Back to Journals » OncoTargets and Therapy » Volume 11
ARHGAP30 suppressed lung cancer cell proliferation, migration, and invasion through inhibition of the Wnt/β-catenin signaling pathway
Received 24 May 2018
Accepted for publication 24 July 2018
Published 24 October 2018 Volume 2018:11 Pages 7447—7457
DOI https://doi.org/10.2147/OTT.S175255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yao Dai
Xiaoliang Mao, Jichun Tong
Department of Thoracic Surgery, Yanghu Branch, Changzhou Second People’s Hospital Affiliated to Nanjing Medical University, Changzhou, China
Objective: Rho GTPase-activating protein 30 (ARHGAP30), a member of the Rho GTPase-activating proteins (Rho GAPs) family, plays an important role in the regulation of cytoskeleton organization and cell adhesion.
Materials and methods: mRNA and protein expression was assessed by quantitative real-time PCR and Western blotting, respectively. Cell Counting Kit-8 (CCK-8) and Transwell assays were conducted to detect cell proliferation, migration, and invasion.
Results: ARHGAP30 expression was downregulated in specimens and cell lines of lung cancer in comparison to non-cancerous specimens and normal bronchial epithelial cell lines, respectively. Moreover, in vitro experiments demonstrated that ARHGAP30 overexpression impeded the proliferative, migratory, and invasive abilities of lung cancer cells. Moreover, bioinformatics analysis with The Cancer Genome Atlas (TCGA) lung cancer dataset showed a negative association between ARHGAP30 expression and the Wnt signaling pathway. Enforced expression of ARHGAP30 decreased the mRNA and protein levels of β-catenin, c-Myc, matrix metalloproteinase-2 (MMP-2) and MMP-9. Besides, the β-catenin inhibitor XAV939 blocked the enhanced cell growth, migration, and invasion caused by ARHGAP30 knockdown. Thus, the Wnt/β-catenin pathway mediated the functions of ARHGAP30 in lung cancer cells.
Conclusion: ARHGAP30 acts as a tumor suppressor in lung cancer by suppressing Wnt/β-catenin signaling.
Keywords: ARHGAP30, Wnt/β-catenin, lung cancer, proliferation, metastasis
Introduction
Lung cancer remains one of the most common malignancies, with more than 1.8 million new cases diagnosed annually, and is the leading cause of cancer-related deaths in both men and women, with approximately 1.59 million deaths each year worldwide.1,2 Non-small-cell lung cancer (NSCLC) accounts for approximately 85% of lung cancer cases and is the major histological subgroup of lung cancer.3 Besides conventional treatments including surgery, chemotherapy, and radiotherapy, targeted therapy has become an important treatment option for lung cancer. However, the 5-year survival of patients with locally advanced lung cancer or metastatic disease remains unsatisfactory.4 Thus, the study of the pathogenesis of lung cancer is still of great significance.
Rho GTPases are a group of guanosine triphosphate (GTP)-binding proteins and belong to the Ras superfamily. Recent studies have found that Rho GTPases are highly expressed in various human tumors and closely related to the development, invasion, and metastasis of tumors.5 Rho GTPases, similarly as all members of the Ras superfamily, cycle between active/GTP-bound and inactive/guanosine diphosphate (GDP)-bound forms.6 The Rho GTPase-activating protein 30 (ARHGAP30) is a member of Rho GTPase-activating proteins (RhoGAPs), which are able to enhance the intrinsic hydrolysis of GTP and act as negative regulators of Rho GTPases. ARHGAP30 is a RhoA- and Rac1-specific Rho GAP, involved in the regulation of cytoskeleton organization and cell adhesion.7 A recent study suggest that ARHGAP30 is a key regulator for p53 acetylation and a potential prognostic marker for colorectal cancer.8 However, no study has focused on the functions of ARHGAP30 in lung cancer.
The Wnt/β-catenin signaling pathway is highly conserved in evolution, and studies have demonstrated that it is involved in embryonic and adult lung development.9 Furthermore, accumulated evidence has demonstrated the important regulatory role of the Wnt/β-catenin signaling pathway in the proliferation, survival, and metastasis of various cancer cells including lung cancer cells.10,11 β-Catenin is a key molecule of the Wnt/β-catenin signaling pathway. A previous article has reported that aberrant β-catenin expression is correlated with the poor prognosis of NSCLC.12,13 Numerous Wnt/β-catenin target genes have been identified, such as proliferation-related gene c-Myc, as well as metastasis key regulators matrix metalloproteinase-2 (MMP-2) and MMP-9.14,15
In the current study, we explored the expression of ARHGAP30 in lung cancer and its function in lung cancer cell proliferation, migration, and invasion. In addition, the Wnt/β-catenin signaling pathway may mediate the tumor suppressor functions of ARHGAP30 in lung cancer.
Materials and methods
Patients and tissue samples
This study was approved and carried out in accordance with the guidelines of the ethics committee of Changzhou Second People’s Hospital Affiliated to Nanjing Medical University (Changzhou, China). Written informed consent was obtained from all study participants. Lung cancer specimens and adjacent noncancerous specimens were obtained from 30 patients with lung cancer who underwent surgical resections at the Department of Thoracic Surgery, Yanghu Branch, Changzhou Second People’s Hospital Affiliated to Nanjing Medical University. Clinical characteristics including age, gender, smoking status, tumor size, and lymph node metastasis were collected from medical records. All specimens were immediately frozen in liquid nitrogen and stored at −80°C for further use.
Human lung cancer dataset and gene set enrichment analysis (GSEA)
A public RNA-sequencing dataset of lung cancer was downloaded from The Cancer Genome Atlas (TCGA; https://tcga-data.nci.nih.gov/tcga/, Version: 7/29/2014). The Student’s t-test was used to compare ARHGAP30 expression between lung cancer (n=488) and non-cancerous specimens (n=58). To explore the signaling pathways associated with ARHGAP30 expression, GSEA was carried out on the dataset with the GSEA software version 2.0 as previously described.16
Cell culture
The cells were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). We cultured NCI-H1299, NCI-H1975, NCI-H358, and NCI-H292 cells in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Hyclone, Logan, UT, USA). A549, 16HBE, and 293 T cells were cultured in DMEM (Thermo Fisher Scientific) containing 10% FBS. All the cell lines were maintained in an atmosphere at 37°C with 5% CO2.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated with the TRIzol reagent (Thermo Fisher Scientific), and cDNA was reverse transcribed with the RevertAid™ First Strand cDNA Synthesis Kit (Thermo Scientific Fermentas, Hanover, MD, USA) in accordance with the manufacturer’s instructions. Real-time PCR (RT-PCR) was subsequently conducted in triplicates on an Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) with Fast SYBR green qPCR kit (Thermo Fisher Scientific). Endogenous GAPDH was used for normalization. The primers used were as follows: ARHGAP30 (NM_001025598.1), 5′-TGAAAGCCAAAGGTAGAG-3′ (forward), and 5′-AGGGTCCAAGAACATAAC-3′ (reverse); β-catenin (NM_001904.3), 5′-CGACACCAAGAAGCAGAGATG-3′ (forward), and 5′-GGGACAAAGGGCAAGATTTCG-3′ (reverse); c-Myc (K02276.1), 5′-AGAGTTTCATCTGCGACCCG-3′ (forward) and 5′-GGCTGCCGCTGTCTTTGC-3′ (reverse); MMP-2 (NM_004530.4), 5′-TTGACGGTAAGGACGGACTC-3′ (forward), and 5′-GGCGTTCCCATACTTCACAC-3′ (reverse); MMP-9 (NM_004994.2), 5′-GTGGCACCACCACAACATCAC-3′ (forward), and 5′-CGCGACACCAAACTGGATGAC-3′ (reverse); and GAPDH (NM_001256799.2), 5′-AATCCCATCACCATCTTC-3′ (forward), and 5′-AGGCTGTTGTCATACTTC-3′ (reverse).
Western blotting
Clinical specimens or cultured cells were lysed with ice-cold RIPA buffer (JRDUN Biotech., Shanghai, China). The extracted proteins were subjected to 10% or 8% SDS-PAGE and then transferred onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked in 5% nonfat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) at room temperature for 1 hour, followed by incubation with the primary antibody overnight at 4°C. After three washes with TBST, the membranes were probed with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. Following three washes with TBST, the membranes were developed with the enhanced chemiluminescent substrate (ECL; Bio-Rad Laboratories Inc., Hercules, CA, USA). Antibodies against ARHGAP30 (Ab103402), c-Myc (Ab39688), MMP-2 (Ab92536), and MMP-9 (Ab38898) were obtained from Abcam (Cambridge, MA, USA). Antibodies against β-catenin (#8480) and GAPDH (#5174) were procured from Cell Signaling Technology (Danvers, MA, USA).
ARHGAP30 expression and RNA interference constructs
The coding sequence of ARHGAP30 was amplified with the following primers and cloned into a linearized pLVX-puro expression vector (Clontech, Palo Alto, CA, USA): ARHGAP30 (NM_001025598.1): forward 5′-GCGAATTCATGAAGTCTCGGCAGAAAGG-3′ (EcoR I); reverse 5′-CGGGATCCTCACAGTCCTTCACCTTTCCC-3′ (BamH I). Underlined sequences denote the restriction sites. RNA interference oligos targeting ARHGAP30 were synthesized and cloned into linearized pLKO.1 plasmids (Addgene, Cambridge, MA, USA). The interference sites were as follows: shARHGAP30#1 (position 2555–2573), CCAGGAGGAGATGAGTATA; shARHGAP30#2 (position 2599–2617), GAGAGAAGAGGATGAACAA; shARHGAP30#3 (position 2608–2626), GGATGAACAAAGAGAGGAA. After verification by DNA sequencing, the constructs were co-transfected into 293 T cells, together with the packaging plasmids psPAX2 and pMD2G (Addgene), using Lipofectamine 2000 (Thermo Fisher Scientific). The lentiviruses were collected at 48–72 hours post transfection for cell transduction.
Cell proliferation assay
Cell proliferation was assessed with the Cell Counting Kit-8 (CCK-8) following the manufacturer’s protocol (SAB Biotech., College Park, MD, USA). In brief, 3×103 cells were seeded in 96-well plates per well and cultured overnight. A549 and NCI-H1299 cells were transduced with ARHGAP30-overexpressing lentivirus (ARH OE) or a control Vector virus. NCI-H292 cells were transduced with ARHGAP30 shRNA (shARH) or control shRNA (shNC) and treated with 10 μM XAV939 (Selleck Chemicals, Houston, TX, USA) or vehicle (diemthyl sulfoxide; DMSO). After different culture periods (0, 24, 48, or 72 hours), the cells were incubated with CCK-8 solution at 37°C for 1 hour. Optical density (OD) at a wavelength of 450 nm was measured in triplicate using a microplate absorbance reader (Bio-Rad Laboratories Inc.).
Cell migration and invasion assay
Transwell migration assay was conducted on Costar Transwell 24-well plates (Corning Incorporated, Corning, NY, USA) to assess cell migration. In brief, 6×104 virus-transduced cells were seeded into the upper chamber with 0.2 mL serum-free medium. Cultured medium supplemented with 10% FBS were added to the lower chamber. After 24 hours at 37°C with 5% CO2, the cells remaining on the upper surface were completely removed. The cells that migrated through the membranes were fixed, stained with crystal violet, and counted under a microscope.
A Transwell invasion assay was carried out in the same manner, except that the membranes were pre-coated with Matrigel (1 μg/μL; BD Biosciences, San Jose, CA, USA).
Statistical analyses
All statistical tests were conducted with GraphPad Prism (Graphpad Software, Inc., La Jolla, CA, USA). Experiments were carried out three times independently. Statistical comparison of ARHGAP30 and β-catenin mRNA expression in clinical specimens was undertaken with the Student’s t-test. Pearson’s correlation analysis was used to check the relationship between mRNA expression of ARHGAP30 and β-catenin in lung cancer specimens. Fisher’s exact test was conducted to analyze the correlation of ARHGAP30 expression with patients’ features. Statistical analyses of in vitro experiments were conducted by ANOVA. Values with P<0.05 were considered statistically significant.
Results
ARHGAP30 is markedly downregulated in lung cancer tissues and cell lines
ARHGAP30 has been reported to be involved in the carcinogenesis of colorectal cancer;8 however, its role in lung cancer remains unclear. Initially, we reanalyzed ARHGAP30 expression on a lung cancer dataset downloaded from the TCGA project. Lung cancer tissues (n=488) exhibited lower expression levels of ARHGAP30 than normal lung tissues (n=58; Figure 1A, P<0.0001). To confirm the decreased expression of ARHGAP30 in lung cancer, we collected 30 pairs of lung cancer tissues and adjacent non-cancerous tissues from our hospital. As evidenced by qRT-PCR, the mRNA levels of ARHGAP30 were significantly decreased in lung cancer tissues (Figure 1B, P<0.001). These 30 patients were divided into either an ARHGAP30 low-expression group or an ARHGAP30 high-expression group, with the median mRNA level as the cutoff value. Correlation analysis showed that ARHGAP30 expression was significantly associated with tumor size, but not with age, gender, smoking status, or lymph node metastasis of lung cancer (Table 1). Western blotting was conducted on eight pairs of available tissue samples, and the results showed that the protein levels of ARHGAP30 were decreased in lung cancer tissues (Figure 1C).
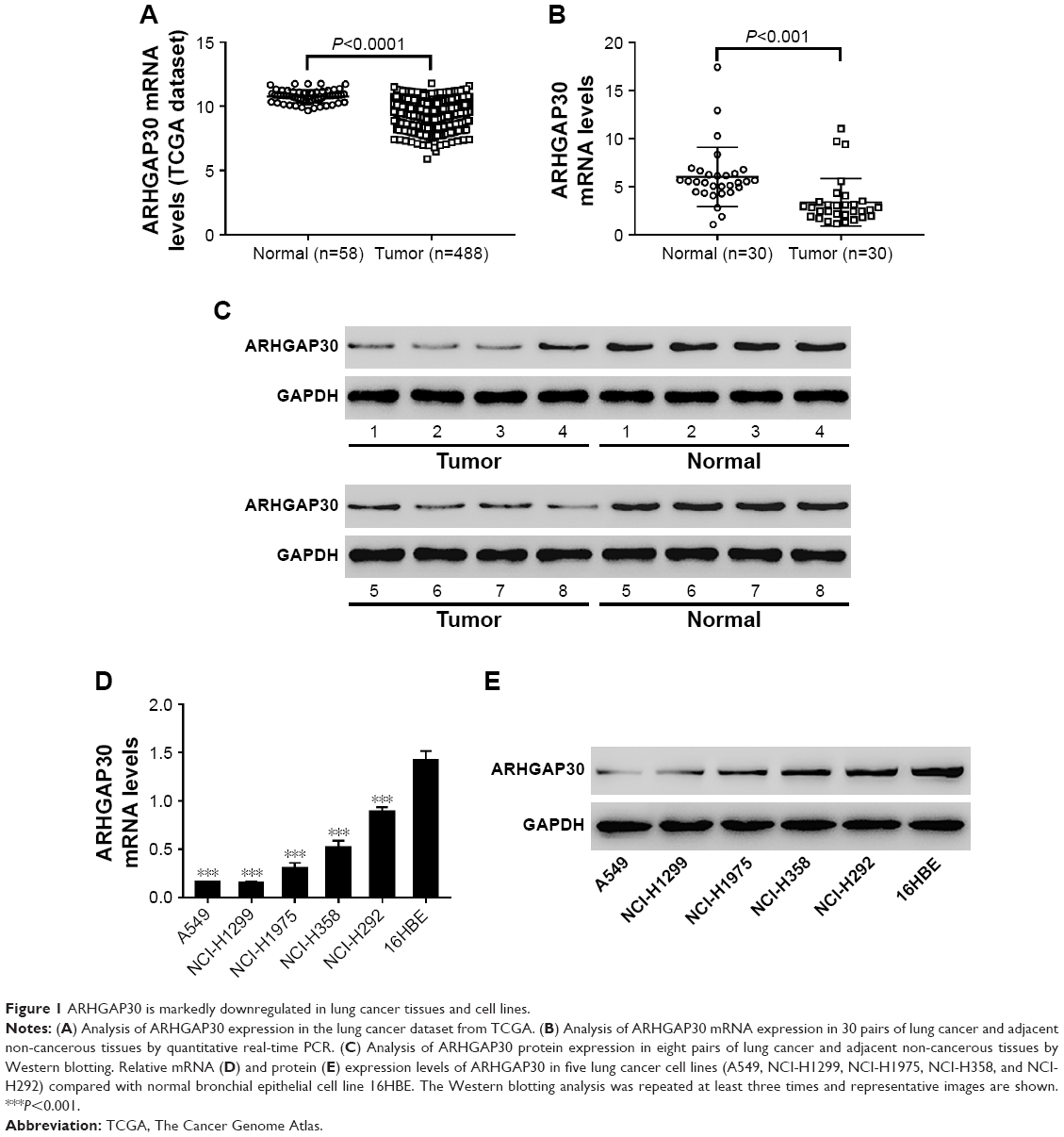 | Figure 1 ARHGAP30 is markedly downregulated in lung cancer tissues and cell lines. |
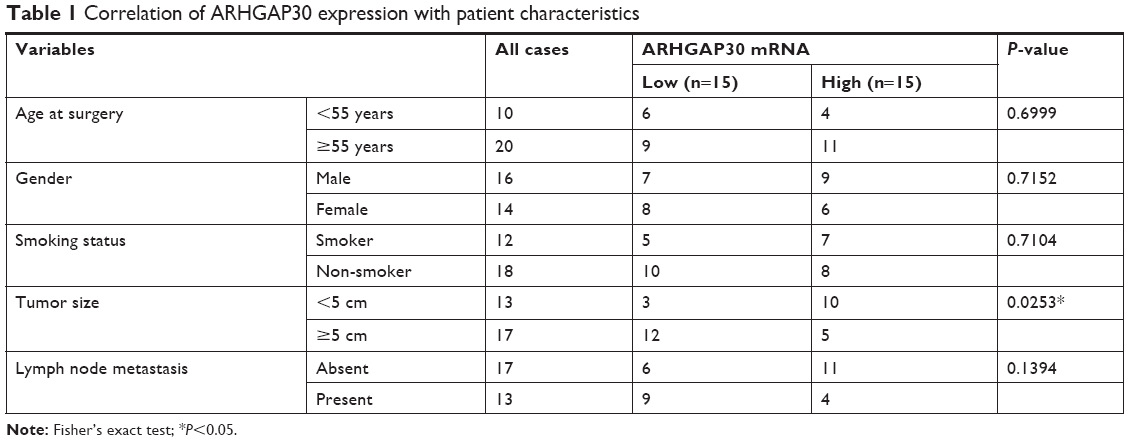 | Table 1 Correlation of ARHGAP30 expression with patient characteristics |
Consistently, ARHGAP30 mRNA and protein levels were significantly downregulated in lung cancer cell lines (A549, NCI-H1299, NCI-H1975, NCI-H358, and NCI-H292) compared with normal bronchial epithelial cell line 16HBE (Figure 1D and E). A549 and NCI-H1299 cells, which had relative low endogenous ARHGAP30 expression, were selected for subsequent experiments. Taken together, we found that ARHGAP30 expression was significantly decreased in lung cancer tissues and cell lines.
ARHGAP30 inhibited lung cancer cell proliferation
To investigate the potential role of ARHGAP30 in lung cancer, A549 and NCI-H1299 cells were transduced with ARH OE or a control Vector virus. Cells in the wild-type (WT) group did not receive any treatment and served as a negative control. As indicated by qRT-PCR (Figure 2A) and Western blotting (Figure 2B) analyses, ARH OE transduction significantly elevated the mRNA and protein levels of ARHGAP30 in both lung cancer cell lines, whereas Vector had no effects on ARHGAP30 expression. As presented in Figure 2C, at 48 and 72 hours post virus transduction, A549 cell proliferation was markedly suppressed by ARH OE compared with the Vector group, whereas cell proliferation was comparable between the Vector and WT groups. Similar results were achieved in NCI-H1299 cells (Figure 2D). Our findings indicated that ARHGAP30 suppressed proliferation of lung cancer cells.
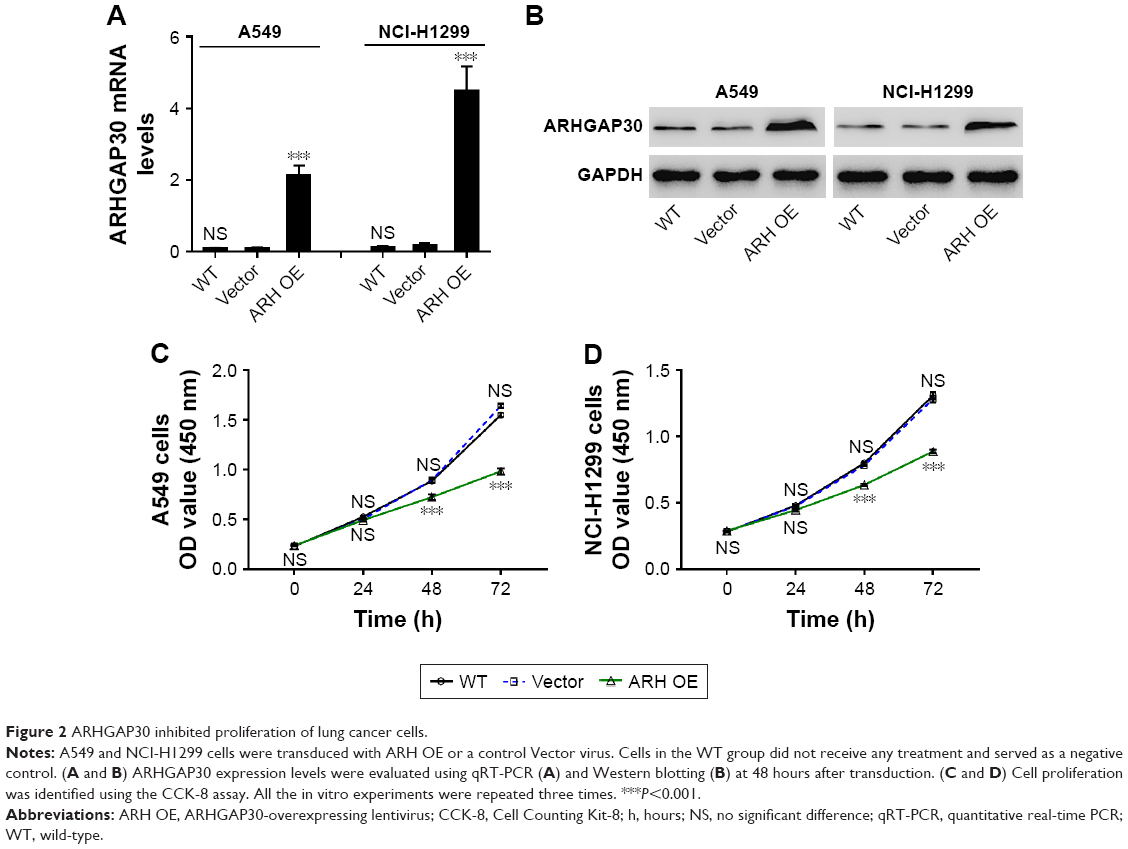 | Figure 2 ARHGAP30 inhibited proliferation of lung cancer cells. |
ARHGAP30 suppressed lung cell migration and invasion
To determine whether ARHGAP30 affects the mobility of lung cancer cells, we assessed the migratory and invasive abilities of A549 and NCI-H1299 cells 24 hours after transduction with ARH OE or Vector. Transwell assays demonstrated that ARHGAP30 overexpression remarkably attenuated the migratory and invasive capabilities of A549 (Figure 3A) and NCI-H1299 (Figure 3B) cells compared with the Vector group. These results indicated that ARHGAP30 repressed lung cancer cell migration and invasion.
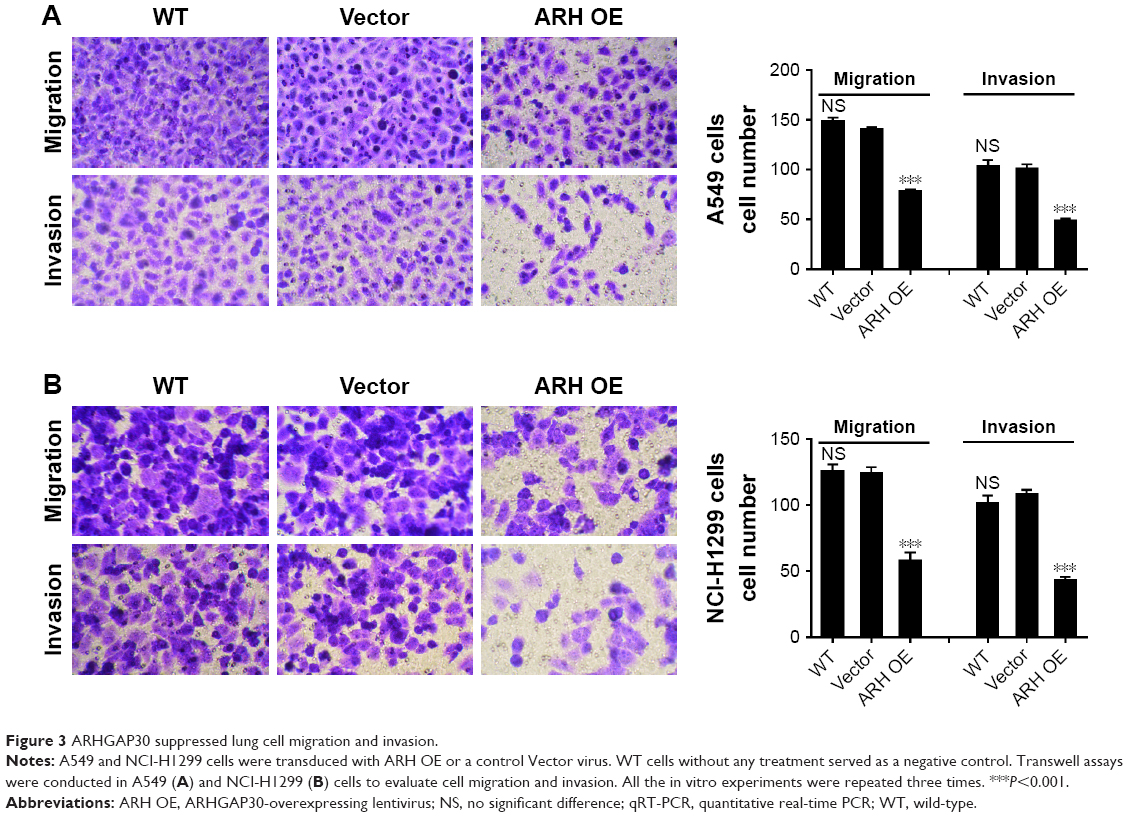 | Figure 3 ARHGAP30 suppressed lung cell migration and invasion. |
ARHGAP30 repressed the Wnt signaling pathway in lung cancer cells
A GSEA on the TCGA lung cancer dataset suggested that ARHGAP30 expression was negatively correlated with the Wnt pathway in lung cancer samples (Figure 4A). As evidenced by qRT-PCR, the mRNA levels of β-catenin were significantly increased in lung cancer tissues collected from our hospital (P<0.001; Figure 4B). Moreover, Pearson’s r correlation analysis revealed a negative correlation between the mRNA expression of ARHGAP30 and β-catenin (Figure 4C, r=−0.6083, P<0.001).
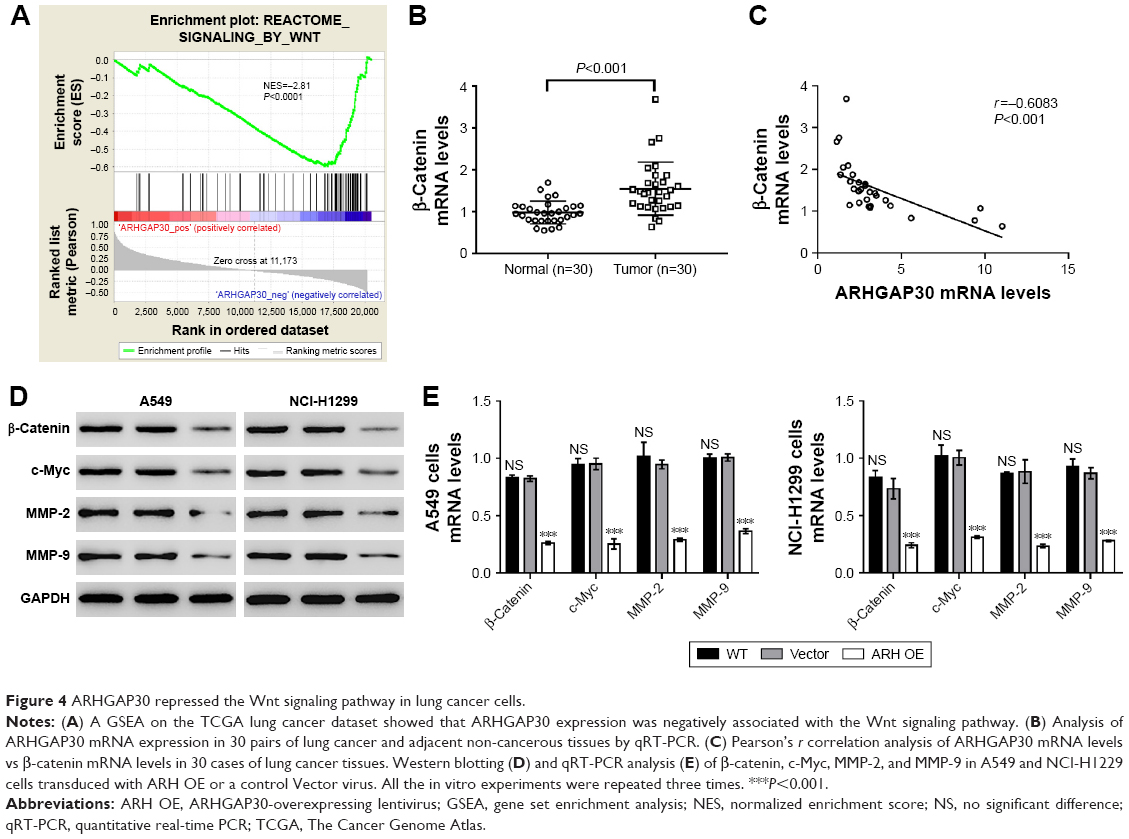 | Figure 4 ARHGAP30 repressed the Wnt signaling pathway in lung cancer cells. |
The expression of β-catenin, c-Myc, MMP-2, and MMP-9 – well-known downstream effectors of the Wnt pathway – was then assessed. Western blotting (Figure 4D) and qRT-PCR (Figure 4E) clearly showed that ARHGAP30 inhibited the Wnt signaling pathway in A549 and NCI-H1299 cells at both protein and mRNA levels. Moreover, the nuclear translocation of β-catenin was reduced in A549 cells with ARHGAP30 overexpression as compared to those with the control Vector virus (Figure S1).
The Wnt signaling pathway mediated the effects of ARHGAP30 on the proliferation, migration, and invasion of lung cancer cells
Copious evidence has highlighted the strong associations between Wnt/β-catenin signaling and the proliferation and metastasis of lung cancer cells.10,11 To further explore the involvement of the Wnt pathway in the functions of ARHGAP30 on lung cancer cells, a specific Wnt/β-catenin pathway inhibitor XAV939 was used to treat cells with ARHGAP30 silence. NCI-H292 cells were chosen because of the relatively higher expression of ARHGAP30 (Figure 1C and D). Three short hairpin RNAs (shRNAs; sh#1, #2, and #3) effectively knocked down ARHGAP30 expression (Figure S2), and the best knockdown efficiency was observed for sh#1, which was used in the subsequent experiments.
We disclosed that ARHGAP30 knockdown significantly strengthened the proliferation (Figure 5A), migration, and invasion (Figure 5B) of NCI-H292 cells, which was compromised when XAV939 was used. In the meantime, ARHGAP30 knockdown upregulated β-catenin, c-Myc, MMP-2, and MMP-9, which was abrogated by XAV939 (Figure 5C). Our data indicated that the inhibitory effects of ARHGAP30 on proliferation, migration, and invasion were mediated by the Wnt signaling pathway.
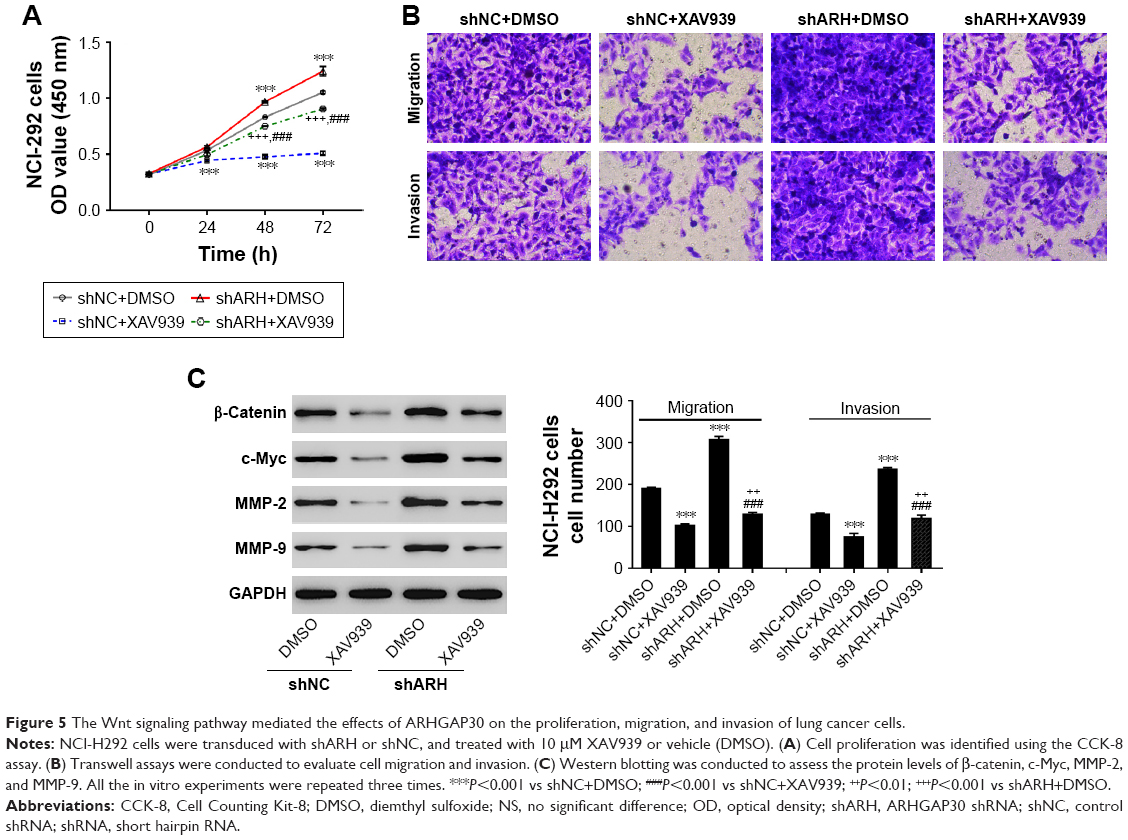 | Figure 5 The Wnt signaling pathway mediated the effects of ARHGAP30 on the proliferation, migration, and invasion of lung cancer cells. |
Discussion
ARHGAP30 – a RhoA- and Rac1-specific Rho GAP7 – has been shown to be significantly correlated with the poor survival of patients with colorectal cancer.8 In this study, we examined the expression of ARHGAP30 in lung cancer tissue samples and found that ARHGAP30 was expressed at lower levels in lung cancer tissues in comparison with normal lung tissues (Figure 1). ARHGAP30 expression was significantly associated with tumor size (Table 1), although these results need to be confirmed by analysis in a larger sample size. We further investigated the role of ARHGAP30 in lung cancer development by using lentivirus-mediated overexpression of ARHGAP30 in lung cancer cell lines. We found that ectopic expression of ARHGAP30 inhibited the proliferation (Figure 2), migration, and invasion (Figure 3) of lung cancer cells, whereas ARHGAP30 knockdown had reverse effects (Figure 5). A lentivirus expressing the RNAi-resistant ARHGAP30 rescued ARHGAP30 expression, and there were promoting effects of ARHGAP30 knockdown on cell proliferation, migration, and invasion (Figure S3). These findings were consistent with a recent study in colorectal cancer.8 These data suggest that ARHGAP30 may serve as a tumor suppressor during the progression of lung cancer.
Furthermore, we tried to explore the mechanism by which ARHGAP30 contributes to lung carcinogenesis. In colorectal cancer, ARHGAP30 binds p53 and promotes p53 acetylation and, thus, functions as a tumor suppressor.8 In the present study, GSEA on TCGA lung cancer dataset showed that there was no significant correlation between ARHGAP30 expression and the KEGG p53 signaling pathway (P=0.113, data not shown), whereas ARHGAP30 expression was negatively correlated with the Wnt signaling pathway (Figure 4A, P<0.0001). According to previous reports, the Wnt/β-catenin signaling pathway plays key roles in the proliferation, survival, and metastasis of lung cancer cells via regulation of the transcription of downstream target genes.10,11 Several Wnt pathway components, such as Wnt-1, Wnt-2, Wnt-3, Wnt-5a, and β-catenin, are overexpressed in lung cancer.10 The aberrant expression of β-catenin is associated with the poor prognosis of NSCLC.12,13 As expected, in the present study, β-catenin mRNA expression was significantly increased in lung cancer tissues (Figure 4B), which was negatively correlated with ARHGAP30 mRNA expression (Figure 4C). Additionally, β-catenin expression was significantly downregulated with ARHGAP30 overexpression in A549 and NCI-H1299 cells (Figure 4). The nuclear translocation of β-catenin was suppressed by ARHGAP30 overexpression (Figure S1). Moreover, the expression of three well-known target genes of the Wnt/β-catenin signaling pathway (c-Myc, MMP-2, and MMP-9)14,15 was simultaneously reduced in ARHGAP30-overexpressed cells at both the transcriptional and translational levels. c-Myc is a well-accepted oncoprotein, implicated in the control of cancer cell proliferation.17 MMP-2 and MMP-9 – responsible for the degradation of the extracellular matrix – play critical roles in cancer cell migration, invasion, and metastasis.18 The decreased expression of these three genes may explain the decreased proliferation, migration, and invasion of lung cancer cells with ARHGAP30 overexpression. Thus, we supposed that ARHGAP30 may inhibit the aggressive phenotypes of lung cancer cells by inhibiting Wnt/β-catenin signaling. We found that the β-catenin inhibitor XAV939 can abrogate the enhanced cell proliferation, migration, and invasion caused by AHGAP30 knockdown (Figure 5). These data suggest that the Wnt signaling pathway may mediate the functions of ARHGAP30 in lung cancer, although further studies are needed to elucidate the exact mechanism.
Conclusion
ARHGAP30 expression was decreased in lung cancer tissues and cell lines. Furthermore, our study preliminarily unveiled that ARHGAP30 suppressed the capacities of cell proliferation, migration, and invasion via inhibition of the Wnt/β-catenin signaling pathway in lung cancer cells. These results may provide new possibilities for therapeutic interventions of lung cancer in the future.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Oser MG, Niederst MJ, Sequist LV, Engelman JA. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015;16(4):e165–e172. | ||
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9(9):690–701. | ||
Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93(1):269–309. | ||
Naji L, Pacholsky D, Aspenström P. ARHGAP30 is a Wrch-1-interacting protein involved in actin dynamics and cell adhesion. Biochem Biophys Res Commun. 2011;409(1):96–102. | ||
Wang J, Qian J, Hu Y, et al. ArhGAP30 promotes p53 acetylation and function in colorectal cancer. Nat Commun. 2014;5:4735. | ||
Pongracz JE, Stockley RA. Wnt signalling in lung development and diseases. Respir Res. 2006;7(1):15. | ||
Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst. 2014;106(1):djt356. | ||
Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. | ||
Xu X, Sun PL, Li JZ, Jheon S, Lee CT, Chung JH. Aberrant Wnt1/β-catenin expression is an independent poor prognostic marker of non-small cell lung cancer after surgery. J Thorac Oncol. 2011;6(4):716–724. | ||
Hommura F, Furuuchi K, Yamazaki K, et al. Increased expression of beta-catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer. 2002;94(3):752–758. | ||
Wu B, Crampton SP, Hughes CC. Wnt signaling induces matrix metalloproteinase expression and regulates T cell transmigration. Immunity. 2007;26(2):227–239. | ||
Zemans RL, Briones N, Campbell M, et al. Neutrophil transmigration triggers repair of the lung epithelium via-catenin signaling. Proc Natl Acad Sci U S A. 2011;108(38):15990–15995. | ||
Wang J, Zhang J, Xu L, Zheng Y, Ling D, Yang Z. Expression of HNF4G and its potential Beyotime, Beijing, China functions in lung cancer. Oncotarget. 2017;9(26):18018–18028. | ||
Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2(10):764–776. | ||
Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. Febs J. 2011;278(1):16–27. |
Supplementary materials
Methods
Immunofluorescence staining
A549 cells were transduced with the ARHGAP30-overexpressing lentivirus (ARH OE) or a control Vector virus. After 48 hours, cells were fixed in 4% paraformaldehyde for 10 minutes at room temperature and then permeabilized with 0.5% Triton X-100 at room temperature for 15 minutes. After blocking in 1% BSA at room temperature for 1 hour, the cells were incubated with primary rabbit anti-β-catenin antibody (Ab16051; Abcam, Cambridge, MA, USA) at 4°C overnight. After washing in PBS, cells were incubated with Alexa-Fluor 488-conjugated goat anti-rabbit immunoglobulin G (IgG; Beyotime) at room temperature for 1 hour. Nuclei were stained with DAPI (Beyotime). The cells were examined under a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
RNAi-resistant ARHGAP30 plasmid construction
To make a RNAi-resistant ARHGAP30 plasmid, the nucleotides targeted by the RNAi (position 2555–2573, CCAGGAGGAGATGAGTATA) of pLVX-ARHGAP30 were partially mutagenized without changing the amino acid sequence (position 2555–2573, CCTGGTGGTGACGAGTATA) by site-directed mutagenesis.
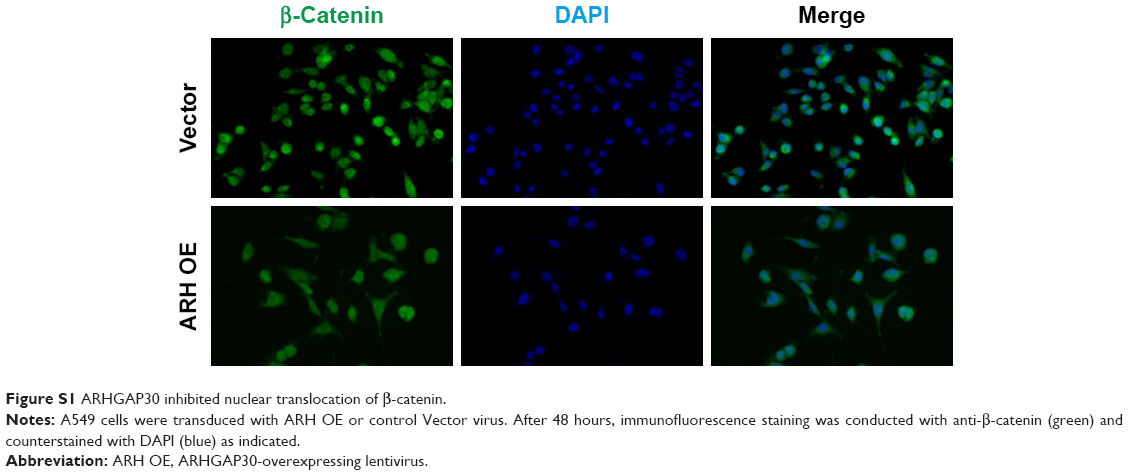 | Figure S1 ARHGAP30 inhibited nuclear translocation of β-catenin. |
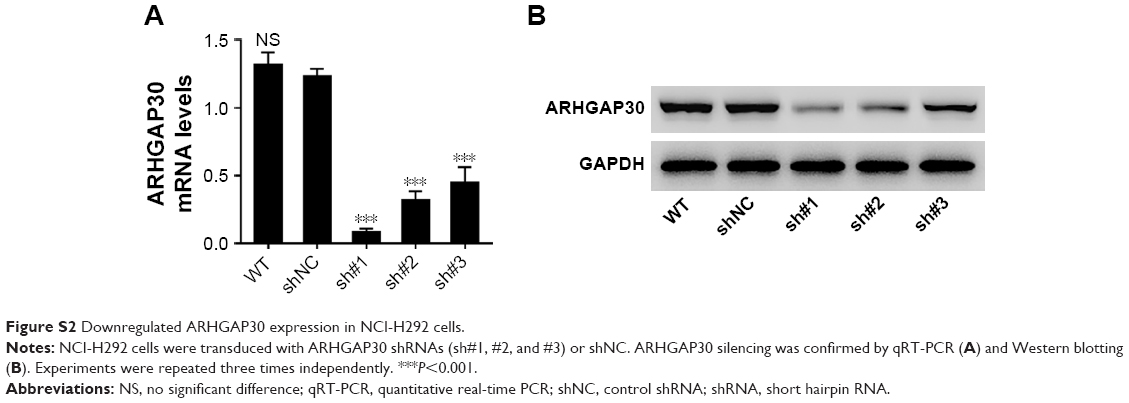 | Figure S2 Downregulated ARHGAP30 expression in NCI-H292 cells. |
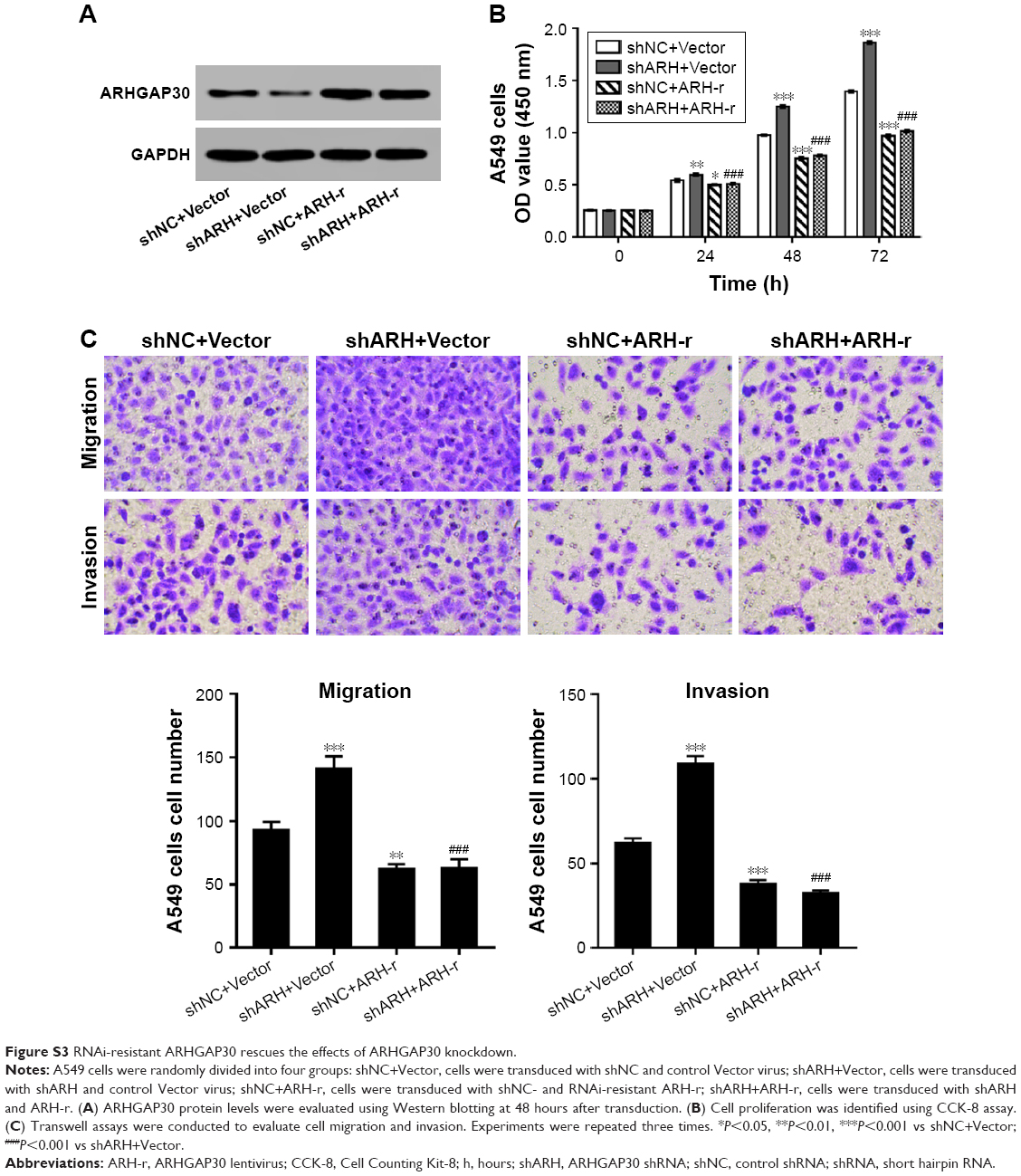 | Figure S3 RNAi-resistant ARHGAP30 rescues the effects of ARHGAP30 knockdown. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.