt A, Taylor D, Patel MX, Kapur S. A systematic review of aripiprazole-dose, plasma concentration, receptor occupancy, and response: implications for therapeutic drug monitoring. J Clin Psychiatry. 2010;71(11):1447–1456. | ||
Gopalakrishna G, Aggarwal A, Lauriello J. Long-acting injectable aripiprazole: how might it fit in our tool box? Clin Schizophr Relat Psychoses. 2013;7(2):87–92. | ||
Shirley M, Perry CM. Aripiprazole (ABILIFY MAINTENA®): a review of its use as maintenance treatment for adult patients with schizophrenia. Drugs. 2014;74(10):1097–1110. | ||
Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. | ||
Gould TD, Manji HK. Glycogen synthase kinase-3: a putative molecular target for lithium mimetic drugs. Neuropsychopharmacology. 2005;30(7):1223–1237. | ||
Schloesser RJ, Huang J, Klein PS, Manji HK. Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 2008;33(1):110–133. | ||
Calabrese F, Luoni A, Guidotti G, Racagni G, Fumagalli F, Riva MA. Modulation of neuronal plasticity following chronic concomitant administration of the novel antipsychotic lurasidone with the mood stabilizer valproic acid. Psychopharmacology (Berl). 2013;226(1):101–112. | ||
Citrome L, Josiassen R, Bark N, Salazar DE, Mallikaarjun S. Pharmacokinetics of aripiprazole and concomitant lithium and valproate. J Clin Pharmacol. 2005;45(1):89–93. | ||
Citrome L, Macher JP, Salazar DE, Mallikaarjun S, Boulton DW. Pharmacokinetics of aripiprazole and concomitant carbamazepine. J Clin Psychopharmacol. 2007;27(3):279–283. | ||
Feltenstein MW, Do PH, See RE. Repeated aripiprazole administration attenuates cocaine seeking in a rat model of relapse. Psychopharmacology (Berl). 2009;207(3):401–411. | ||
Brunetti M, Di Tizio L, Dezi S, Pozzi G, Grandinetti P, Martinotti G. Aripiprazole, alcohol and substance abuse: a review. Eur Rev Med Pharmacol Sci. 2012;16(10):1346–1354. | ||
Voronin K, Randall P, Myrick H, Anton R. Aripiprazole effects on alcohol consumption and subjective reports in a clinical laboratory paradigm–possible influence of self-control. Alcohol Clin Exp Res. 2008;32(11):1954–1961. | ||
Martinotti G, Di Nicola M, Di Giannantonio M, Janiri L. Aripiprazole in the treatment of patients with alcohol dependence: a double-blind, comparison trial vs naltrexone. J Psychopharmacol. 2009;23(2):123–129. | ||
Barbui C, Signoretti A, Mule S, Boso M, Cipriani A. Does the addition of a second antipsychotic drug improve clozapine treatment? Schizophr Bull. 2009;35(2):458–468. | ||
Miller DD. Review and management of clozapine side effects. J Clin Psychiatry. 2000;61 Suppl 8:14–17, discussion 18–19. | ||
Shim JC, Shin JG, Kelly DL, et al. Adjunctive treatment with a dopamine partial agonist, aripiprazole, for antipsychotic-induced hyperprolactinemia: a placebo-controlled trial. Am J Psychiatry. 2007;164(9):1404–1410. | ||
Byerly MJ, Marcus RN, Tran QV, Eudicone JM, Whitehead R, Baker RA. Effects of aripiprazole on prolactin levels in subjects with schizophrenia during cross-titration with risperidone or olanzapine: analysis of a randomized, open-label study. Schizophr Res. 2009;107(2–3):218–222. | ||
Englisch S, Zink M. Combined antipsychotic treatment involving clozapine and aripiprazole. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(6):1386–1392. | ||
Fleischhacker WW, Heikkinen ME, Olie JP, et al. Effects of adjunctive treatment with aripiprazole on body weight and clinical efficacy in schizophrenia patients treated with clozapine: a randomized, double-blind, placebo-controlled trial. Int J Neuropsychopharmacol. 2010;13(8):1115–1125. | ||
Chernoloz O, El Mansari M, Blier P. Electrophysiological studies in the rat brain on the basis for aripiprazole augmentation of antidepressants in major depressive disorder. Psychopharmacology (Berl). 2009;206(2):335–344. | ||
Croxtall JD, Scott LJ. Olanzapine/fluoxetine: a review of its use in patients with treatment-resistant major depressive disorder. CNS Drugs. 2010;24(3):245–262. | ||
Weiden PJ, Buckley PF. Reducing the burden of side effects during long-term antipsychotic therapy: the role of “switching” medications. J Clin Psychiatry. 2007;68 Suppl 6:14–23. | ||
Fagiolini A, Brugnoli R, Di Sciascio G, De Filippis S, Maina G. Switching antipsychotic medication to aripiprazole: position paper by a panel of Italian psychiatrists. Expert Opin Pharmacother. 2015;16(5):727–737. | ||
Newcomer JW, Campos JA, Marcus RN, et al. A multicenter, randomized, double-blind study of the effects of aripiprazole in overweight subjects with schizophrenia or schizoaffective disorder switched from olanzapine. J Clin Psychiatry. 2008;69(7):1046–1056. | ||
Weiden PJ. Switching antipsychotic medications: not enough, too often, or just right? Am J Psychiatry. 2011;168(9):882–884. | ||
Buckley PF, Correll CU. Strategies for dosing and switching antipsychotics for optimal clinical management. J Clin Psychiatry. 2008;69 Suppl 1:4–17. | ||
Kane JM, Sanchez R, Perry PP, et al. Aripiprazole intramuscular depot as maintenance treatment in patients with schizophrenia: a 52-week, multicenter, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2012;73(5):617–624. | ||
Fleischhacker WW, Sanchez R, Perry PP, et al. Aripiprazole once-monthly for treatment of schizophrenia: double-blind, randomised, non-inferiority study. Br J Psychiatry. 2014;205(2);135–144. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.