")
Back to Journals » International Journal of General Medicine » Volume 17
Application of Newly Customized Myeloid NGS Panel in the Diagnosis of Myeloid Malignancies
Authors Alkhatabi HA, Alqahtani W, Alsolami R, Elaimi A , Hazzazi MS , Almashjary MN, Alkhatabi HA, Alghuthami ME, Daous YM, Yasin EB , Barefah A
Received 27 August 2023
Accepted for publication 14 December 2023
Published 6 January 2024 Volume 2024:17 Pages 37—48
DOI https://doi.org/10.2147/IJGM.S437327
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Heba A Alkhatabi,1,2 Wejdan Alqahtani,3 Reem Alsolami,1,2 Aisha Elaimi,1,4 Mohannad S Hazzazi,1,2 Majed N Almashjary,1,2 Hind A Alkhatabi,5 Mohammad E Alghuthami,6 Yara M Daous,2,7 Elrashed B Yasin,8 Ahmed Barefah2,9
1Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah, 22254, Saudi Arabia; 2Hematology Research Unit (HRU), King Fahad Medical Research Center, King Abdulaziz University, Jeddah, 22254, Saudi Arabia; 3Department of Medical Laboratory, King Abdulaziz University Hospital, Jeddah, Saudi Arabia; 4Center of Excellence in Genomic Medicine Research (CEGMR), King Abdulaziz University, Jeddah, 22254, Saudi Arabia; 5Department of Biochemistry, College of Science, University of Jeddah, Jeddah, 21589, Saudi Arabia; 6GCC Accreditation Center GAC, Jeddah, Saudi Arabia; 7Department of Pathology, Faculty of Medicine, King Abdulaziz University, Jeddah, 22254, Saudi Arabia; 8Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Rabigh, 25732, Saudi Arabia; 9Hematology Department, Faculty of Medicine, King Abdulaziz University, Jeddah, 21589, Saudi Arabia
Correspondence: Elrashed B Yasin, Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Rabigh, 25732, Saudi Arabia, Tel +966 5 60910199, Email [email protected]
Purpose: Genetic mutations are major factors in the diagnosis and prognosis of leukemia, and it is difficult to assess these variants using single-gene analysis. Therefore, this study aimed to develop a fast and cost-effective method for genetic screening of myeloid malignancies using a customized next-generation sequencing (NGS) panel.
Patients and Methods: A customized myeloid panel was designed and investigated in 15 acute myeloid leukemia patients. The panel included 11 genes that were most commonly mutated in myeloid malignancies. This panel was designed to sequence the complete genome of CALR, IDH1, IDH2, JAK2, FLT3, NPM1, MPL, TET2, SF3B1, TP53, and MLL.
Results: Among the 15 patients, 14 actual pathogenic variants were identified in nine samples, and negative results were found in six samples. Positive findings were observed for JAK2, FLT3, SF3B1, and TET2. Interestingly, non-classical FLT3 mutations (c.1715A>C, c.2513delG, and c.2507dupT) were detected in patients who were negative for FLT3-ITD and TKD by routine molecular results. All identified variants were pathogenic, and the high coverage of the assay allowed us to predict variants at a low frequency (1%) with 1000x coverage.
Conclusion: Utilizing a custom panel allowed us to identify variants that were not detected by routine tests or those that were not routinely investigated. Using the costuming panel will enable us to sequence all genes and discover new potential pathogenic variants that are not possible with other commercially available panels that focus only on hotspot regions. This study’s strength in utilizing NGS and implanting a customized panel to identify new pathogenic variants that might be common in our population and important in routine diagnosis for providing optimal healthcare for personalized medicine.
Keywords: myeloid malignancies, myeloid leukemia, FLT3, NGS, genomic sequence
Introduction
Acute Myeloid leukemia (AML) is a phenotypically heterogeneous disorder that can occur due to genetic changes, cytogenetic alterations, and epigenetic abnormalities. Genetics is one of the main causes of Acute Myeloid Leukemia (AML) and contributes to its diagnosis and prognosis.1 Understanding the heterogeneity of myeloid malignancies and the presence of multiple genetic abnormalities is the greatest challenge in disease management. This has increased the demand for improved diagnostic method.2 The available molecular methods rely on a single test for examining a single abnormality, and searching for or examining multiple variants is laborious and cost-effective. Therefore, with advances in laboratory methods and the evolution of next-generation sequencing (NGS), it has become useful and easy to implement in routine diagnostic laboratories. NGS has demonstrated its utility as a diagnostic platform and value in clinical applications in several countries.3–6 Given techniques other than NGS, comprehensive analysis using single-gene assays is cumbersome and requires large amounts of DNA; thus, timely compilation of results for clinical purposes is not feasible.6 In the last decade, many targeted panels for myeloid malignancies have been developed and have become commercially available. These panels differ in their technology and the covered gene, but mostly cover genes involved in splicing machinery, epigenetic modifiers, cohesions, transcription factors, signaling molecules, and chromatin modifiers.6
Utilizing targeted panels with NGS has an advantage over whole-exome sequencing. First, cost will be lower and decreases over time with technological improvements. Furthermore, with an increase in the number of investigated genes, it will take more time to analyze, and it will be challenging to implement some molecular variant interpretation of the results and reporting. To manage the time and decrease the turnaround time of patients, especially when using targeted therapy for patients with positive FLT3 or IDH mutations using traditional molecular analysis in parallel with NGS, it takes a few days to reach the same result as a traditional molecular method. Therefore, we avoided duplicate studies and considered labor-intensive technique.7 Moreover, the smaller panel size had higher coverage and depth in somatic analysis for the highly sensitive detection of variants at 1% allele frequencies.
Therefore, the clinical application of a customized focused NGS panel will demonstrate its usefulness and robustness of the NGS panel as a fast and accurate detection method for genetic mutation screening in myeloid malignancies.
Materials and Methods
Patients
This study was performed on 15 AML patients with Acute Myeloid Leukemia using a myeloid-customized panel for next-generation sequencing applications. Patient samples (Bone marrow samples) and clinical information were obtained from different hospitals in the Makkah region, including King Abdulaziz University Hospital in Jeddah and King Abdullah Medical City in Makkah along with ethical approval number (17–358). Informed consent was obtained from all patients as per as the rules of the Helsinki Declaration. The inclusion criteria were as follows: Saudi Arabian patients who were not treated at presentation of the disease. The exclusion criteria were undertreatment or post-treatment and patients with other malignancies or metastases.
Custom Myeloid Panel Design
The customized myeloid panel included 11 genes that are the most commonly mutated genes in AML and was designed with the collaboration of the Illumina company. This panel was designed to sequence the full sequences of CALR, IDH1\2, FLT3, MPL, SF3B1, TP53, JAK2, MLL, NPM1, and TET2 Table 1.
 |
Table 1 Specification of the Myeloid Custom Panel |
Next-Generation Sequencing Using Myeloid Customized Panel
All the DNA samples were quantified using a fluorometry-based Qubit dsDNA HS Assay. The required starting concentration for the DNA sample was 5 ng/µL, and all the samples were diluted to reach the optimal concentration. DNA was amplified using 5X AmpliSeq HiFi mix (Illumina, AmpliSeq library plus), 2X AmpliSeq DNA Panel 1, 2X AmpliSeq DNA Panel 2 (Illumina, AmpliSeq custom myeloid panel), and nuclease-free water and then incubated in a thermal cycler (Applied Biosystems) according to the manufacturer’s recommendations. The samples were then partially digested using the FuPa enzyme and ligated with unique adaptors (i5) and (i7) as identifiers for each sample. Library clean-up was performed using Agencourt AMPure XPn and 70% ethanol to clean the pooled samples, and a LoBind tube was used on the magnetic stand to isolate a clear mixture. The cleaned library was amplified using 1x Lib Amp Mix and 10X Library Amp Primers and incubated in the master mix following the manufacturer’s recommendations. Subsequently, a second clean-up was performed, and the library was quantified using the Qubit 2.0 or 3.0 Fluorometer with the Qubit DNA HS Assay Kit.
In addition, library quality was assessed using a Fragment Analyzer with a high-sensitivity NGS fragment analysis kit (diluted to 1–5 ng/ μL). Subsequently, the library concentration was adjusted to 2nM, denatured using 0.2 N NaOH, diluted to 8 pM, and loaded onto the reagent cartridge to start sequencing on a MiSeq sequencer (Illumina). According to the manufacturer’s recommendation, the 20 pM library was diluted to 8 pM, the library was loaded onto the reagent cartridge, and sequencing was set up to start the run.
Data Analysis
The MiSeq Reporter Software 1.3.17. was used to convert the signals generated from the raw data to nucleotides in the FASTQ format. Integrative Genomics Viewer (IGV) was used to visualize read alignment and confirm variant calls. IGV was used to align short reads generated from FASTQ to reference the genome in Binary Alignment Map (BAM) format. Viewer Interpreter software was used to analyze the Variant Calling File (VCF) files of each sample. Hundreds of variants were detected on each sample; these variants are filtered according to their quality, clinical significance, and frequencies. The applied criteria for filtration in our study cohort included heterozygote genotype, SNVs, and insertion and deletion variants. All variants were passed through a filter with > 30 score in quality, > 20 read depth, and > 2% allele frequency. Pathogenicity was determined using algorithm software (polyphen “damaging”, SIFT “deleterious”) and Varsome databases (Figure 1).
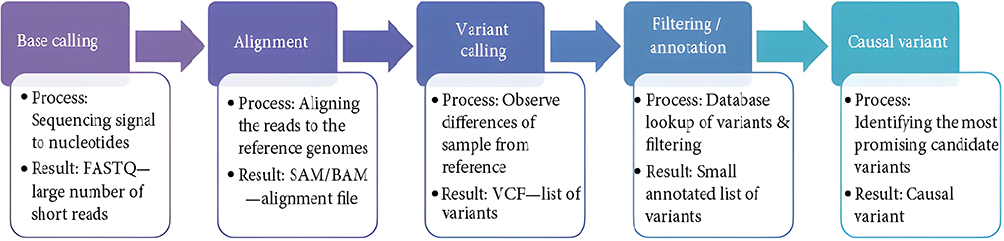 |
Figure 1 Bioinformatic pipeline in the analysis of AML patients. |
Results
Patients Sample
This study included 15 Acute Myeloid Leukemia cases collected from King Abdulaziz University Hospital in Jeddah and King Abdullah Medical City in Makkah. The subjects included 11 males and 4 females with a median age of 48 years (range, 20–69 years).
Cytogenetic and Molecular Results
Cytogenetic and molecular data were collected from the medical records of the Center of Excellence in Genomic Medicine Research, CEGMR, Jeddah, and King Abdullah Medical City in Makkah. The patient data are summarized in Table 2.
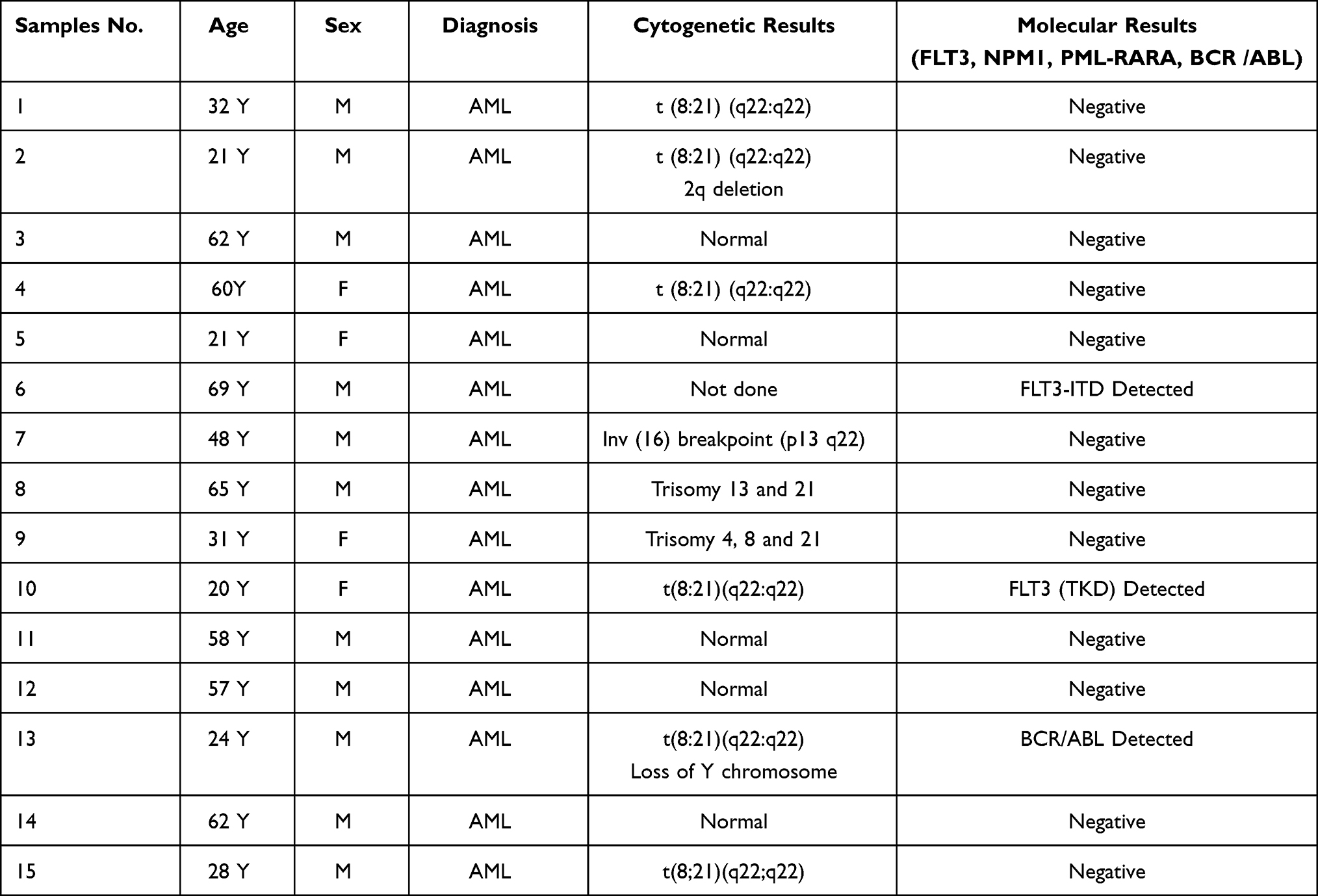 |
Table 2 Clinical and Molecular Characteristics of AML Patients |
The cytogenetic results for the patients showed that five patients had a normal karyotype (33%), another five patients had a single chromosomal abnormality (33%), four patients had a complex karyotype (27%), and one patient was not tested for cytogenetic abnormality (7%).
For the molecular results, all samples were analyzed for a single gene mutation or gene fusion, according to the clinical phenotype and the requested test. The molecular tests performed on the patient samples included BCR/ABL, PML/RARA fusions, and mutation screening for FLT3 and NPM1. The results showed that two patients were positive for FLT3 mutations (13%), and one patient had BCR-ABL fusion (7%). The remaining samples tested negative in molecular tests (80%).
NGS Analysis Result
NGS was performed using the Myeloid Customized Panel, which includes 11 genes that are the most screened genes in myeloid malignancies and the panel design covers the entire exonic region of the investigated genes.
Variant interpreter software (online software from Illumina) was used to analyze the VCF files. Hundreds of variants were detected on each sample, and that requires further filtration to identify the real and clinically relevant variants. The implemented filtration pipeline includes a passing filter, pathogenic and likely pathogenic variants, excluding non-coding variants, and benign and likely benign exonic variants.
Sixty-nine variants were identified among the examined cases, 69 variants were annotated. The IGV software was used to confirm the presence of variants in the BAM files of the 15 samples. After importing the reference genome (hg19) onto the IGV, Mapped files were loaded into the IGV to observe the alignment reads. Following BAM validation, 14 real variants were confirmed in 9 patients, and no variants were detected in 6 patients; examples of positive and negative variants detected by IGV are illustrated in Table 3 and (Figures 2 and 3).
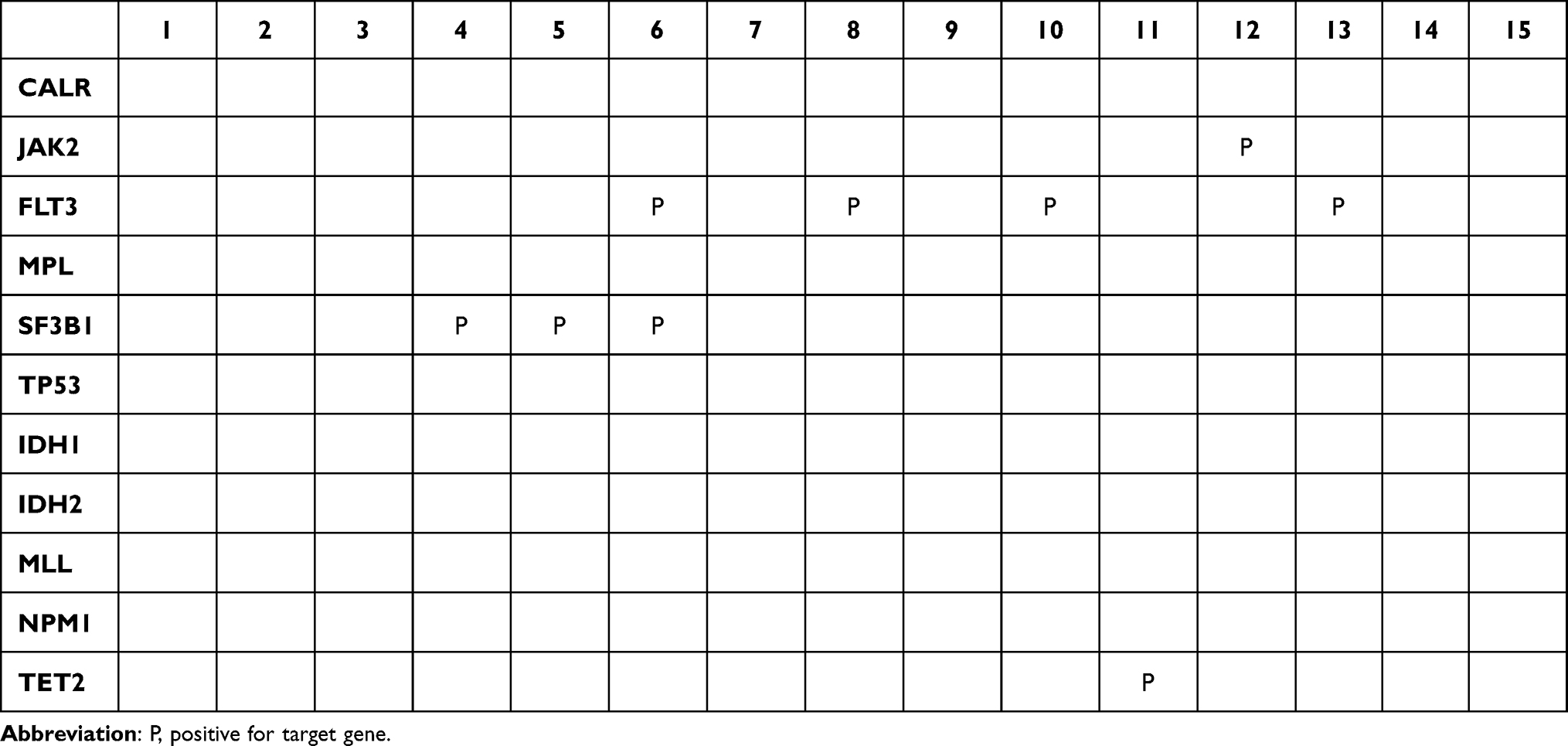 |
Table 3 Positive Sample Among Customized Genes List |
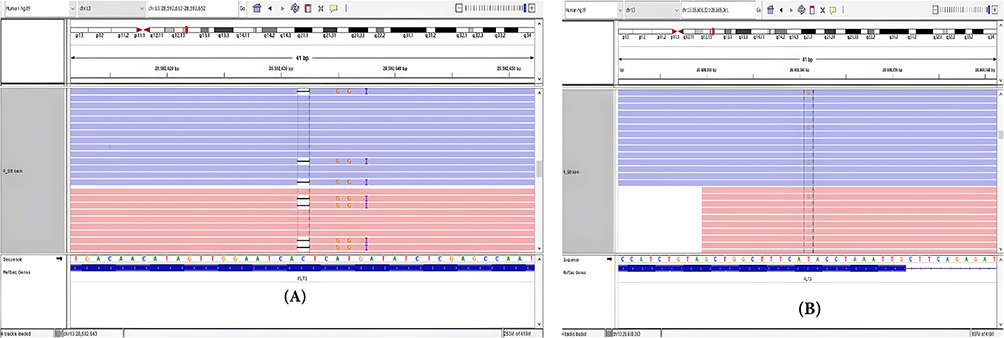 |
Figure 2 BAM confirmation results for FLT-3 mutation with a coordinate number (ch13: 28592632) (A) and FLT-3 mutation with a coordinate number (ch13: 28608341) (B) (in sample #8) by using IGV. |
 |
Figure 3 BAM validation of mutation in sample # 8 by using IGV and it showed false negative result of the analysis detected mutation and this will be called wrong event and the result will be negative. |
The most recurrent mutation was found on the Splicing factor 3b subunit 1 (SF3B1) gene in samples 4, 5, and 6. The detected variant was a nonsense mutation (NM_012433:c.3797G>A) in which the G base was changed to A on exon 25 and was reported as a pathogenic variant based on ClinVar (Clinical Variant).
In addition, different mutations have been detected in the FMS-like tyrosine kinase 3 (FLT3) gene in different samples. Some of these mutations were not detected using the conventional molecular method for FLT3 mutation detection, including (NM_004119.2c.1780_1781insGGAACA) and (NM_004119:c.1780T>G), in which GGAACA was inserted and the T base was changed to G on exon 14. These mutations were detected in the same patient (sample 13) and were likely pathogenic and pathogenic variants based on the genome interpreter, respectively. In addition, FLT3 missense mutation (NM_004119:c.2504A>T), in which the A base was replaced with T in exon 20, was found in sample 10, and it is a pathogenic variant. Furthermore, two missense variant mutations in FLT3 were found in one patient (sample 6) (NM_004119.2c.1807T>C) and (NM_004119.2c.2501G>A), in which the T base was changed to A on exon 14 and the G base was changed to A on exon 20, and they are likely pathogenic variants in the ClinVar and Varsome databases. Finally, (NM_004119: c.1715A>C), (NM_004119: c.2513delG) and (NM_004119:c.2507dupT) in which A base was replaced to C base on exon 14, G base was deleted and T base was duplicated on exon 20 respectively, all detected in sample 8 and based on varsome and ClinVar the first two variants are likely pathogenic and the last one is pathogenic respectively.
Furthermore, in Janus kinase 2 (JAK2) missense mutation was identified on patient number 12, the detected mutation is (NM_004972: c.1849G>T) in which G base was replaced to T on exon 14, and it is a pathogenic variant.
The Tet Methylcytosine Dioxygenase 2 (TET2) variant stop-gain mutation was found in patient number 12, and it is a pathogenic variant recognized as (NM_001127208: c.4393C>T) in which the C base was replaced with T on exon 10.
An isocitrate dehydrogenase 1 (IDH1) mutation was detected in patient 9, and this mutation was a missense mutation (NM_005896: c.395G>A) in which the G base was changed to A on exon 4, which was reported as a pathogenic variant. Finally, no variants were detected in samples 1, 2, 3,7,14, and 15 (Table 4).
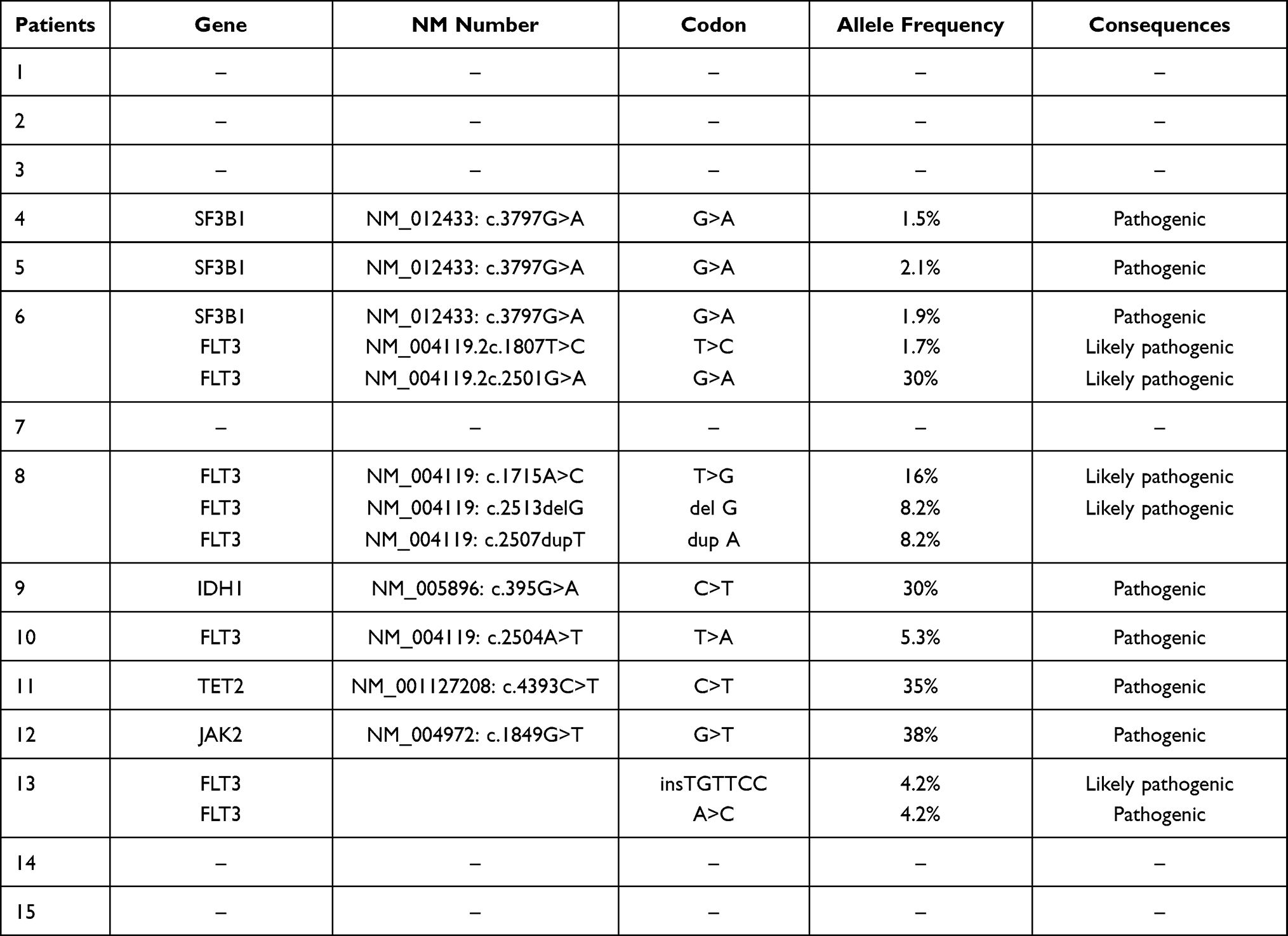 |
Table 4 Confirmed Variants Details Among Samples |
Comparison of Custom Kit and Ampliseq Myeloid Kit
Two different samples were processed using the Ampliseq myeloid kit from Illumina and validated using a custom kit. Quality metrics were assessed between the two kits, including amplicon mean coverage, percentage of Q30 reads, and total passing filter reads. In sample #1, the amplicon mean coverage was 4461.8, Q30% was 91.93% using a custom kit, and in the myeloid kit, the mean coverage of the amplicon was 4416.1, and Q30% was 96.60%.
In sample #2 from the custom kit, the amplicon mean coverage was 3590.9, and 92.82% for Q30. For the myeloid kit, 5995.6 was the mean coverage, and the Q30 was 96.49% (Table 5).
 |
Table 5 Quality Metrics of Myeloid Kit and Custom Panel on Two Repeated Samples |
Comparison Between NGS and Molecular Results
As explained previously, molecular alterations were detected in 3 out of 15 cases. FLT3-ITD, which was detected by molecular testing, was also detected using NGS. However, NGS analysis revealed additional FLT3 mutations in other samples that were negative in molecular analysis.
Samples 8 and 13 were negative for FLT3-ITD in the molecular tests, and NGS allowed for the detection of mutations in different exonic regions with different allele frequencies. Sample 8 was positive for FLT3 in (NM_004119:c.1715A>C), (NM_004119:c.2513delG), and (NM_004119:c.2507dupT) with allele frequencies of 16, 8.2, and 8.2%, respectively. Sample 13 was positive for FLT3 gene (NM_004119.2c.1780_1781insGGAACA) and (NM_004119: c.1780T>G) with a 4.2% allele frequency, respectively. About FLT3 positive molecular and NGS tests, sample 6 was positive (NM_004119.2: c.1807T>C) and (in M_004119.2: c.2501G>A) with a 1.7% and 30% allele frequency, respectively. In addition, sample 10 was positive (NM_004119: c.2504A>T) with an allele frequency of 5.3%.
Finally, there was only one BCR-ABL fusion in sample 13 detected by molecular analysis, which had an additional mutation in FLT3, as detected by NGS. However, NPM1 mutations were negative in both the molecular and NGS tests.
Comparison Between NGS and Cytogenetics
Different NGS results were identified, and the most commonly associated variants with a normal karyotype were mutations in JAK2, SF3B1, and TET2. A single abnormality was also associated with the unique variants of SF3B1 and FLT3. Finally, a complex karyotype was commonly associated with mutations in FLT3 and IDH1 genes, as shown in Figure 4.
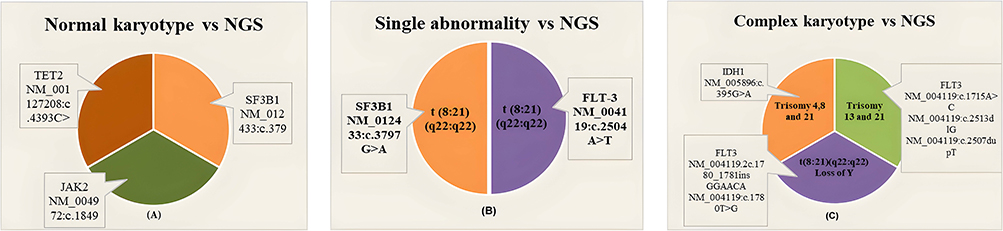 |
Figure 4 This is a figure. Comparison between Cytogenetic (Normal karyotype (A), single abnormality (B) and Complex karyotype (C)) and NGS results. |
Discussion
Myeloid malignancies are a group of diseases that includes acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPNs), chronic myeloid leukemia (CML), myelodysplastic/myeloproliferative neoplasms (MDS/MPN), and other rare diseases.8 These malignancies have different phenotypes owing to their genetic, epigenetic, and cytogenetic abnormalities. Mutational characterization is used in the diagnostic and prognostic stratification of myeloid neoplasia, and may further inform personalized treatment strategies using targeted and selective therapies.9
Using next-generation sequencing, a large number of novel mutations can be detected in various diseases, including myeloid malignancies.10 Therefore, this study aimed to develop a fast and cost-effective method for genetic screening of myeloid malignancies using a customized next-generation sequencing (NGS) panel. The difference between the designed panel and commercially available panels for myeloid diagnosis is that it covers the entire exonic region of the selected genes, not only the hotspot region, as in the other panel. In addition, the panel is small and includes only clinically relevant genes, which will allow for highly sensitive detection of small clonal sizes owing to the achievement of high coverage.
Quality validation is an essential step in verifying the utility of a panel in assessing the validity of a customized panel. Five samples were sequenced using different NGS kits, Ampliseq myeloid panel from Illumina, which covers the host spot region for all genes on the custom panel. The results were used to verify the overall quality, reads, and variants in the shared regions. The outcome of this comparison was that the Q30 was >90% using both kits, and the coverage was above 1000x, which is important in somatic analysis for the detection of low allele frequencies. Moreover, the read depth and coverage of the genes on the custom panel were higher and covered the entire exonic region, which is attributed to the small size of the custom panel, leading to high sensitivity in the detection of low variant frequencies.
In addition, a sample with a known result, FLT3-ITD, detected using the fragment analysis method, was used as a control to check the validity and accuracy of the panel in detecting this variant, and 100% accuracy was identified.
Another quality assessment was performed to examine the reproducibility of nucleotide variant calls in replicate sequencing experiments, including library preparation and target region capture of the same starting genomic DNA. The variants generated in the vcf file were compared with highly confident variants (SNVs and indels), and analytical performance metrics (Precision, Sensitivity, and Specificity) were evaluated. Based on these results, the custom panel was assessed with very high accuracy, sensitivity, and precision. Therefore, a custom panel was approved for further genomic analysis of all the samples in this study.
Sequencing of 15 AML samples was performed and compared with cytogenetic results and common molecular diagnostic markers such as FLT3-ITD, BCR-ABL, PML-RARA, and NPM1, which were investigated in the molecular diagnostic laboratory at CEGMR. Among the 15 patients, two were positive for FLT3 and one was positive for BCR-ABL, according to the molecular diagnosis. In the cytogenetic test, five patients had normal karyotypes, five patients had a single chromosomal abnormality, four patients had complex karyotypes, and one patient had no cytogenetic results.
Sequencing results showed 14 variants that were detected in nine samples, and negative results were found in six samples. We detected mutation on SF3B1 (c.3797G>A) gene in three samples at low frequencies ranging from (1.5–2%) in association with normal karyotype (33%) in one patient and translocation between 8 and 21(33%) in the second patient, and the third case did not have cytogenetic results for it. Molecular testing showed that three samples were negative for NPM1, PML-RARA, and BCR-ABL, but only one sample was positive for FLT3-ITD, and the patient had no cytogenetic results. Based on clinical databases (VarSome and ClinVar), this variant is pathogenic. SF3B1 encodes a protein that regulates transcription The prognosis of patient no.5, who had a normal karyotype with a mutation in SF3B1, was good, which disagrees with the findings of Hou et al, who showed a poor prognosis of patients with SF3B1 mutation in an intermediate-risk group with a normal karyotype.11 In our study, one patient had a poor prognosis associated with SF3B1 gene, but the patient had t(8:21) (q22:q22), which is considered a favorable risk group, and there was no supporting evidence for this finding.
The last sample with the same variant in SF3B1 was associated with FLT3-ITD mutation. FLT3-ITD is an unfavorable diagnostic marker and is associated with poor prognosis; however, patient no. 6 showed a good response to treatment and improved. There are no published articles about the prognosis of patients with combined mutations in SF3B1 and FLT3-ITD in de novo AML; however, Jeromin et al published an article about the incidence of SF3B1 with FLT3-ITD mutations in acute myeloid leukemia with ring sideroblasts.12
Mutations in FLT3 were detected in samples 8, 10, and 13. FLT-3 encodes for a tyrosine kinase receptor that plays an important role in cell growth. In sample 8, different mutations were detected in FLT3 by NGS analysis; none of them were FLT3-ITD, which matches the molecular result. Despite the absence of FLT-3 ITD, the panel allowed for the discovery of new variants of the gene that have never been discovered using another method that will help in changing the management and diagnosis of the disease. Based on the database, two variants (NM_004119: c.1715A>C) and (NM_004119: c.2513delG) were likely pathogenic and the third variant (NM_004119:c.2507dupT) had allele frequencies of 16%, 8.2%, and 8.2%, respectively. The cytogenetic findings for this patient were trisomies 13 and 12, and the patient did not survive. Therefore, despite the presence of an intermediate-risk group in this patient (trisomies 13 and 12 with mutations in FLT3 gene), the patient had a poor prognosis. This finding is supported by the findings of Canaani et al regarding the poor outcome of patients with FLT3 mutations in the intermediate-risk group.13 They used the ALWP/EBMT registry to determine the impact of the FLT-ITD mutation in the intermediate-risk group of 8558 patients who underwent allotransplantation. Although they focused on FLT3-ITD, examining the impact of other FLT3 mutations in association with prognostic scoring criteria is mandatory.
In sample 10, FLT3-TKD mutation was detected using NGS analysis, which was similar to the molecular test results. The cytogenetic finding was t(8:21) (q22:q22), which was a good prognostic marker. Based on ClinVar and VarSome, the variant (NM_004119: c.2504A>T) was pathogenic. Despite the presence of a mutation in FLT3-TKD, the patient’s prognosis was good, suggesting a less harmful impact of the mutation on disease progression. This is identical to the findings of Elyamany et al regarding FLT3-TKD and its unaggressive effect on patient prognosis of patients.14 However, our results disagree with those published by Yamamoto et al regarding the effect of the presence of FLT-TKD in patients with AML conferring a bad prognosis.15 Further screening should consider all prognostic markers to verify which has a stronger effect on disease prognosis.
In sample 13, two different variants were detected by NGS of FLT3 gene, which also had a BCR-ABL fusion gene, and cytogenetic findings showed t(8:21) (q22:q22) and a loss of the Y chromosome. The identified variants of FLT3 are (NM_004119: c.1780T>G), which is known for its pathogenic effect, and (NM_004119.2c.1780_1781insGGAACA), which is likely pathogenic and associated with a good prognosis.
Considering all previously discussed variants in FLT3 that suggested the prominent effect of the favorable prognostic marker (t(8:21)) when it presented with any FLT3 mutation other than ITD, all investigated patients showed a good prognosis despite the presence of FLT3, which is a bad marker with t(8;21). This suggestion is supported by Boissel et al, who found a poor prognosis in patients with t(8;21) with FLT3-ITD mutation.16
Furthermore, TET2 mutations were detected in sample 11 using NGS analysis. The molecular result was negative, and the cytogenetic findings were normal. Based on VarSome and ClinVar, the TET2 (NM_001127208: c.4393C>T) is pathogenic. TET2 encodes a protein that plays an important role in epigenetic modifications. The prognosis of the patient was good, despite the presence of TET2, a poor marker. Our findings disagree with what Chou et al published about the poor prognosis of AML patients who had an intermediate-risk group with a normal karyotype with TET2.17
In addition, IDH1 mutation was detected in Sample 9. The molecular result of the patient was negative, and the cytogenetic findings were trisomies 4, 8, and 21. Based on VarSome and ClinVar, the variant of IDH1 (NM_005896: c.395G>A) is pathogenic with a 30% allele frequency. IDH encodes proteins that play important roles in the Krebs cycle. The patient was a young female (30 years old) with IDH1 gene mutation, showed a poor prognosis, and did not survive. Our findings are supported by a published article that discussed the association between poor prognosis IDH1 mutation in younger patients aged <60 years.18
Furthermore, JAK2 mutations were detected in Sample 12. The molecular results of the patient were negative, and the cytogenetic findings were normal. Based on VarSome and ClinVar, the JAK2 variant (NM_004972:c.1849G>T) is pathogenic, with a high frequency of 38%. This gene was not examined in a molecular lab, which offers the advantage of using a panel test, as it covers different genes in a single test. Molecular tests can easily detect this variant specialty with a high allele frequency, but not requesting this test might lead to misdiagnosis. JAK2 encodes for a protein that controls hematopoietic cell production. The patient had a poor prognosis, which could be attributed to other unknown genetic markers because JAK2 plays an important role in transforming myeloproliferative to AML and has not been reported in association with disease progression, which is in accordance with other studies.19
Finally, regarding negative NGS results, six samples did not show any significant mutations based on the panel used. Negative findings were also observed in the molecular tests. Different cytogenetic findings were detected in each of the six samples. Samples 1 and 14 had t(8;21) (q22;q22) deletions, but sample 2 had t(8;21)(q22;q22) and 2q deletions. Samples 3 and 15 had normal karyotypes and sample 7 had Inv (16) breakpoints (p13 q22). The prognosis of these patients improved, except for patient 15, who did not survive. In this study, we focused on certain genes, but perhaps the patients had other mutations that were not covered by our panel, and saying that there were negative findings does not mean that the patient did not have any molecular changes. Further investigation is required to confirm the negative involvement of molecular variants and disease pathogenicity in these patients.
Utilizing NGS as a molecular test in the diagnosis of oncology is very important because of the limitations of single-gene analysis. This allowed us to detect novel variants that may be associated with our population, which requires further investigation. Customizing certain panels was helpful in terms of time, labor, and cost of the test in comparison to using whole-exome sequencing technology. Therefore, it is an alternative option for improving the diagnosis of the disease at a lower cost and in a shorter time.
Conclusion
Utilizing a custom panel allowed us to identify variants that were not detected by routine tests or those that were not routinely investigated. Using the costuming panel will enable us to sequence all genes and discover new potential pathogenic variants that are not possible with other commercially available panels that focus only on hotspot regions. This study’s strength in utilizing NGS and implanting a customized panel to identify new pathogenic variants that might be common in our population and important in routine diagnosis for providing optimal healthcare for personalized medicine.
Institutional Review Board Statement
The study was conducted, and patient samples were collected under ethical approval by the KAMC IRB number (17-358) registered at the National BioMedical Ethics Committee, King Abdulaziz City for Science and Technology (KACST) (registration no. H-02-K-001). Informed consent was obtained from all patients as per as the rules of the Helsinki Declaration.
Acknowledgments
This research work was funded by Institutional Fund Projects under grant no (IFPRC-034-140-2020); therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, Jeddah, Saudi Arabia. Also, we would like to thank King Abdulaziz University Hospital and King Abdullah medical city for facilitating sample collection and for approval to collect the samples.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research work was funded by Institutional Fund Projects under grant no (IFPRC-034-140-2020); therefore, the authors gratefully acknowledge technical and financial support from the Ministry of Education and King Abdulaziz University, Jeddah, Saudi Arabia.
Disclosure
The authors declare no conflict of interest.
References
1. Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi:10.1056/NEJMoa1112304
2. Alserihi R, Hameeda Ahmad HA, Heba Alkhatabi HA, et al. Analysis of NPM1 and FLT3 mutations in patients with acute myeloid leukemia in Jeddah, Saudi Arabia: a Pilot Study. Int J Biomed. 2023;13(1):73–83. doi:10.21103/Article13(1)_OA9
3. Kohlmann A, Grossmann V, Klein H-U, et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol. 2010;28(24):3858–3865. doi:10.1200/JCO.2009.27.1361
4. Grossmann V, Tiacci E, Holmes AB, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118(23):6153–6163.
5. Grossmann V, Schnittger S, Kohlmann A, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012;120(15):2963–2972.
6. Thol F, Kölking B, Damm F, et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3 -ITD or NPM1 mutations. Genes Chromosomes Cancer. 2012;51(7):689–695. doi:10.1002/gcc.21955
7. Bacher U, Shumilov E, Flach J, et al. Challenges in the introduction of next-generation sequencing (NGS) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018;8(11):113. doi:10.1038/s41408-018-0148-6
8. Patnaik MM, Tefferi A. Atypical chronic myeloid leukemia and myelodysplastic/myeloproliferative neoplasm, not otherwise specified: 2023 update on diagnosis, risk stratification, and management. Am J Hematol. 2023;98(4):681–689. doi:10.1002/ajh.26828
9. Duncavage EJ, Bagg A, Hasserjian RP, et al. Genomic profiling for clinical decision making in myeloid neoplasms and acute leukemia. Blood. 2022;140(21):2228–2247.
10. Wang L, Swierczek SI, Drummond J, et al. Whole-exome sequencing of polycythemia vera revealed novel driver genes and somatic mutation shared by T cells and granulocytes. Leukemia. 2014;28(4):935–938. doi:10.1038/leu.2014.7
11. Hou H-A, Liu C-Y, Kuo -Y-Y, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7(8):9084. doi:10.18632/oncotarget.7000
12. Jeromin S, Bacher U, Bayer K, et al. SF3B1 mutations are detectable in 48.9% of acute myeloid leukemia with normal karyotype (AML-NK) and≥ 15% ring sideroblasts and are closely related to FLT3-ITD and RUNX1 mutations. Blood. 2012;120(21):406.
13. Canaani J, Labopin M, Itälä-Remes M, et al. Prognostic significance of recurring chromosomal abnormalities in transplanted patients with acute myeloid leukemia. Leukemia. 2019;33(8):1944–1952. doi:10.1038/s41375-019-0439-3
14. Elyamany G, Awad M, Fadalla K, et al. Frequency and prognostic relevance of FLT3 mutations in Saudi acute myeloid leukemia patients. Adv Hematol. 2014;2014:1–7. doi:10.1155/2014/141360
15. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439.
16. Boissel N, Leroy H, Brethon B, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia. 2006;20(6):965–970. doi:10.1038/sj.leu.2404188
17. Chou W-C, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118(14):3803–3810.
18. Schnittger S, Haferlach C, Ulke M, et al. IDH1 mutations are detected in 6.6% of 1414 AML patients and are associated with intermediate risk karyotype and unfavorable prognosis in adults younger than 60 years and unmutated NPM1 status. Blood. 2010;116(25):5486–5496.
19. Vicente C, Vázquez I, Marcotegui N, et al. JAK2-V617F activating mutation in acute myeloid leukemia: prognostic impact and association with other molecular markers. Leukemia. 2007;21(11):2386–2390. doi:10.1038/sj.leu.2404812
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.