")
Back to Journals » OncoTargets and Therapy » Volume 12
ARHGAP4 mediates the Warburg effect in pancreatic cancer through the mTOR and HIF-1α signaling pathways
Authors Shen Y, Chen G, Zhuang L, Xu L, Lin J, Liu L
Received 4 March 2019
Accepted for publication 20 May 2019
Published 28 June 2019 Volume 2019:12 Pages 5003—5012
DOI https://doi.org/10.2147/OTT.S207560
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr XuYu Yang
Yehua Shen,1,2,* Gang Chen,3,* Liping Zhuang,1,2 Litao Xu,1,2 Junhua Lin,1,2 Luming Liu1,2
1Department of Integrative Oncology, Fudan University Shanghai Cancer Center, Shanghai 200032, People’s Republic of China; 2Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, People’s Republic of China; 3Department of Pediatric Cardiothoracic Surgery, Children’s Hospital of Fudan University, Shanghai 201102, People’s Republic of China
*These authors contributed equally to this work
Objective: The phenomenon that cancer cells avidly exhibit glycolysis with lactate secretion and decrease in mitochondrial activity under aerobic conditions is known historically as the Warburg effect. Rho GTPase-activating protein 4 (ARHGAP4) is an important negative regulator of the Rho signaling pathway that was associated with the tumorigenesis. Our study aims to determine the function of ARHGAP4 in controlling the glycolytic process of pancreatic cancer in vitro and possible molecular mechanism involved.
Methods: ARHGAP4 and PKM2 expressions in pancreatic cancer tissues were measured by immunohistochemistry. Human pancreatic cancer cells transfected with ARHGAP4 expressing lentivirus or siRNA were treated with either mTOR inhibitor (Rapamycin) or HIF-1α inhibitor (YC-1), and the effects were analyzed on cell viability, glucose uptake, lactate release, and the levels of ARHGAP4, p-mTOR, mTOR, PKM2, and HIF-1α expression.
Results: Our findings showed that ARHGAP4 and PKM2 expressions were, respectively, down-regulated and up-regulated in pancreatic cancer tissues. Overexpression of ARHGAP4 significantly inhibited cell viability, glucose uptake, lactate release, PKM2 expression, and activation of mTOR and HIF-1α signaling pathways in pancreatic cancer cells while ARHGAP4 silencing and treatment of Rapamycin or YC-1 showed inverse effects. Additionally, ARHGAP4 downregulation induced cell morphology of pancreatic cancer was inhibited by Rapamycin or YC-1 treatment.
Conclusion: These findings suggest that mTOR and HIF-1α signaling pathways can regulate the ARHGAP4-mediated glycolytic process of pancreatic cancer.
Keywords: pancreatic cancer, ARHGAP4, glycolysis, cell viability
Introduction
Pancreatic cancer is the leading cause of cancer-related deaths with a high risk of malignancy and poor prognosis. Because of its difficulty in early diagnosis, it is usually only detected in the late stages, and the 5-year survival rate for patients with pancreatic cancer is less than 5%.1,2 At present, the treatment of pancreatic cancer mainly includes surgical resection, chemotherapy, and radiotherapy. Radical resection can be performed in less than 20% of the patients who will also face the risk of metastasis and recurrence3 while the other patients are only able to undergo palliative radiotherapy and chemotherapy due to tumor local infiltration or distant metastasis.4 Meanwhile, pancreatic cancer is not sensitive to current treatments, which do not significantly prolong survival.5 It is therefore important to explore new therapeutic methods to further improve the curative effect of pancreatic cancer.
The rapid proliferation, invasion, and migration of tumor cells are closely related to their unique energy metabolism. Normal cells undergo glycolysis only under hypoxic conditions while tumor cells prefer glycolysis even under aerobic conditions, consuming large numbers of glucose and leading to lactate secretion, a phenomenon termed “the Warburg effect”,6,7 which is considered to be a basal metabolic change in malignant transformation.8 This metabolic adaptation benefits cancer cells in surviving through hypoxic conditions, commonly found in tumors, and to support their anabolic requirements, which differs remarkably from normal cells, thus providing a new therapeutic window. PI3K signaling pathway with mTOR as the core kinase plays a critical role in cell metabolism, growth, and autophagy.9,10 Transcription factor HIF-1α as a downstream effector of mTOR signaling regulates cell adaptability to hypoxia and promotes glycolysis metabolism by regulating the production of various proteins and enzymes.11 Inactivating mTOR signaling suppressed the HIF-1α activity, resulting in suppression of glycolytic metabolism in pancreatic cancer and colon cancer cells,12,13 suggesting that the function of mTOR and HIF-1α may promote the glycolytic progress in cancer tumorigenesis.
PKM2 is an isoenzyme of the glycolytic enzyme pyruvate kinase. Increased PKM2 expression is required for cancer cells undergo aerobic glycolysis, thus playing a critical role in the regulation of cancer metabolism.14,15 Inhibition of the mTOR and HIF1α signaling pathways induces impairment of glucose uptake and decreases PKM2 expression, suppressing the glycolytic phenotype in melanoma cells.16 Up-regulation of PKM2 expression through PI3K/mTOR-mediated HIF1α induction enhances aerobic glycolysis in cancer cells.17 We, therefore, figure out the hypothesis that mTOR and HIF-1α signaling pathways positively regulate glycolytic phenotype in pancreatic cancer. However, the regulation of mTOR and HIF-1α signaling pathways remains unclear.
Rho GTPase-activating protein 4 (ARHGAP4) belongs to the small GTPase family, which can hydrolyze the active GTP into inactive GDP and negatively regulate RhoA protein, therefore, being strongly associated with the onset and progression of cancer. ARHGAP4 mediates axon outgrowth and cell motility18 and is up-regulated in colorectal cancer and correlated with metastasis, T/N stage, and clinical stage.19 Except for our previous report suggesting ARHGAP4 regulates the cell migration and invasion of pancreatic cancer by the HDAC2/β-catenin signaling pathway,20 the cellular functions of ARHGAP4 in pancreatic cancer still not fully understood. We for the first time investigated the role of ARHGAP4 in regulating pancreatic cancer metabolism and reported that mTOR and HIF-1α signaling pathways modulated the weakened effects of ARHGAP4 on aerobic glycolysis.
Materials and methods
Study subjects
The human pancreatic cancer and normal tissue microarrays used in this study were prepared by Shanghai Outdo Biotech Co., Ltd. (Shanghai, China). The study was approved by the Ethics Committee of Fudan University Shanghai Cancer Center.
Bioinformatics
RNA-sequencing dataset of pancreatic cancer cohort was obtained from The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/). To identify the pathways that were significantly enriched between ARHGAP4 high and low, gene set enrichment analysis (GSEA) algorithm was used.21
Immunohistochemistry
Tissue slides (4–7 μm) were initial treatment for deparaffinization, rehydration, and antigen-retrieval, followed by 3% H2O2 incubation for 10 mins. Sections were incubated with anti-ARHGAP4 (1:100; ab198696; Abcam, USA) or anti-PKM2 primary antibody (1:500; ab150377; Abcam), and then incubated with horseradish peroxidase (HRP)-labeled IgG secondary antibody (Beyotime Institute of Biotechnology, Shanghai, China). Fields from each slide were examined and photographed under a light microscopy (×200).
Cell culture
Human pancreatic cancer cell lines PANC-1, BxPC-3, and SW1990 were obtained from the American Type Culture Collection (ATCC, Wesel, Germany). Cells were maintained in RPMI1640 (Hyclone, USA; for BxPC-3 cells) or DMEM (high glucose; Hyclone; for SW1990 and PANC-1 cells) containing 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin in an atmosphere of 5% CO2 and 95% air at 37°C.
Cell transfection
Three siRNAs targeting human ARHGAP4 (siRNA-1, position 42–60, 5ʹ-GGCTGAGTATGAGACGCAA-3ʹ; siRNA-2, position 48–66, 5ʹ-GTATGAGACGCAAGTCAAA-3ʹ; siRNA-3, position 1697–1715, 5ʹ-GCTGCATTCGCTTCATCAA-3ʹ) were produced and transfected into the PANC-1 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instruction. The coding sequence of ARHGAP4 was synthesized and integrated into pLVX-Puro lentivirus. To generate high-titer lentivirus, the lentivirus encoding the target gene and the pMD2G and psPAX2 packaging plasmids were cotransfected into HEK 293T cells using Lipofectamine 2000. Forty-eight hours after transfection, viral particles in cell culture medium were collected and transduced SW1990 and BxPC-3cells. SW1990 and BxPC-3 cells with blank pLVX-Puro lentivirus transduction and PANC-1 cells with scramble siRNA (siNC) transfection were used as a negative control.
Group
SW1990 and BxPC-3 cells were transduced with pLVX-Puro-ARHGAP4 or blank pLVX-Puro. PANC-1 cells were transfected with ARHGAP4 siRNA or siNC in the presence of either mTOR inhibitor (Rapamycin, 10 nM; Selleck Chemicals, Houston, TX, USA) or HIF-1α inhibitor (YC-1, 20 μM; Selleck Chemicals).
CCK-8 assay
PANC-1, SW1990, and BxPC-3 cells (3×103 cell/well) were cultured in the 96-well plate and maintained at 37°C overnight. After 0, 24, 48, and 72 hrs treatment, Cell Counting Kit-8 (CCK-8) solution (10 μL) was added into each well as the manufactory protocol for another 1 hr, and the cell viability was determined using a microplate reader at OD450nm.
Flow cytometry analysis
BxPC-3, SW1990, and PANC-1 cells (5×105 cell/well) were cultured in the six-well plate and maintained for one day at 37°C. Forty-eight hours after treatment, cells were cultured in RPMI1640 or DMEM for 3 hrs, and then washed with Krebs-Ringer Bicarbonate Buffer containing 2% bovine serum albumin (BSA), followed by incubation with RPMI1640 or DMEM containing 100 μM of 2-NBDG (Cayman Chemical, Ann Arbor, Michigan, USA) for 45 mins. Glucose uptake in cells was measured by flow cytometry (FACSCalibur, BD Biosciences, USA).
Metabolic assays
BxPC-3, SW1990, and PANC-1 cells (5×105 cell/well) were grown in the six-well plate and maintained for one day at 37°C. Forty-eight hours after treatment, lactate release from the cells was determined by Lactic Acid assay kit (Nanjing Jiancheng Bioengineering Institute, China) following the manufacturer’s instruction.
Quantitative RT-PCR
Total RNA was extracted from the BxPC-3, SW1990, and PANC-1 cells using TRIzol reagent (Life Technologies, Inc., Waltham, MA, USA). cDNA was synthesized using the PrimeScript kit (Takara Biotechnology, Dalian, China) based on the manufacturer’s protocol. The Quantitative RT-PCR using SYBR green PCR master mix (Applied Biosystems, Foster, CA, USA) was performed in an ABI 9700 real-time PCR system (Applied Biosystem). The primers used for PCR are as follows: ARHGAP4-F: 5ʹ-CTACAACCTGGCCGTGTGCTTC-3ʹ, ARHGAP4-R: 5ʹ-CTTCCAGCTCCGGCTCATTGTC-3ʹ; HIF-1α-F: 5ʹ-CTGGCTACAATACTGCACAAAC-3ʹ, HIF-1α-R: 5ʹ-ATGCTACTGCAATGCAATGG-3ʹ; PKM2-F: 5ʹ-AGCAAGAAGGGTGTGAAC-3ʹ, PKM2-R: 5ʹ-CGGATGAATGACGCAAAC-3ʹ; GAPDH-F: 5ʹ-AATCCCATCACCATCTTC-3ʹ, GAPDH-R: 5ʹ-AGGCTGTTGTCATACTTC-3ʹ. The fold changes of ARHGAP4, HIF-1α, and PKM2 mRNA were determined by the 2−ΔΔCT method.
Western blotting
Total protein was extracted from pancreatic cancer tissues and cell lines by RIPA buffer (Solarbio, Beijing, China) and separated by 10% SDS-PAGE before being transferred onto a PVDF membrane (Millipore, Bedford, MA, USA). The membrane was then blocked with 5% BSA and probed using the primary antibodies against ARHGAP4 (Abcam), p-mTOR (Cell Signaling Technology, Beverly, MA, USA), mTOR (Cell Signaling Technology), PKM2 (Cell Signaling Technology), HIF-1α (Abcam), and GAPDH (Cell Signaling Technology). The detection was performed using HRP-labeled secondary antibody (Beyotime Institute of Biotechnology). GAPDH levels are shown as loading controls.
Statistical analysis
All the results were represented as the mean ± SD. All experiments were independently repeated thrice. Data were performed by one-way ANOVA analysis followed by Dunnett post-test, two-way ANOVA analysis followed by Bonferroni post-test, and Student’s t-test using SPSS version 23.0 (SPSS Inc., Chicago, IL, USA). The level of statistical significance was set at P<0.05.
Results
ARHGAP4 expression in pancreatic cancer tissues
We first investigated ARHGAP4 protein expressions on tissue microarrays by immunohistochemistry. It showed that ARHGAP4 protein expressions in pancreatic cancer tissues were markedly down-regulated (Figure 1A), and 50% (40/80) pancreatic cancer tissues displayed the negative or lower expression of ARHGAP4 (less than 25% of the tumor cells were positively stained) (Figure 1B). Western blot and Real-time PCR analysis demonstrated PANC-1 cells with a higher ARHGAP4 expression when compared with SW1990 and BxPC-3 cells (Figure 1C and D).
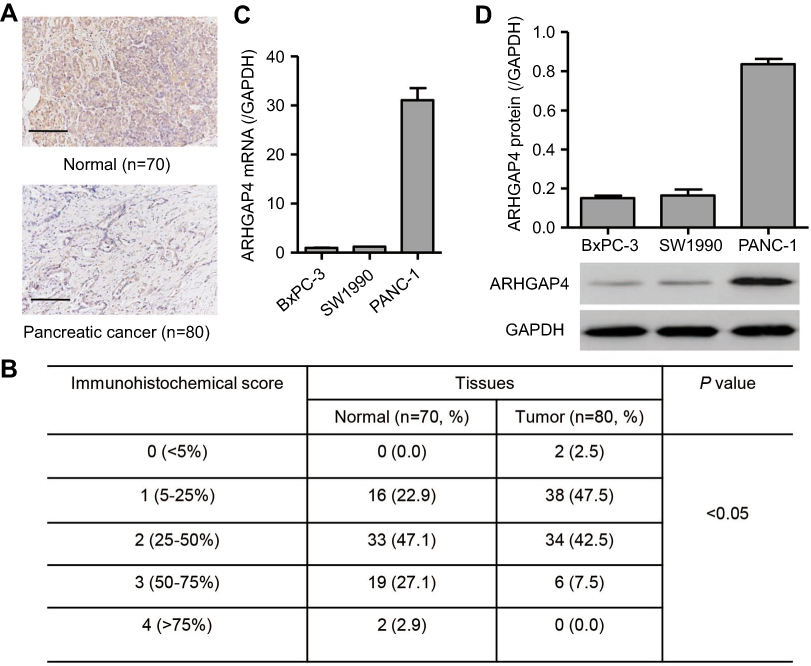 |
Figure 1 ARHGAP4 expression in pancreatic cancer. Immunohistochemical analysis (A, B) was performed to examine the expression of ARHGAP4 on tissue microarrays containing pancreatic cancer tissues (n=80) and adjacent-normal pancreatic tissues (n=70). Scale bars: 100 μm. Real-time PCR (C) and Western blot analysis (D) were performed to detect the expression of ARHGAP4 in pancreatic cancer cell lines, including BxPC-3, SW1990, and PANC-1 cells. |
ARHGAP4 up-regulation inhibited the cell viability
To investigate the function of ARHGAP4 in pancreatic cancer, pLVX-Puro-ARHGAP4 lentivirus or blank pLVX-Puro lentivirus (vector) was transduced into the SW1990 and BxPC-3 cells, and the cell viability was measured. As shown in Figure 2A and B, pLVX-Puro-ARHGAP4 lentivirus transduction in BxPC-3 and SW1990 cells showed 1.74-fold and 1.50-fold increase in ARHGAP4 protein expression, respectively, compared with vector groups. Moreover, CCK-8 analysis demonstrated that 24, 48, and 72 hrs after pLVX-Puro-ARHGAP4 lentivirus transduction resulted in 10.1%, 28.5%, and 33.8% decrease in cell viability of BxPC-3 cells and in 7.6%, 23.4%, and 27.0% decrease in cell viability of SW1990 cells, compared to the vector groups (Figure 2C and D).
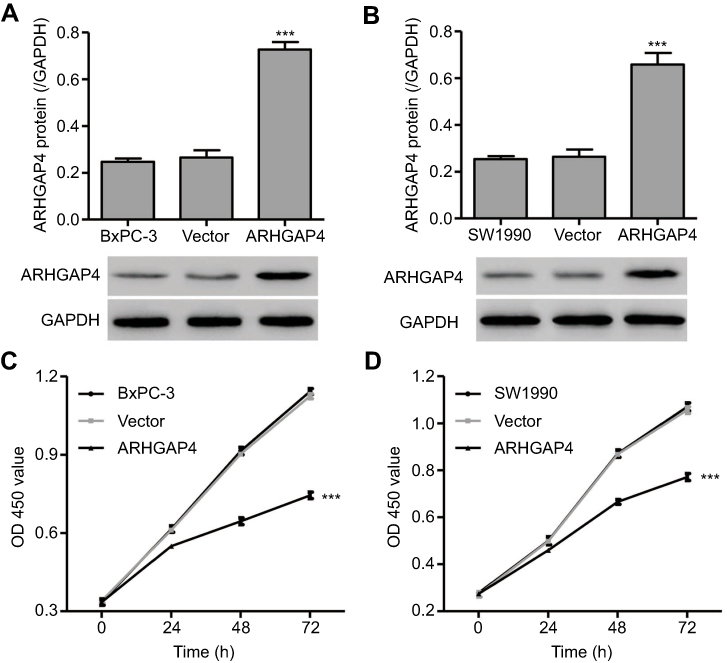 |
Figure 2 ARHGAP4 up-regulation inhibited cell viability of BxPC-3 and SW1990. BxPC-3 and SW1990 cells were transduced with pLVX-Puro-ARHGAP4 lentivirus or blank lentivirus (vector), and ARHGAP4 expression and cell viability were measured by Western blot analysis (A, B) and CCK-8 assay (C, D), respectively. ***P<0.001 compared with vector. |
ARHGAP4 up-regulation inhibited glucose uptake and lactate release
Active glucose uptake and more lactate production are characteristics of elevated glycolysis and were therefore measured in pancreatic cancer cells. Flow cytometry analysis revealed that pLVX-Puro-ARHGAP4 lentivirus transduction significantly inhibited glucose uptake by 36.4% and 36.2% in BxPC-3 and SW1990 cells, respectively, compared with vector groups (Figure 3A, B, D, and E). Additionally, pLVX-Puro-ARHGAP4 lentivirus transduction also significantly inhibited lactate release by 52.2% and 50.7% in BxPC-3 and SW1990 cells, respectively, compared with vector groups (Figure 3C and F).
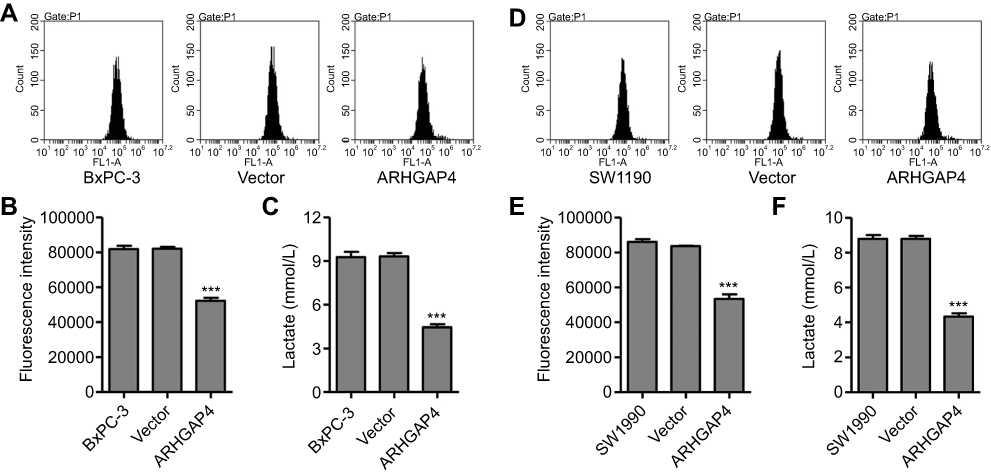 |
Figure 3 ARHGAP4 up-regulation inhibited glucose uptake and lactate release in BxPC-3 and SW1990 cells. BxPC-3 and SW1990 cells were transduced with pLVX-Puro-ARHGAP4 lentivirus or blank lentivirus (vector), and glucose uptake (A, B, D, E) and lactate release (C, F) were measured. ***P<0.001 compared with vector. |
ARHGAP4 up-regulation inhibited mTOR and HIF-1α signaling pathways
In view of the GSEA data that higher expressions of ARHGAP4 were negatively correlated with mTOR and HIF signaling pathways (Figure 4A), we then investigated possible mechanisms by which ARHGAP4 inhibited glycolytic process in pancreatic cancer. We found that pLVX-Puro-ARHGAP4 lentivirus transduction in SW1990 and BxPC-3 cells showed the decrease in the expression of p-mTOR compared with vector groups, but had no effect on that of mTOR (Figure 4B and E). Moreover, it also resulted in the decreases in the HIF-1α and PKM2 expressions in both SW1990 and BxPC-3 cells, compared with vector groups (Figure 4C, D, F, and G). These results suggest that ARHGAP4 is associated with mTOR and HIF-1α signaling pathways.
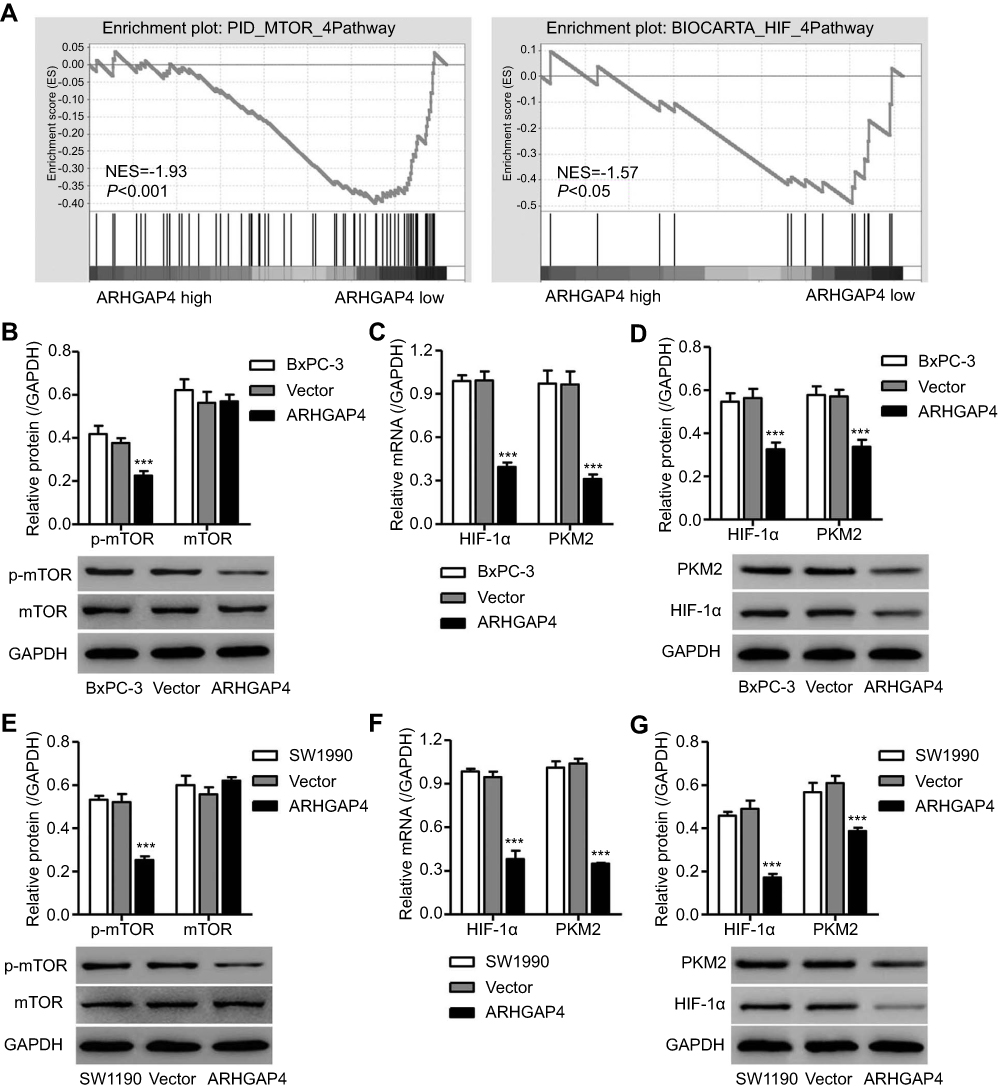 |
Figure 4 ARHGAP4 up-regulation inhibited mTOR and HIF-1α signaling pathways in BxPC-3 and SW1990 cells. (A) GSEA demonstrated the genes of mTOR and HIF signaling pathways were more correlated with patients with ARHGAP4 low vs ARHGAP4 high. NES, normalized enrichment score. BxPC-3 and SW1990 cells were transduced with pLVX-Puro-ARHGAP4 lentivirus or blank lentivirus (vector), and the p-mTOR, mTOR, HIF-1α, and PKM2 expressions were measured by Western blot (B, D, E, G) and Real-time PCR (C, F). ***P<0.001 compared with vector. |
Involvement of mTOR and HIF-1α signaling pathways in the functions of ARHGAP4
To clarify the contribution of mTOR and HIF-1α signaling pathways, PANC-1 cells, which displayed a relatively high level of ARHGAP4, were transfected with ARHGAP4 siRNA or siNC and treated with either mTOR inhibitor Rapamycin or HIF-1α inhibitor YC-1. As shown in Figure 5A, siARHGAP4-1, siARHGAP4-2, or siARHGAP4-3 transfection significantly inhibited ARHGAP4 expression by 79.2%, 34.2%, and 24.6% compared with siNC groups, respectively, and siARHGAP4-1 was therefore chosen for further experiments. siARHGAP4-1 transfection in PANC-1 cells significantly induced cell viability (Figure 5B), glucose uptake, (Figure 5C and D), lactate release (Figure 5E), PKM2 expression, and activation of mTOR and HIF-1α signaling pathways (Figure 5F–H) while Rapamycin or YC-1 had inverse effects. Additionally, Rapamycin or YC-1 treatment attenuated the effects of siARHGAP4-1 transfection in PANC-1 cells (Figure 5B–H). These data suggest that mTOR and HIF-1α signaling pathways may be therefore potential downstream regulators of ARHGAP4 in pancreatic cancer.
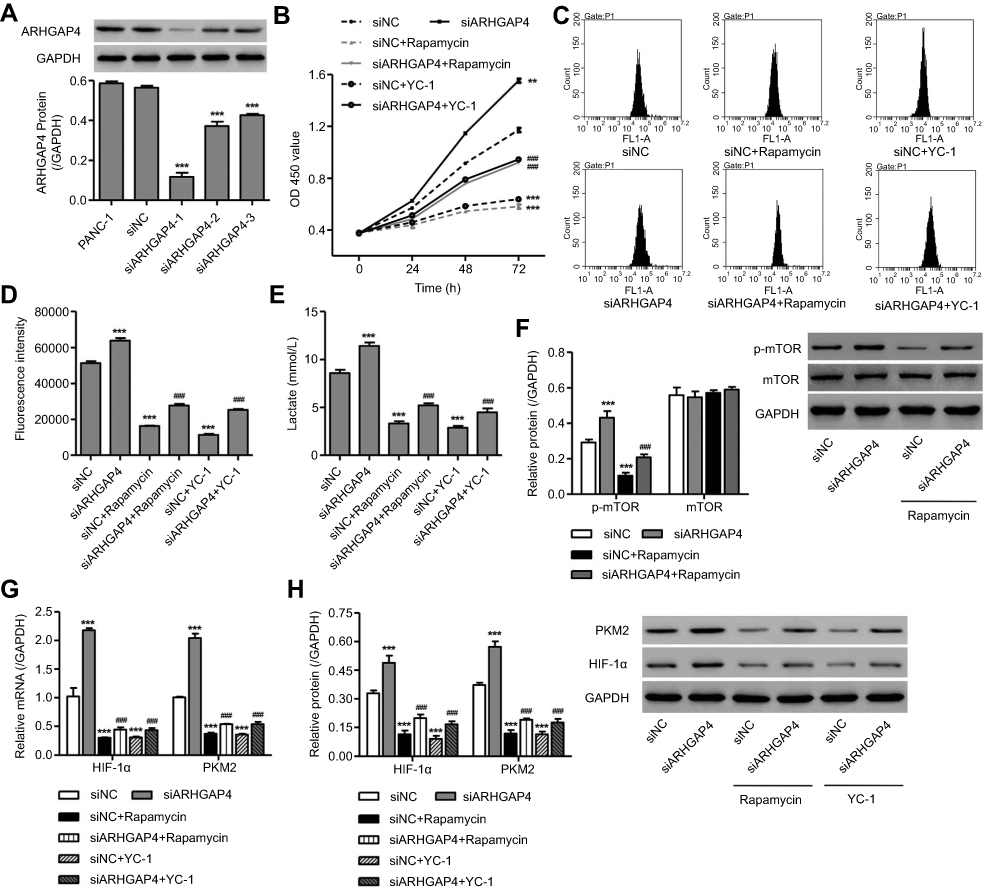 |
Figure 5 ARHGAP4 downregulation induced cell viability, glucose uptake, and lactate release through mTOR and HIF-1α signaling pathways in PANC-1 cells. PANC-1 cells were transfected with three siRNAs targeting human ARHGAP4 or scramble siRNA (siNC), and the ARHGAP4 expression was measured by Western blot (A). Cell viability (B), glucose uptake (C, D), lactic acid production (E) and protein levels of p-mTOR, mTOR, HIF-1α, and PKM2 (F–H) in PANC-1 cells with siRNA-1 or siNC transfection were determined in the presence of either mTOR inhibitor (Rapamycin, 10 nM) or HIF-1α inhibitor (YC-1, 20 μM). ***P<0.001 compared with siNC. ###P<0.001 compared with siARHGAP4. |
PKM2 expression in pancreatic cancer tissues
We next determined PKM2 protein levels on tissue microarrays using immunohistochemistry. It showed that PKM2 protein expressions were markedly up-regulated in pancreatic cancer tissues (Figure 6A), and 95% (76/80) pancreatic cancer tissues displayed a higher expression of PKM2 (more than 25% of the tumor cells were positively stained) (Figure 6B). These data further supported the findings in pancreatic cancer cells.
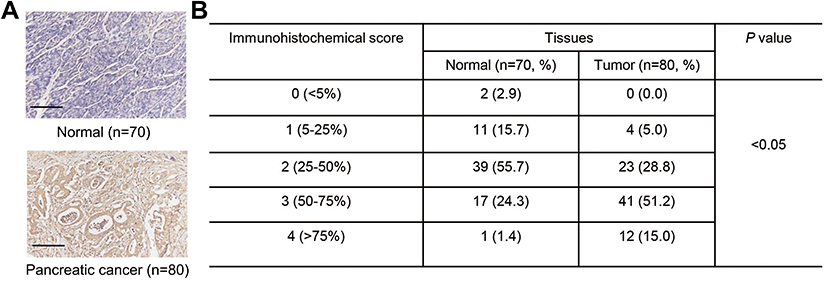 |
Figure 6 PKM2 expression in pancreatic cancer tissues. Immunohistochemical analysis (A, B) was performed to examine the expression of PKM2 on tissue microarrays containing pancreatic cancer tissues (n=80) and adjacent-normal pancreatic tissues (n=70). Scale bars: 100 μm. |
Discussion
In recent years, the metabolic transformation of tumor cells has attracted much attention due to its great potential as a therapeutic target. It is necessary to screen for more negative effects of tumor suppressors on cancer metabolism. Metabolic enzymes may be the focus of treatment since they play an important role in regulating metabolic pathways in cancer cells. In this study, we investigated the role of ARHGAP4 in the regulation of aerobic glycolysis in pancreatic cancer cells and the underlying mechanism involved. We provided three pancreatic cancer cell lines to support a crucial role for ARHGAP4 in regulating aerobic glycolysis by the silenced ARHGAP4 and overexpressed ARHGAP4. In pancreatic cancer tissues, ARHGAP4 expression was decreased while PKM2 expression was increased. Overexpression of ARHGAP4 inhibited cell viability, glucose uptake, lactate release, and expression of p-mTOR, HIF-1α, and PKM2. However, ARHGAP4 silencing demonstrated an opposite effect, which was reversed by inactivation of mTOR and HIF-1α signaling pathways.
Rho GTPase-activating protein (ARHGAP) family of genes is associated with cancer because their genetic changes cause carcinogenesis through the dysregulation of Rho/Rac/Cdc42-like GTPases.22 Results presented here unravel decreased ARHGAP4 protein expression in tissues of pancreatic cancer, and overexpression of ARHGAP4 inhibited cell viability while silence of ARHGAP4 promoted it, suggesting its important role in the development of pancreatic cancer. ARHGAP6 overexpression inhibited cell proliferation and induced cell cycle arrest and cell apoptosis in cervical carcinoma.23 Lower ARHGAP24 expression was found in renal cell carcinoma tissues, promoted cell growth, and inhibited cell apoptosis.24 However, their effects on cancer metabolism remained un-elucidated.
Abnormal glucose metabolism is an important feature of tumor cells, which is different from normal cells. Warburg effect is considered to be the underlying metabolic change in malignant transformation,25 and the enhancement of glycolysis in tumor cells is the main way to generate ATP as energy metabolism and has gradually become an important point for cancer diagnosis and treatment.26 RhoA deficiency prevented the development of allergic airway inflammation, impaired TH2 cell differentiation, and diminished glycolysis in activated T cells.27 Given the function of ARHGAPs in regulating RhoA, we hypothesize that ARHGAP4 may also involve in the development of pancreatic cancer through regulating aerobic glycolysis. Overexpression of ARHGAP4 significantly inhibited the aerobic glycolysis, evidenced by the decreases in glucose uptake and lactate release while silence of ARHGAP4 promoted it. Some of the previous evidences for the correlation between aerobic glycolysis and oncogenes are the stimulation of lactate release and glucose uptake.28 High glucose uptake and lactate production were observed in cancer cells with curcumin treatment, showing decreased Warburg effect.7
Considering the GSEA data that ARHGAP4 was correlated with mTOR and HIF signaling pathways, which have been found to be associated with cell growth and aerobic glycolysis,7,29 we further investigated the underlying mechanism in the content of mTOR and HIF signaling pathways. Overexpression of ARHGAP4 significantly inhibited PKM2, HIF-1α, and p-mTOR expression while the silence of ARHGAP4 promoted it. Importantly, mTOR inhibitor Rapamycin and HIF-1α inhibitor YC-1 significantly inhibited glucose uptake and lactate release as well as PKM2, HIF-1α, and p-mTOR expressions induced by ARHGAP4 silencing, suggesting that ARHGAP4 inhibits aerobic glycolysis through inactivation of mTOR and HIF-1α signaling pathways. mTOR has been recently found to promote tumor growth and mediate cancer cell aerobic glycolysis by up-regulating PKM2,7,30 which functions as a transcriptional coactivator to promote aerobic glycolysis and proliferation in cancer.31 Hyperactivation of HIF-1α by modulating mTOR signaling activates the expression of PKM27 and is accompanied by the increase in aerobic glycolysis in tamoxifen-resistant breast cancer.29 The modulation of mTOR on the levels of HIF-1α and PKM2 underlines the important role of mTOR signaling in the regulation of cancer metabolism. PKM2 was highly expressed in tissues of pancreatic cancer,32 and its downregulation suppressed the migration, proliferation, and glycolytic activities of pancreatic ductal adenocarcinoma,33 suggesting the involvement of PKM2 in pancreatic cancer. Our data also demonstrated the increased PKM2 expression in pancreatic cancer tissues; however, its role in regulation of ARHGAP4 mediated aerobic glycolysis needs to be further confirmed.
Conclusion
In conclusion, we revealed that ARHGAP4 was expressed lowly while PKM2 was expressed highly in pancreatic cancer tissues, and ARHGAP4 regulated cell viability and aerobic glycolysis in pancreatic cancer through mTOR and HIF-1α signaling pathways. Our work could help to inspire research for targeted therapies against pancreatic cancer.
Acknowledgments
The study was supported by funding from the National Natural Science Foundation of China (81573757/H2902).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21:7988–8003. doi:10.3748/wjg.v21.i26.7988
2. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–1049. doi:10.1056/NEJMra1404198
3. Rucki AA, Foley K, Zhang P, et al. Heterogeneous stromal signaling within the tumor microenvironment controls the metastasis of pancreatic cancer. Cancer Res. 2017;77:41–52. doi:10.1158/0008-5472.CAN-16-1383
4. Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318–348. doi:10.3322/caac.21190
5. Paulson AS, Tran Cao HS, Tempero MA, Lowy AM. Therapeutic advances in pancreatic cancer. Gastroenterology. 2013;144:1316–1326. doi:10.1053/j.gastro.2013.01.078
6. Pathria G, Scott DA, Feng Y, et al. Targeting the Warburg effect via LDHA inhibition engages ATF4 signaling for cancer cell survival. Embo J. 2018;37:e99735. doi:10.15252/embj.201899735
7. Siddiqui FA, Prakasam G, Chattopadhyay S, et al. Curcumin decreases Warburg effect in cancer cells by down-regulating pyruvate kinase M2 via mTOR-HIF1alpha inhibition. Sci Rep. 2018;8:8323. doi:10.1038/s41598-018-25524-3
8. Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007;39:267–274. doi:10.1007/s10863-007-9086-x
9. Manthari RK, Tikka C, Ommati MM, et al. Arsenic-induced autophagy in the developing mouse cerebellum: involvement of the blood-brain barrier’s tight-junction proteins and the PI3K-Akt-mTOR signaling pathway. J Agric Food Chem. 2018;66:8602–8614. doi:10.1021/acs.jafc.8b02654
10. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi:10.1016/j.cell.2017.03.035
11. Cheng SC, Quintin J, Cramer RA, et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi:10.1126/science.1250684
12. Liu W, Zhang B, Hu Q, et al. A new facet of NDRG1 in pancreatic ductal adenocarcinoma: suppression of glycolytic metabolism. Int J Oncol. 2017;50:1792–1800. doi:10.3892/ijo.2017.3938
13. Ban HS, Kim BK, Lee H, et al. The novel hypoxia-inducible factor-1alpha inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 2017;8:e2843. doi:10.1038/cddis.2017.518
14. Vander Heiden MG, Locasale JW, Swanson KD, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329:1492–1499. doi:10.1126/science.1188015
15. Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43:969–980. doi:10.1016/j.biocel.2010.02.005
16. Laurenzana A, Chilla A, Luciani C, et al. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int J Cancer. 2017;141:1190–1200. doi:10.1002/ijc.30817
17. Iqbal MA, Siddiqui FA, Gupta V, et al. Insulin enhances metabolic capacities of cancer cells by dual regulation of glycolytic enzyme pyruvate kinase M2. Mol Cancer. 2013;12:72. doi:10.1186/1476-4598-12-72
18. Vogt DL, Gray CD, Young WS
19. Xuehu XU, Yuandong XU, Zhu Z, Shuling LI, Xiaobing WU, Liu X. Expression and clinical significance of ARHGAP4 in colorectal cancer. J Pract Med. 2017;33:705–708.
20. Shen Y, Xu L, Ning Z, et al. ARHGAP4 regulates the cell migration and invasion of pancreatic cancer by the HDAC2/beta-catenin signaling pathway. Carcinogenesis. 2019. doi:10.1093/carcin/bgz067
21. Yang X, Zhong X, Tanyi JL, et al. mir-30d Regulates multiple genes in the autophagy pathway and impairs autophagy process in human cancer cells. Bioche Biophys Res Commun. 2013;431:617–622. doi:10.1016/j.bbrc.2012.12.083
22. Katoh Y, Katoh M. Identification and characterization of ARHGAP27 gene in silico. Int J Mol Med. 2004;14:943–947.
23. Li J, Liu Y, Yin Y. Inhibitory effects of Arhgap6 on cervical carcinoma cells. Tumour Biol. 2016;37:1411–1425. doi:10.1007/s13277-015-4502-z
24. Xu G, Lu X, Huang T, Fan J. ARHGAP24 inhibits cell cycle progression, induces apoptosis and suppresses invasion in renal cell carcinoma. Oncotarget. 2016;7:51829–51839. doi:10.18632/oncotarget.10386
25. Wilde L, Roche M, Domingo-Vidal M, et al. Metabolic coupling and the reverse Warburg effect in cancer: implications for novel biomarker and anticancer agent development. Semin Oncol. 2017;44:198–203. doi:10.1053/j.seminoncol.2017.10.004
26. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152. doi:10.1186/1476-4598-12-152
27. Yang JQ, Kalim KW, Li Y, et al. RhoA orchestrates glycolysis for TH2 cell differentiation and allergic airway inflammation. J Allergy Clin Immunol. 2016;137:231–245.e234. doi:10.1016/j.jaci.2015.05.004
28. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi:10.1038/nrc3038
29. Woo YM, Shin Y, Lee EJ, et al. Inhibition of aerobic glycolysis represses Akt/mTOR/HIF-1alpha axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS One. 2015;10:e0132285. doi:10.1371/journal.pone.0132285
30. Li W, Wang J, Chen QD, et al. Insulin promotes glucose consumption via regulation of miR-99a/mTOR/PKM2 pathway. PLoS One. 2013;8:e64924. doi:10.1371/journal.pone.0064924
31. Luo W, Semenza GL. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget. 2011;2:551–556. doi:10.18632/oncotarget.299
32. Mohammad GH, Olde Damink SW, Malago M, Dhar DK, Pereira SP. Pyruvate kinase M2 and lactate dehydrogenase A are overexpressed in pancreatic cancer and correlate with poor outcome. PLoS One. 2016;11:e0151635. doi:10.1371/journal.pone.0151635
33. Yokoyama M, Tanuma N, Shibuya R, et al. Pyruvate kinase type M2 contributes to the development of pancreatic ductal adenocarcinoma by regulating the production of metabolites and reactive oxygen species. Int J Oncol. 2018;52:881–891. doi:10.3892/ijo.2018.4258
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.