Back to Journals » OncoTargets and Therapy » Volume 11
Are ovarian cancer stem cells the target for innovative immunotherapy?
Received 28 October 2017
Accepted for publication 26 February 2018
Published 8 May 2018 Volume 2018:11 Pages 2615—2626
DOI https://doi.org/10.2147/OTT.S155458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Liang Wang, Tianmin Xu, Manhua Cui
Department of Gynecology and Obstetrics, The Second Hospital of Jilin University, Changchun, Jilin, People’s Republic of China
Abstract: Cancer stem cells (CSCs), a subpopulation of cancer cells with the ability of self-renewal and differentiation, are believed to be responsible for tumor generation, progression, metastasis, and relapse. Ovarian cancer, the most malignant gynecological cancer, has consistent pathology behavior with CSC model, which suggests that therapies based on ovarian cancer stem cells (OCSCs) can gain a more successful prognosis. Much evidence has proved that epigenetic mechanism played an important role in tumor formation and sustainment. Since CSCs are generally resistant to conventional therapies (chemotherapy and radiotherapy), immunotherapy is a more effective method that has been implemented in the clinic. Chimeric antigen receptor (CAR)- T cell, an adoptive cellular immunotherapy, which results in apparent elimination of tumor in both hematologic and solid cancers, could be used for ovarian cancer. This review covers the basic conception of CSCs and OCSCs, the implication of epigenetic mechanism underlying cancer evolution considering CSC model, the immunotherapies reported for ovarian cancer targeting OCSCs currently, and the relationship between immune system and hierarchy cancer organized by CSCs. Particularly, the promising prospects and potential pitfalls of targeting OCSC surface markers to design CAR-T cellular immunotherapy are discussed here.
Keywords: cancer stem cells, ovarian cancer, epigenetics, tumor cell surface marker, immunotherapy, CAR
Introduction
Cancer stem cells (CSCs, also named tumor-initiating cells [TICs] or tumor-propagating cells [TPCs]) are a small subsection of cancer cells which can self-renew and differentiate into heterogeneous tumor cells and are believed to be the culprit for tumor initiation, growth, and recurrence. Many CSCs have been identified from a variety of human tumors including brain cancer, melanoma, breast cancer, liver cancer, pancreatic cancer, colon cancer, prostate cancer, and ovarian cancer.1,2 Epigenetics have been found to have crucial function in cancer development as researches continue, which connect cancer cellular states and the tumor microenvironment in a tumor. Among gynecologic malignancies, ovarian cancer is the second most common and the first cause of death but it has the highest mortality rate.3 Unfortunately, the majority of ovarian cancer patients who have complete remission by surgery and chemotherapy will ultimately have the cancer reoccur. Ovarian cancer stem cells (OCSCs) have been identified for more than a decade, and many kinds of OCSCs are thought to exist due to the variety of ovarian cancer subtypes or/and the heterogeneity within a tumor, and many surface markers of OCSCs have been targeted for therapy. CSCs are universally resistant to chemotherapy and radiotherapy; hence, immunotherapy is a promising approach for the elimination of CSCs, and a lot of clinical data can support the immunotherapy. Chimeric antigen receptor (CAR)-T cells are reconstructed T cells, which can use the receptors attached to themselves that can specifically recognize the specific antigen on cancer cells, CAR-T cells do not depend on HLA to kill cancer cells. HLA deficiency is a way of immune evasion for some cancer cells. The chimeric receptor is a designed receptor on T cell which does not depend on the T cell existing receptor. Anti-CSC CAR-T cells have been reported in some types of cancers, such as prostate cancer,4 glioblastoma multiforme (GBM),5 nasopharyngeal carcinoma,6 and cholangiocarcinoma (CCA).7 To date, only one study using CAR-natural killer (NK) cells to target OCSCs in vitro has been reported in a tiny but significant step in this field.8 Targeting OCSCs to design CAR has the prerequisite of known antigens on OCSCs; hence, it is possible to construct CAR to combat ovarian cancer by targeting OCSCs. Based on the CSC model to explore ovarian cancers and the correlative epigenetic mechanism under this process, the current immunotherapies are reviewed here and the recapitulation of CAR-T cellular immunotherapy targeting OCSCs is also discussed in this context.
The conception of CSCs and OCSCs
CSCs are a subpopulation in a tumor which differentiate into CSCs and non-CSCs possessing self-renewal ability which can also survive in conventional therapy. They were first found in acute myeloid leukemia (AML) in 1997, termed as severe combined immunodeficiency (SCID) which can differentiate into leukemic blasts revealing the hierarchy organization of AML clone.9 They have the characterization resembling normal stem cells, that is, differentiation into themselves and progeny cells with a slow cycling states. Traditional gene model means that tumor is a mutation of single gene cell subset. Indeed, a tumor does not contain sole single tumor genome, instead comprises of multiple genomes that have heterogeneity. “Gene model” and “CSC model” are harmonized when synthesizing genetic diversity, non-genetic influences like epigenetic modification, and nontumor cell elements (tumor microenvironment) which are all involved in intratumoral heterogeneity.10 The origins of CSCs are putative by three theories: a normal stem cell, a normal committed progenitor cell, or a transit-amplifying cell which possesses limited differentiation potential and self-renewal capability similarity with a stem cell.11
CSCs are at the helm of tumor hierarchy owning functional traits, unlimited self-renewal potential, quiescent or slow-cycling states, and increased resistance to conventional therapies, which contribute to tumorigenesis, recurrence, and metastasis.12 The dormancy of CSCs can explain the undetectable persistence of a tumor for a period of time even though a patient responds clinically after therapy until malignancy relapse exists in primary or secondary tumor site.13 Plasticity of CSCs confers the reversible dynamic change between non-CSC state and CSC state mediated by tumor microenvironment and tumor niche which facilitates the intricacy and heterogeneity of a tumor.14 The theories involved in CSCs resulting in carcinogenesis include chemical carcinogenesis, infection, mutation, or epigenetic changes.15 Cancer is an ecosystem, where CSC is on the apex of the pyramid of this ecosystem that is controlled by intricate hierarchical organization of CSCs. The clinical significance of CSC refers to many fields including detecting prognostic outcome, chemoresistance, radiation resistance, dissemination/anoikis resistance, immune system evasion, dormancy, and field cancerization which enlarge the dimensionality of cancer research.14
To date, many surface markers have been identified on CSCs, secreting specific molecular, interacting specific signal pathway. Many signal pathways mediate various CSC traits including the Janus-activated kinase/signal transducer and activator of transcription, Hedgehog, Wnt, Notch, phosphatidylinositol, 3-kinase/phosphatase and tensin homolog, and nuclear factor-kB signaling pathways.16
Ovarian cancer is the most lethal gynecologic malignancy. Globally, it is a significant reason of morbidity and mortality in females, with rising rates in economically underdeveloped countries and increasing case numbers in high-income countries as a result of population aging, with sharp recurrence and improving malignancy and 5-year survival rates below 45%.17 OCSCs have been found in human epithelial cancer more than a decade ago.2 Both biological and clinical characterizations of ovarian cancer prove it as a prototypical example of CSC-driven disease.18 OCSCs can explain primary tumor growth, metastasis, relapse, and conventional therapy resistance of ovarian cancer.19 There is also a relationship between OCSCs and epithelial–mesenchymal transformation (EMT), which leads to the genetic alteration in cancer and is a mechanism under angiogenesis and chemotherapy resistance.20 Some surface markers have been found in OCSCs, including CD133, CD44, CD47. Some functional markers such as ALDH1 are under intensive research, too. Indeed, there is no stringent distinction between surface markers and functional markers because they all participate in tumorigenesis and progression and are called “marker.” Various signal pathways that play a significant role in ovarian cancer have been researched, including Wnt/β-catenin, NOTCH, IL6/JAK/STAT3, Hedgehog, NFκB, PI3K/AKT, TLR2-MyD88-NFκB, HMGA1, PKCι/Ect2/ERK, YAP/TEAD, and hypoxia/NOTCH1/SOX2.18 Traditional therapy strategies based on cytoreductive surgery, chemotherapy, and radiotherapy are effective to some extent; however, recurrence and resistance to them are inevitable consequences in advanced ovarian cancer. Nowadays, immunotherapy has been used in ovarian cancer showing prospect. OCSCs are subpopulation in a tumor, and are regarded as the initiation cells with the ability to induce resistance to conventional therapy, relapse, and dissemination and should be major target for therapy.
The epigenetic modifications involved in the CSCs and OCSCs
Epigenetics refers to heritable traits that can be inherited from parent cells by daughter cells and are not caused by changes in the DNA sequence through meiotic or mitotic division. Epigenetic regulation mechanisms include DNA methylation, histone modification, chromatin remodeling, and modulation by noncoding RNAs.21 During cancer growth, the epigenome of cancer cells is consistently determined or regulated by cell-intrinsic/extrinsic mechanisms. Meanwhile, a subpopulation of cells (CSCs) in the tumor are conferred stemness, which refers to self-renewal and differentiation. First, epigenetic dysregulation plays an important role in the formation of CSCs, and then epigenetic mechanisms maintain the hierarchical organization of cancer controlled by CSCs.22 Epigenetic modifications play two distinct roles, namely inhibiting the majority of cancer cells to self-renew to constitute hierarchies and sustaining the subsection of CSC stemness. Dynamic epigenetic states have been implicated in the reversible transition from CSCs to non-CSCs and vice versa which is called plasticity and may be a confusion of the deterministic identity of CSCs. Among epigenetic mechanism, DNA methylation is integral to the formation of CSCs in cancer biology. For example, promoter methylation and histone modifications controlled the transcription of CD133, which is a cell surface marker protein of CSCs in many cancer types and is widely detected to identify OCSCs. The extent of DNA methylation of P2 promoter of CD133 is inversely relevant for CD133 transcription level, and CD133− cells sorted from epithelial ovarian cancer are treated with DNA methyltransferase and histone deacetylase inhibitors expressed increasing CD133 surface markers.23 Methylation abnormalities are essential in the process of CSC formation in the first step of cancer.
The surface markers or functional markers on OCSCs as the immunotherapy targets
Many markers on OCSCs have been reported so far, and some of them have been targets for immunotherapy. Considering the physiological and pathological traits of these markers and their current implication is stable evidence for further innovated therapy.
CD133
CD133 (prominin-1) is a transmembrane glycoprotein and was first described as a specific marker of human hematopoietic progenitor cells.24 It is a surface marker to identify various cancer types, such as brain cancer,25 colorectal cancer,26 breast cancer,27 neck and head cancer,28 and liver cancer.29 It can define a TIC population in human ovarian cancer as CD133+ and CD133− cells are sorted, respectively and primary uncultured ovarian cancer cells are injected into non-obese diabetes/SCID mice and then the CD133+ cells increasingly generated a heterogeneous tumor as the primary cancer.30 In addition, CD133 mRNA levels were inversely correlated to the DNA methylation status of the CpG sites for ovarian cancer which is regulated by epigenetic mechanism.31 CD133 expression is an independent predictor of poor prognosis which can reduce disease-free survival time for patients with ovarian cancer.32 Other investigations also described an inconsistent phenomenon that CD133− cells can induce the same characteristics as CD133+ cells, which questions whether the specificity of CD133 to identify CSCs and the glycosylation status of CD133 may be more relevant to the function of CD133 in CSCs.24
CD44
CD44 is a cell surface glycoprotein and the receptor of hyaluronan, a component of the extracellular matrix and the interaction of them has been proved to be associated with a variety of signal transduction pathways such as Nanog and EGFR-Ras-ERK.18 CD44 can be expressed on both somatic cells and CSCs, which is associated to the sphere forming, self-renewing, chemo-resistance, tumor-initiating, proliferation, and invasiveness of CSCs in preclinical experiments.33,34 The expression of CD44 is promoted during EMT, which is involved in the acquisition of stemness of epithelial tumor cells.35 CD44/CD44v isoforms have been identified to isolate and enrich CSCs in various types of malignancies, such as head and neck cancer,36 lung cancer,37 gastric cancer,38 pancreatic cancer,39 colon cancer,40 and ovarian cancer.41–43 As for ovarian cancer, a study had suggested that CD44+CD24− identifies a group of CSC-like cells from ovarian adenocarcinoma cell line 3AO, and these cells can differentiate into CD44+CD24+ cells and are obviously resistant to carboplatin and paclitaxel therapy.42 Especially, CD44 splice variants have exclusive roles in tumorigenesis, such as CD44v6 expression is associated with poor prognosis and metastasis.44 CD44v7/8 has been employed as engineered cytotoxic T-cell immunotherapy target for cervical cancer and has shown a promising prospect.45 However, some studies have suggested that CD44+ tumor cells induce a significantly shorter disease-free survival than the CD44− group. On the contrary, other studies have suggested high CD44 expression resulting in improved ovarian cancer outcome, although these inconsistences could be attributed to technical factors, and there is still an ambiguous inherence of CD44 in the CSC model.33
CD47
CD47 is a transmembrane protein which is the ligand for signal regulatory protein-alpha (SIRPα) and secretes matricellular protein thrombospondin-1.46 Substantially, CD47 is an overexpressed surface marker on all human cancers, because its combination with SIRPα sends a “don’t attack me” signal for phagocytic cells, which inhibits macrophage-mediated destruction to evade immune surveillance that is the foremost step in CSC tumorigenesis.47 Higher CD47 expression is significantly correlated with poor prognosis of high-grade serous ovarian carcinoma patients.48
ALDH1
Aldehyde dehydrogenases (ALDHs) are enzymes that promote the oxidation of aldehyde substrates to their corresponding carboxylic acids. ALDH1 is a functional marker on OCSCs, while there is no obvious barrier when considering the specificity to identify OCSCs provided this marker can be detected by modern assays, but the functional marker which is not expressed on the cell surface may not be adapted to direct target therapies. Within the ALDH family, which is composed of 19 isoforms with similar catalytic functions, the ALDH1 subgroup is particularly active in normal cells and CSCs. Particularly, ALDH1A1 is exploited more widely in many different cancer types of CSCs than ALDH1A2 and ALDH1A3 in ALDH1 isozymes.49 ALDH1 is expressed on the cancer cell surface as a molecular pump to efflux chemotherapeutic agents which is the mechanism underlying chemo-resistance in CSCs.18 A recent study has shown that the evolution of surface markers (CD133 and ALDH1) expressed on high-grade serous OCSCs has changed from primary ovarian cancers to recurrent ones and CD133 and ALDH1 coexpression is an independent factor for poor prognosis.50 However, a study suggested that ALDH is superior to CD133, serving as a marker to identify OCSCs, as ALDH is correlated with spheroid formation and tumor forming capability.51
Others
There are some other surface markers to distinguish OCSCs such as epithelial cell adhesion molecule (EpCAM), CD117, and CD24 or functional factors such as transcription factor SOX2 and VAV3.52–55 EpCAM (also named CD326) is a Ca2+-independent cell adhesion molecule expressed on the basolateral surfaces of most epithelial cells.56 It can distinguish CSCs of colon cancer,57 prostate cancer,4 ovarian cancer,58 pancreatic cancer,59 lung cancer, breast cancer, and gastric cancer.60
A marker found to distinguish OCSCs is never limited in ovarian cancer but overlaps many cancer types. In another context, to identify OCSCs always needs combined coexpression “marker pairs,” such as Lgr5/ALDH1,61 CD44/CD117,41 and ALDH/CD133.62 The dual diversity reveals the commonness of various malignancies and the heterogeneity within one tumor. It is a drawback that none of these markers are exclusively within CSCs/OCSCs, but nowadays taking advantage of great disparity of expression levels on CSCs/OCSCs when compared with normal tissues gives greater knowledge into immunotherapeutic strategies targeting these markers which is discussed in “Immune system and immunotherapeutic strategies relevant to CSCs/OCSCs”.
Immune system and immunotherapeutic strategies relevant to CSCs/OCSCs
Immune surveillance is the barrier against tumor initiation. CSCs can use immune evasion to grow, differentiate, spread, and generate primary lesion and metastatic lesion. Nowadays, many immune-suppressive molecules have been identified including programmed cell death 1 (PD-1), programmed cell death 1 ligand 1 (PD1-L1), transforming growth factor β (TGF-β), cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), B- and T-lymphocyte attenuator, and CD200.11,63 There is a hypothesis called “immunoediting,” which indicates that the immune system plays a dual contradictory role in cancer not only suppressing tumor growth but also promoting tumor outgrowth with the ability of host-protection and tumor promotion.64,65 The innate mechanism underlying cancer and immune system needs more researches, and the role of CSCs in the immunoediting hypothesis has not been revealed yet. Because of the resistance to conventional therapeutic strategies of CSCs and the ability to recapitulate original tumor to be the source of recurrent tumor, the immunotherapy may be a processing way and has been partly proved in the clinic. There are many immunotherapeutic strategies targeting CSCs in various types of cancers, such as NK cell, cancer therapeutic vaccine, monoclonal antibody (mAb) immunotherapy, and blockade of immune checkpoints.
NK cells
NK cells can target and eliminate CSCs in an major histocompatibility complex (MHC) unrestricted fashion. CSCs can upregulate NK cell ligands (MICA and MICB) after treatment and conceal those ligands for immune evasion. NK cell immunotherapy targeting CSCs has been researched in many cancers, such as colorectal, glioblastoma, melanoma, and breast cancer.66 A study reported that NK cells activated by interleukin-2 (IL-2) and IL-15 are effective for eliminating CD44+CD24− human breast CSCs with the upregulation of NKG2D (the receptor on NK cell) ligands ULBP1, ULBP2, and MICA on these CSCs.67
Cancer therapeutic vaccine
Cancer therapeutic vaccine requires the participation of innate and adaptive immunity of a patient by first stimulating the host by a tumor-specific antigen to activate large amounts of tumor antigen-specific cytotoxic T lymphocytes (CTLs) to remove tumor.68 Cancer vaccine is superior to other therapy because the immunological memory will prevent cancer recurrence for a period. It has implication in the field of OCSCs therapy now. For example, Wu et al reported that the SKOV3 CD117+CD44+ CSC vaccine depressed ovarian cancer growth in xenograft mice. This base-CD177/CD44 vaccine can reduce the CD117+CD44+ CSC and ALDH1-positive cell populations in the immunized mice with enhanced serum interferon-γ (IFN-γ), decreased TGF-β levels, and increased cytotoxic activity of NK cells.69 CD117, CD44, and ALDH are specific surface markers of many types of cancers which can be targeted in ovarian cancer therapy.
Monoclonal antibody
Monoclonal antibody (mAb) immunotherapy uses antigen–antibody response to exploit the immunocompetence of the host to remove the targeted cells, which resulted in mAb is widely detected for decades. The mechanism underlying it is to activate antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity, inhibit receptor-mediated signaling, prime antigen-presenting cells, effector and memory T cells.70 Many preclinical and clinical research studies have been exploring this therapy targeting CSCs, considering ovarian cancer is an aggressive malignancy, and mAbs targeting OCSCs are under intense scrutiny. For example, a study has examined the reactivity of anti-CD133 Mab CC188 to CD133+ ovarian cancer cells which are believed to be OCSCs by using immunofluorescence staining methods and tissue microarray technique, and the results showed that Mab CC188-based imaging and therapeutic reagents may provide a promising method to detect ovarian tumors in an earlier stage and also in treatment.71 Besides mAbs, antibody constructs that are designed flexibly can induce a more effective outcome, for instance, Catumaxomab, a trifunctional antibody construct consisting of two half antibodies, each with one light and one heavy chain that originate from parental mouse IgG2a and rat IgG2b isotypes which can bind to antigens CD133 and EpCAM. In a Phase II/III clinical study of patients suffering malignant ascites with advanced ovarian, gastric, pancreatic cancer and other origins, the results showed that CD133+/EpCAM+ CSCs are vanished in the catumaxomab samples.72
Blockade of immune checkpoints
Immune checkpoints are cell surface molecules that are crucial for maintaining self-tolerance and regulating physiological immune responses by mediating co-inhibitory signaling pathways, such as CTLA-4, PD-1, and PD-L1 the antibodies of which can be blockers of immunosuppression.73 These regulatory pathways result in a suppressive tumor microenvironment and tumor cell niche which is entrenched especially in CSCs that protect cancer cells.74 Nowadays, clinical trials of antibodies of immune checkpoints in ovarian cancer have been partly completed and partly ongoing, for example, ipilimumab (anti-CTLA-4 antibody), nivolumab (anti-PD-1 antibody), avelumab (anti-PD-L1 antibody), which are clearly effective but still limited especially with some adverse effects, and those are reviewed by Mittica et al.75 Recently, MYC, an oncogene code for a transcription factor which regulates the expression of CD47 (innate immune regulator and discussed above because of its role in cancer evading immune surveillance in the CSC tumorigenesis) and PD-L1 (adaptive immune checkpoint) are inactivated to enhance the antitumor immune response, and it will be an important implication in immunotherapy.76 Until now, immune checkpoints in CSCs/OCSCs carcinogenesis are still interesting to be studied.
All those immunotherapeutic strategies have been shown to be effective in ovarian cancer to some extent, while there is a long way to go for an apparent cure in clinical practice. Nowadays, adoptive cellular immunotherapy has shed light on both hematologic and solid cancers. CAR-T cells have been a relatively successful method to mediate tumor rejection, especially in B-cell malignancies targeting CD19.77 In the present study, the basic conception of CAR, the current progression, and the possibility to employ this method in OCSCs are reviewed.
The promising immunotherapy CARs targeting OCSCs
CARs are recombinant receptors for specific antigen, which can reprogram the specificity and function of T lymphocytes or other immune cells such as NK cells.8,78 The basic approach of CAR is to redirect tumor targeted T cells, which is called CAR-T cells (CAR-T lymphocyte), bypassing major histocompatibility complex inducing immune reactions and leading to cytotoxicity. As an adoptive transfer immunotherapy, CAR-T has three primary components including the extracellular domain, transmembrane, and intracellular domain. The primary feature of the extracellular domain is the single-chain variable fragment (scFv) region which is a fusion protein of the variable regions of the heavy (VH) and light chains (VL) of an antibody specific for the antigen. The hinge/spacer and transmembrane domains are usually CD8α or CD28, which links the extramembrane domain and intramembrane and may contribute to the interaction with antigen and recruitment of signals resulting in immune activation of CAR.79 Transmembrane domains can influence the immunogenicity depending on its length or flexibility.80 The intracellular domain is to lead to T-cell activation.81 It comprises CD3ζ chain and is often incorporated with costimulatory molecules that include CD27, CD28, CD134 (OX-40), and CD137 (4-1BB). These costimulatory molecules aid the signals that influence the proliferation and the persistence of the T cells.82 CAR-T cells are separated by generation as the field has progressed. The first generation of CAR-T cells include only CD3ζ as an intracellular signaling domain. The second generation modifies the surface of the CAR and includes an additional costimulatory molecule like CD28. The third generation is on the basement of the second to add multiple costimulatory molecules on CAR such as OX-40/4-1BB (CD134/CD137). The fourth generation is significantly different in its function of releasing cytokines (IL-12) and is also known as T-cell redirected universal cytokine killing (TRUCK) (Figure 1).83
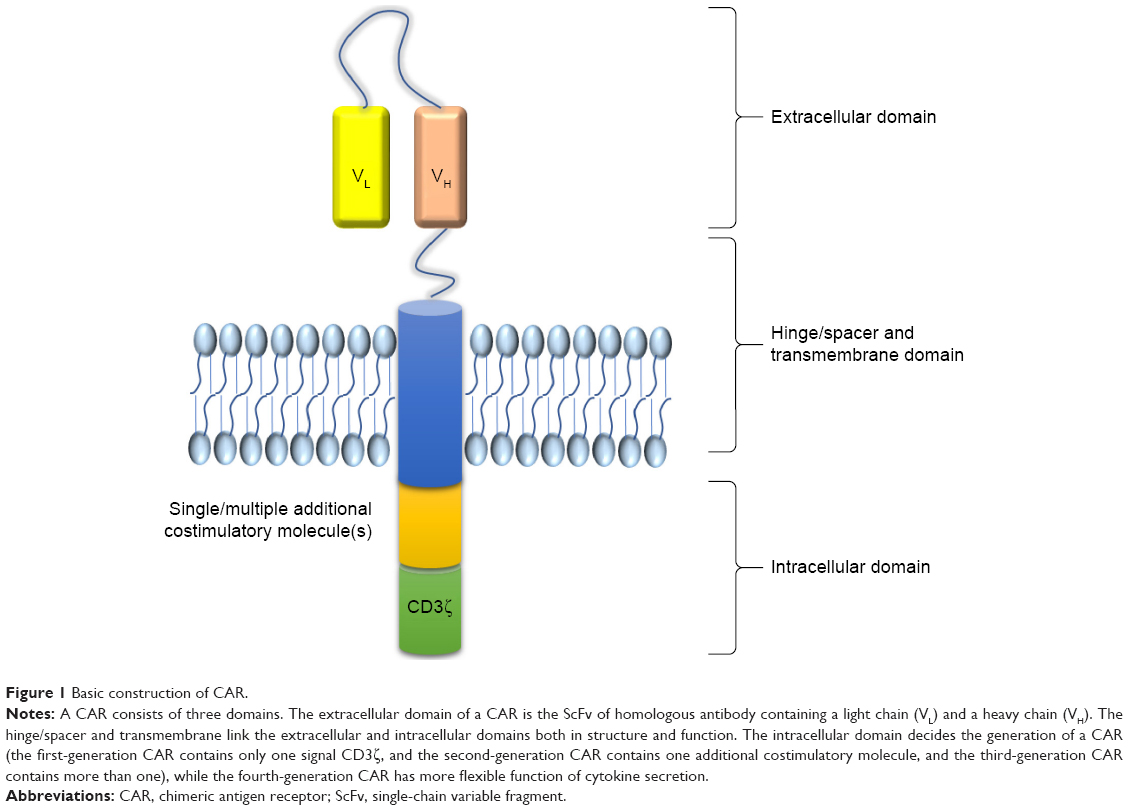 | Figure 1 Basic construction of CAR. |
The primary standard protocol of CAR-T design implemented from bench to bedside is of several steps. First, T cells are extracted from the patient. Second, the T cells that can recognize the specific antigen of the cancer cells are engineered in vitro and called CAR-T cells. Then, the CAR-T cells are cultured millions and billions fold. Next, these CAR-T cells are injected back into the patient. Finally, with the antitumor ability of the T cells, the patient is expected to be healed (Figure 2). In addition, many advanced reconstruction strategies have solved the branch problems of CAR-T cells. For example, the traditional autologous adoptive transfer strategy is not appropriate for patients with a distempered immune system to isolate enough T cells in both quantity and quality; hence, recently allogeneic adoptive cell transfer strategy is developing. The CAR-T cells with two genes (TRAC and B2M) disrupted can make allogeneic T cells (these T cells come from healthy donors) adapt in acceptors. Mutation in T-cell receptor α constant (TRAC) leads to loss of αβ TCR on T-cell surface to avoid graft-versus-host-disease. Destruction of B2M can interfere with the expression of HLA-Is.84 Such gene reconstruction method enlarges the source of T cells for engineering. A study showed that disruption of endogenous PD1 pathway enhances the efficacy of gene disrupted allogeneic CAR-T cells by CRISPR/cas9 system, that is, triple simultaneous ablation of TCR, B2M, and PD1 of the engineered CAR-T cells improves antitumor ability.85 Using CRISPR/Cas9 system to direct a CD19-specific CAR to TRAC locus results in uniform CAR expression in human peripheral blood T cells and enhances T-cell potency in a mouse model of acute lymphoblastic leukemia which has avoided random CAR transduction compared with conventional retrovirus or lentivirus transfection methods.86 Nowadays, the success of CAR-T cells in hematological malignancies is inspiring, however is less in solid cancers, which is mainly due to the heterogeneity of a solid tumor and the complex protection of tumor microenvironment that can reduce T-cell trafficking or killing kinetics, loss of CAR expression, or exhaustion of CAR-T cells.87
 | Figure 2 The process diagram of CAR-T cellular immunotherapy targeting OCSCs. |
The current application of CAR-T cells in CSCs
The CSCs on the top of the hierarchy cancer are the basement of the heterogeneity and interfere in many signals to communicate with microenvironment. Targeting CSCs can evade the barriers against solid cancers. Many preclinical and clinical research studies have been employed by designing CAR-T cells to target CSCs in cancer immunotherapy. A study using CAR-NK cells to target OCSCs in vitro which target CD133 on ovarian cancer cell lines and are combined with chemotherapy showed a strong antitumor capability.8 A study showed that anti-AC133/CD133+-specific CAR-T cells targeting glioblastoma stem cells have therapeutic efficacy against GBM both in vitro and in vivo, while CD57 expression on T cells is upregulated which CD57 is not a bona fide CSC marker for GBM.5 EpCAM was targeted by CARs and the data demonstrated that anti-EpCAM-specific CARs had apparently antitumor capabilities in prostate cancer and peritoneal carcinomatosis in vitro and in vivo.4,88 Oncofetal antigen 5T4 is predominately expressed in nasopharyngeal carcinoma stem cell-like cells. CAR-T cells targeting 5T4 have anti-nasopharyngeal carcinoma ability when CARs are combined with cytokine-induced killer cells.6 A CAR-T cocktail immunotherapy has been implied on a 52-year-old female with advanced CCA targeting EGFR and CD133 who had gained an 8.5-month partial response (PR) from anti-EGFR CAR and a 4.5-month PR from anti-CD133 CAR suffering from toxicities in the meanwhile (Table 1).7
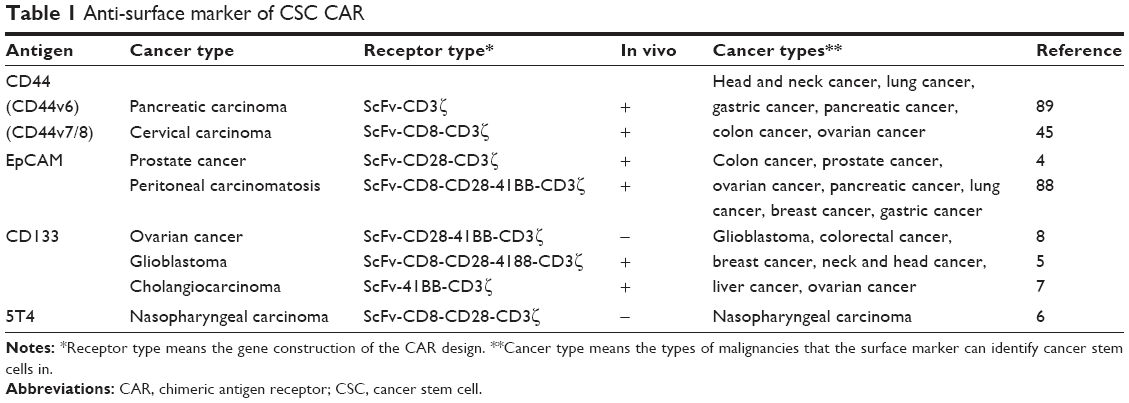 | Table 1 Anti-surface marker of CSC CAR |
As for CAR adoptive cellular immunotherapy in ovarian cancers, the first-generation CAR design targeting α-folate receptor (FR) was first practiced clinically in patients with ovarian cancer in 2006, although no reduction in tumor burden was seen in any patient.90 There have been many studies on ovarian cancer using CAR, and the targets are various and targets are including MUC16,91,92 FR-α,90 MUC1,93 NKG2D ligands,94 and mesothelin95,96 all with some barriers. It is noteworthy that anti-CD133 CAR-NK cells combined with chemotherapy have effect on ovarian cancer cell lines in vitro, which tests the determinacy of CD133 as a surface marker of OCSCs, represents the possibility in CAR therapy targeting OCSCs but also lacks powers of persuasion in vivo, and endows universal and exclusive limitation in CAR application.8
The promise and potential pitfalls of targeting OCSCs for CAR-T design
Targeting CSC markers to design CAR to eradicate tumor in vivo or in vitro, is there any advantage to capitalize it on OCSC markers to design CAR? In this scenario, there are three advantages: 1) Ovarian cancer has the tendency of recurrence after a period of clinical recovery from the primary foci, the biological behavior of which can be explained by “tumor stem cell model”.97 A myriad of research studies and practices in the last decades have identified the existence of OCSC population in ovarian cancer which is at the top of the pyramid of cancer ecosystem. Elimination of OCSCs and therapy targeting OCSCs are necessary to combat ovarian cancer; 2) CSCs can resist conventional therapy and in certain cancers have been observed to be enriched after conventional therapy which results in more dismal prognosis of chemotherapy and radiotherapy.98 Immunotherapy has brought major clinical progressing in antitumor therapy. Adoptive cellular immunotherapy labeled by “targeting cancer” is the optimal method, in that, on the one hand it can capture tumor cells by specific reorganization, and on the other hand, it may avoid unpredictable non-cancer cell toxicities to protect normal tissues; and 3) The CAR-T cell is one of the adoptive cellular immunotherapies which come from the bench to the bedside in a more directive reconstruction pathway guided by clinical need in a shorter experimental period. It is independent of MHC-I presentation which is always downregulated in tumor cells especially in CSCs to evade immune system surveillance. Allogeneic T-cell transfer strategies can enlarge the source of T cells for CAR manufacture,84 which will expand the production scale resulting in generic drugs for different patients. Furthermore, the immunogenicity of CARs should be averted to make CAR tools of choice for targeted cancer immunotherapies.99
So far, only one study in vitro has reported about specifically targeting OCSCs by CAR design.8 The paucity of specific markers to identify OCSCs is still pending urgent resolution. Ovarian cancer is intratumoral heterogeneous and harbors many pathology types which have different origins,100 and there may be more than one group of OCSCs in a tumor; hence, the heterogeneity of OCSCs cannot be identified by the only marker or group of markers, in that, even the most common markers of OCSCs are found in only <40% of tumors.18 In addition, unexpected toxicities emerge even though CAR-T cells have been tested in vitro tests and then were applied in clinical trials including cytokine release syndrome, neurologic toxicity, “on target/off tumor” recognition, and anaphylaxis, which calls for more safe administration undoubtedly.101
Although the CAR-T therapy has been hard-hitting therapy for numerous cancers, considering it as regimen for patients with ovarian cancer still has pitfalls. 1) Immunotherapy must combine other conventional therapies. Primary cytoreductive surgery should be considered as the gold standard to eliminate macroscopic foci for patients with advanced epithelial ovarian cancer.102 Chemotherapy and radiotherapy have decreased mortality rate of ovarian cancer in last decades. Meanwhile, ovarian cancer cytoreduction can induce transformation of T-cell ratio in the tumor which will enhance immune function against cancer.103 Epigenetic inhibitors such as DNA demethylation agents were examined in the clinic against chemotherapy-resistant ovarian cancer.104 Since the management of ovarian cancer needs is a multidisciplinary teamwork,105 “combination” is the headline of individual therapy referring to surgery, chemotherapy, epigenetic drugs, and immunotherapy. 2) OCSC is a rare population of the cancer; CAR designed for CSCs must combine with other immunotherapies such as the blockade of immune checkpoints, antibodies, and innate immunotherapy. Within CAR design, modifying more function parts is under hot investigation. For example, “combinatorial antigen recognition with balanced signaling” is an engineered T cell designed with a CAR and a chimeric costimulatory receptor (CCR). The CAR and CCR can recognize respective antigens which suggests that only when the two antigens are on the tumor cell surface, this CAR-T can kill the tumor cell.106 This way could suit OCSCs well because there are coexpression markers on OCSCs such as CD133+/EpCAM+, and this way will improve the specificity of CAR-T cells by preventing healthy cells from killing which is called “on antigen off target” effect. Different CAR-T cells can be used in the same patient targeting antigens sequentially which is called cocktail therapy, such as the case reported on a female with advanced CCA who was treated with targeting EGFR CAR-T cells and anti-CD133 CAR-T cells and had mitigation when considering the disease progression.7 3) A certain marker or a group of markers specific for CSCs that can be used to identify CSCs as an exact standard has not yet been found. Different markers can be found in the same cancer type which may imply that heterogeneity is not only in a tumor but also exists inside CSCs, so what is the earliest ancestor of CSCs is not certain yet or the current pathology cancer type standard does not suit for CSCs to match the disease. Some surface markers of stem cells are not always carried on the CSCs, which may be replaced by non-CSCs making up the bulk of tumor, so “relying on markers will fool you,” suggesting that the identification of CSC and non-CSC is not consistent indeed.107 It is probably due to that some non-CSCs can dedifferentiate into CSCs with a dynamic transformation. The therapy targeting CSCs should be combined not only mutually in the same treatment course but also in a scheme containing different steps of the tumor progressing in macro and various cell cycles in micro which is rigorously individual for every patient. 4) The most ideal markers on OCSCs as CAR-T antigens have not yet been discovered to our knowledge. For instance, CD133 which is the most widely investigated in CSCs of many cancer types is expressed in normal brain tissues, hematopoietic stem cells, and endothelial progenitor cells.24 Anti-CD133 CAR-T may result in toxicity for normal tissues (“on antigen off target”) without combined balance modification in CAR design to avoid side effects. More safe and effective strategies about CAR-T cells and immunotherapy by combination with other conventional therapy to eliminate cancer need further research.
Conclusion
The knowledge of cellular and molecular mechanisms underlying CSC characterizations is still very limited to explain the whole process of architecting the cancer bulk. CSCs in ovarian cancer (OCSCs) are a rare and elusive population which cannot be used to explain all the pathology behaviors of ovarian cancer although many markers have been identified and used in therapy such as CD133, CD47, CD44, ALDH1. Studies of epigenetic mechanisms and signal pathways may explore the concealing information behind the markers of CSCs/OCSCs, which will reveal the pathogenesis and prognosis of cancer. A novel marker identifying OCSCs may emerge in the future ideally meanwhile the current markers should be taken into account to optimize the genetic and epigenetic mechanisms underlying ovarian cancer primary formation, aggressive progression, and unanticipated relapse and metastases. The connection between immune system and ovarian cancer is not constant when synthetizing immune surveillance, immune evasion, and immune editing which can be taken advantage of for therapy. CAR-T cell adoptive cellular immunotherapy shows efficacy in some cancers when targeting CSC markers for ovarian cancer which is an optional candidate based on CAR construction method and specificity of OCSC antigen. Immunotherapies will not be isolated from traditional therapies like surgery, chemotherapy, and radiotherapy, and epigenetic drugs. Meanwhile, protection management to reduce adverse events and administration of regimens must be stringent in further research studies.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (81772772 and 81302242), Jilin Province Science and Technology Funds (20150204007YY, 20140204022YY, 20150204041YY, and 20160101043JC), and Jilin Province Development and Reform Commission Funds (2016C046-2 and 2014G073).
Disclosure
The authors report no conflicts of interest in this work.
References
Koury J, Zhong L, Hao J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017;2017:2925869. | ||
Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65(8):3025–3029. | ||
Desai A. Epithelial ovarian cancer: an overview. World J Transl Med. 2014;3(1):1–8. | ||
Deng Z, Wu Y, Ma W, Zhang S, Zhang YQ. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16:1. | ||
Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6(1):171–184. | ||
Guo X, Zheng H, Luo W, Zhang Q, Liu J, Yao K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci Rep. 2017;7(1):4859. | ||
Feng KC, Guo YL, Liu Y, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10(1):4. | ||
Klapdor R, Wang S, Hacker U, et al. Improved killing of ovarian cancer stem cells by combining a novel CAR-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28(10):886–896. | ||
Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. | ||
Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14(3):275–291. | ||
Kawasaki BT, Farrar WL. Cancer stem cells, CD200 and immunoevasion. Trends Immunol. 2008;29(10):464–468. | ||
Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012;198(3):281–293. | ||
Patel P, Chen EI. Cancer stem cells, tumor dormancy, and metastasis. Front Endocrinol (Lausanne). 2012;3:125. | ||
Talukdar S, Emdad L, Das SK, Sarkar D, Fisher PB. Evolving strategies for therapeutically targeting cancer stem cells. Adv Cancer Res. 2016;131:159–191. | ||
Sell S. On the stem cell origin of cancer. Am J Pathol. 2010;176(6):2584–2494. | ||
Matsui WH. Cancer stem cell signaling pathways. Medicine (Baltimore). 2016;95(1 Suppl 1):S8–S19. | ||
Webb PM, Jordan SJ. Epidemiology of epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2017;41:3–14. | ||
Lupia M, Cavallaro U. Ovarian cancer stem cells: still an elusive entity? Mol Cancer. 2017;16(1):64. | ||
Aguilar-Gallardo C, Rutledge EC, Martinez-Arroyo AM, Hidalgo JJ, Domingo S, Simon C. Overcoming challenges of ovarian cancer stem cells: novel therapeutic approaches. Stem Cell Rev. 2012;8(3):994–1010. | ||
Ottevanger PB. Ovarian cancer stem cells more questions than answers. Semin Cancer Biol. 2017;44:67–71. | ||
Shah M, Allegrucci C. Stem cell plasticity in development and cancer: epigenetic origin of cancer stem cells. Subcell Biochem. 2013;61:545–565. | ||
Wainwright EN, Scaffidi P. Epigenetics and cancer stem cells: unleashing, hijacking, and restricting cellular plasticity. Trends Cancer. 2017;3(5):372–386. | ||
Baba T, Convery PA, Matsumura N, et al. Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene. 2008;28(2):209–218. | ||
Irollo E, Pirozzi G. CD133: to be or not to be, is this the real question? Am J Transl Res. 2013;5(6):563–581. | ||
Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. | ||
Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445(7123):111–115. | ||
Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010;70(11):4624–4633. | ||
Wei XD, Zhou L, Cheng L, Tian J, Jiang JJ, Maccallum J. In vivo investigation of CD133 as a putative marker of cancer stem cells in Hep-2 cell line. Head Neck. 2009;31(1):94–101. | ||
Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132(7):2542–2556. | ||
Curley MD, Therrien VA, Cummings CL, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27(12):2875–2883. | ||
Grosse-Gehling P, Fargeas CA, Dittfeld C, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol. 2013;229(3):355–378. | ||
Zhang J, Guo X, Chang DY, Rosen DG, Mercado-Uribe I, Liu J. CD133 expression associated with poor prognosis in ovarian cancer. Mod Pathol. 2012;25(3):456–464. | ||
Sacks JD, Barbolina MV. Expression and function of CD44 in epithelial ovarian carcinoma. Biomolecules. 2015;5(4):3051–3066. | ||
Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11(4):254–267. | ||
Xu H, Tian Y, Yuan X, et al. The role of CD44 in epithelial-mesenchymal transition and cancer development. Onco Targets Ther. 2015;8:3783–3792. | ||
Bourguignon LY, Wong G, Earle C, Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. J Biol Chem. 2012;287(39):32800–32824. | ||
Shi Y, Liu C, Liu X, Tang DG, Wang J. The microRNA miR-34a inhibits non-small cell lung cancer (NSCLC) growth and the CD44hi stem-like NSCLC cells. PLoS One. 2014;9(3):e90022. | ||
Yoon C, Park DJ, Schmidt B, et al. CD44 expression denotes a subpopulation of gastric cancer cells in which hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res. 2014;20(15):3974–3988. | ||
Li L, Hao X, Qin J, et al. Antibody against CD44s inhibits pancreatic tumor initiation and postradiation recurrence in mice. Gastroenterology. 2014;146(4):1108–1118. | ||
Su YJ, Lai HM, Chang YW, Chen GY, Lee JL. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011;30(15):3186–3199. | ||
Zhang S, Balch C, Chan MW, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68(11):4311–4320. | ||
Shi MF, Jiao J, Lu WG, et al. Identification of cancer stem cell-like cells from human epithelial ovarian carcinoma cell line. Cell Mol Life Sci. 2010;67(22):3915–3925. | ||
Meng E, Long B, Sullivan P, et al. CD44+/CD24− ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin Exp Metastasis. 2012;29(8):939–948. | ||
Tjhay F, Motohara T, Tayama S, et al. CD44 variant 6 is correlated with peritoneal dissemination and poor prognosis in patients with advanced epithelial ovarian cancer. Cancer Sci. 2015;106(10):1421–1428. | ||
Dall P, Herrmann I, Durst B, et al. In vivo cervical cancer growth inhibition by genetically engineered cytotoxic T cells. Cancer Immunol Immunother. 2005;54(1):51–60. | ||
Guo Y, Feng K, Wang Y, Han W. Targeting cancer stem cells by using chimeric antigen receptor-modified T cells: a potential and curable approach for cancer treatment. Protein Cell. Epub 2017 Mar 13. | ||
Willingham SB, Volkmer JP, Gentles AJ, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109(17):6662–6667. | ||
Li Y, Lu S, Xu Y, et al. Overexpression of CD47 predicts poor prognosis and promotes cancer cell invasion in high-grade serous ovarian carcinoma. Am J Transl Res. 2017;9(6):2901–2910. | ||
Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7(10):11018–11032. | ||
Ruscito I, Cacsire Castillo-Tong D, Vergote I, et al. Exploring the clonal evolution of CD133/aldehyde-dehydrogenase-1 (ALDH1)-positive cancer stem-like cells from primary to recurrent high-grade serous ovarian cancer (HGSOC). A study of the Ovarian Cancer Therapy-Innovative Models Prolong Survival (OCTIPS) Consortium. Eur J Cancer. 2017;79:214–225. | ||
Ishiguro T, Sato A, Ohata H, et al. Establishment and characterization of an in vitro model of ovarian cancer stem-like cells with an enhanced proliferative capacity. Cancer Res. 2016;76(1):150–160. | ||
Wen Y, Hou Y, Huang Z, Cai J, Wang Z. SOX2 is required to maintain cancer stem cells in ovarian cancer. Cancer Sci. 2017;108(4):719–731. | ||
Kwon AY, Kim GI, Jeong JY, et al. VAV3 overexpressed in cancer stem cells is a poor prognostic indicator in ovarian cancer patients. Stem Cells Dev. 2015;24(13):1521–1535. | ||
Yang B, Yan X, Liu L, Jiang C, Hou S. Overexpression of the cancer stem cell marker CD117 predicts poor prognosis in epithelial ovarian cancer patients: evidence from meta-analysis. Onco Targets Ther. 2017;10:2951–2961. | ||
Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29(18):2672–2680. | ||
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–768. | ||
Dalerba P, Dylla SJ, Park IK, et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104(24):10158–10163. | ||
Zavesky L, Jandakova E, Kohoutova M. [Cancer stem cells and ovarian cancer Characteristics, importance and potential applications in clinical practice]. Ceska Gynekologie. 2013;78(2):169–174. Czech. | ||
Cioffi M, Dorado J, Baeuerle PA, Heeschen C. EpCAM/CD3-bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin Cancer Res. 2012;18(2):465–474. | ||
Kwiatkowska-Borowczyk EP, Gabka-Buszek A, Jankowski J, Mackiewicz A. Immunotargeting of cancer stem cells. Contemp Oncol (Pozn). 2015;19(1A):A52–A59. | ||
Sun Y, Jia X, Wu X. High expressions of Lgr5 and ALDH1 in primary epithelial ovarian cancer correlate with advanced tumor stage and grade as well as poor prognosis of the patients. Gynecol Obstet Invest. Epub 2015 Jun 20. | ||
Kryczek I, Liu S, Roh M, et al. Expression of aldehyde dehydrogenase and CD133 defines ovarian cancer stem cells. Int J Cancer. 2012;130(1):29–39. | ||
Santarpia M, Gonzalez-Cao M, Viteri S, Karachaliou N, Altavilla G, Rosell R. Programmed cell death protein-1/programmed cell death ligand-1 pathway inhibition and predictive biomarkers: understanding transforming growth factor-beta role. Transl Lung Cancer Res. 2015;4(6):728–742. | ||
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (New York, NY). 2011;331(6024):1565–1570. | ||
Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–848. | ||
Luna JI, Grossenbacher SK, Murphy WJ, Canter RJ. Targeting cancer stem cells with natural killer cell immunotherapy. Expert Opin Biol Ther. 2017;17(3):313–324. | ||
Yin T, Wang G, He S, Liu Q, Sun J, Wang Y. Human cancer cells with stem cell-like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL-2 and IL-15 activated natural killer cells. Cell Immunol. 2016;300:41–45. | ||
Krishnan V, Berek JS, Dorigo O. Immunotherapy in ovarian cancer. Curr Probl Cancer. 2017;41(1):48–63. | ||
Wu D, Wang J, Cai Y, et al. Effect of targeted ovarian cancer immunotherapy using ovarian cancer stem cell vaccine. J Ovarian Res. 2015;8:68. | ||
Naujokat C. Monoclonal antibodies against human cancer stem cells. Immunotherapy. 2014;6(3):290–308. | ||
Xu M, Rettig MP, Sudlow G, et al. Preclinical evaluation of Mab CC188 for ovarian cancer imaging. Int J Cancer. 2012;131(6):1351–1359. | ||
Jager M, Schoberth A, Ruf P, et al. Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM x anti-CD3). Cancer Res. 2012;72(1):24–32. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Pan Q, Li Q, Liu S, et al. Concise review: targeting cancer stem cells using immunologic approaches. Stem Cells. 2015;33(7):2085–2092. | ||
Mittica G, Genta S, Aglietta M, Valabrega G. Immune checkpoint inhibitors: a new opportunity in the treatment of ovarian cancer? Int J Mol Sci. 2016;17(7):pii:E1169. | ||
Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (New York, NY). 2016;352(6282):227–231. | ||
Geyer MB, Brentjens RJ. Review: current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy. 2016;18(11):1393–1409. | ||
Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor (CAR) design. Cancer Discov. 2013;3(4):388–408. | ||
Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625–2635. | ||
Guest RD, Hawkins RE, Kirillova N, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. | ||
Smith AJ, Oertle J, Warren D, Prato D. Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: summary and perspective. J Cell Immunother. 2016;2(2):59–68. | ||
Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. | ||
Abken MCH. TRUCKs: the fourth generation of CARs. Blood. 2012;119(18):10. | ||
Liu X, Zhang Y, Cheng C, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–157. | ||
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23(9):2255–2266. | ||
Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. | ||
Davenport AJ, Jenkins MR, Ritchie DS, et al. CAR-T cells are serial killers. Oncoimmunology. 2015;4(12):e1053684. | ||
Ang WX, Li Z, Chi Z, et al. Intraperitoneal immunotherapy with T cells stably and transiently expressing anti-EpCAM CAR in xenograft models of peritoneal carcinomatosis. Oncotarget. 2017;8(8):13545–13559. | ||
Hekele A, Dall P, Moritz D, et al. Growth retardation of tumors by adoptive transfer of cytotoxic T lymphocytes reprogrammed by CD44v6-specific scFv:zeta-chimera. Int J Cancer. 1996;68(2):232–238. | ||
Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20):22. | ||
Chekmasova AA, Rao TD, Nikhamin Y, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. 2010;16(14):3594–3606. | ||
Ramos CA, Dotti G. Chimeric antigen receptor (CAR)-engineered lymphocytes for cancer therapy. Expert Opin Biol Ther. 2011;11(7):855–873. | ||
Shi H, Liu L, Wang Z. Improving the efficacy and safety of engineered T cell therapy for cancer. Cancer Lett. 2013;328(2):191–197. | ||
Spear P, Barber A, Rynda-Apple A, Sentman CL. NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunol Cell Biol. 2013;91(6):435–440. | ||
Tanyi JL, Stashwick C, Plesa G, et al. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T cells. J Immunother. 2017;40(3):104–107. | ||
Beatty GL, Haas AR, Maus MV, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2(2):112–120. | ||
Foster R, Buckanovich RJ, Rueda BR. Ovarian cancer stem cells: working towards the root of stemness. Cancer Lett. 2013;338(1):147–157. | ||
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. | ||
Sadelain M, Brentjens R, Riviere I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–223. | ||
Karnezis AN, Cho KR, Gilks CB, Pearce CL, Huntsman DG. The disparate origins of ovarian cancers: pathogenesis and prevention strategies. Nat Rev Cancer. 2017;17(1):65–74. | ||
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. | ||
Hacker NF, Rao A. Surgery for advanced epithelial ovarian cancer. Best Pract Res Clin Obstet Gynaecol. 2017;41:71–87. | ||
Napoletano C, Bellati F, Landi R, et al. Ovarian cancer cytoreduction induces changes in T cell population subsets reducing immunosuppression. J Cell Mol Med. 2010;14(12):2748–2759. | ||
Natanzon Y, Goode EL, Cunningham JM. Epigenetics in ovarian cancer. Semin Cancer Biol. Epub 2017 Aug 3. | ||
Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384(9951):1376–1388. | ||
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotech. 2013;31(1):71–75. | ||
Kaiser J. The cancer stem cell gamble. Science. 2015;347(6219):226–229. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.