Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 5
Apolipoprotein E ε-4 as a genetic determinant of Alzheimer’s disease heterogeneity
Authors Kotze M, Luckhoff H, Brand T, Pretorius J, J van Rensburg S
Received 1 April 2014
Accepted for publication 8 May 2014
Published 12 January 2015 Volume 2015:5 Pages 9—18
DOI https://doi.org/10.2147/DNND.S41721
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Kotze MJ.
Views: 2866
MJ Kotze,1 HK Lückhoff,1 T Brand,1 J Pretorius,1 SJ van Rensburg2
1Division of Anatomical Pathology, Department of Pathology, Faculty of Medicine and Health Sciences, Stellenbosch University, Tygerberg, South Africa; 2Division of Chemical Pathology, Department of Pathology, Faculty of Medicine and Health Sciences, Stellenbosch University and the National Health Laboratory Service, Tygerberg Hospital, Tygerberg, South Africa
Abstract: Alzheimer’s disease (AD) displays a high degree of heterogeneity in terms of its etiology, presentation, prognosis, and treatment response. This can partly be explained by high-penetrance mutations in the amyloid precursor protein, presenilin 1 and presenilin 2 genes causing amyloid beta aggregation, which is a major pathogenic mechanism in the development of early-onset AD in a small subgroup of patients. Late-onset AD is considered a polygenic disorder in which cumulative risk resulting from interaction with modifiable environmental risk factors may be responsible for the majority of cases. The ε-4 allele of the apolipoprotein E (APOE) gene has emerged as the most significant genetic risk factor for late-onset AD, influencing nearly every pathogenic domain affected in AD. It is a major risk factor for cerebral amyloid angiopathy, recognized as a common pathological finding in an AD subtype associated with white matter dysfunction. The APOE ε-4 allele is also a known risk factor for ischemic stroke, which can result in vascular dementia or contribute to subcortical vascular dysfunction. In this review, we evaluate the clinical relevance of APOE genotyping in relation to cholesterol metabolism and available evidence on risk reduction strategies applicable to AD.
Keywords: Alzheimer’s disease, heterogeneity, APOE, cholesterol, polymorphism, pharmacogenetics
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder associated with a significant disease burden that places tremendous emotional and financial strain on families, caregivers, and society at large. AD is largely underreported on death certificates and medical records, with the figure in the United States found to be six times higher than previously estimated.1 Between 2006 and 2010, the global prevalence of AD increased by nearly 35% (going from 25.6 million to 35.6 million), and it is expected to increase threefold by 2050.2 It is anticipated that AD will then affect ~1% of the global population, the majority (71%) of whom will reside in low- or middle-income countries.3,4 The growing prevalence of AD globally has been attributed to a worldwide increase in life expectancy across different population groups. Intervention strategies aimed at delaying disease onset and progression by merely 1 year may result in more than 9 million people being spared this devastating diagnosis by 2050.2
In sub-Saharan Africa, dementia is set to become one of the major public health care challenges of the third millennium, as its prevalence is expected to increase dramatically from the current rate of ~2%–4% in the coming decades.5,6 AD further places a significant strain on the global economy. In 2010, the direct and indirect health expenditure for AD worldwide exceeded 1% of the global gross domestic product, at $600 billion. Its total contribution to global health expenditure, including medical services and hospice care, is anticipated to reach $1.1 trillion in the near future.7,8
Diagnosis of AD
AD has conventionally been defined as a clinicopathological entity, with extracellular amyloid beta (Aβ) aggregation and intracellular neurofibrillary tangle formation being associated with its clinical symptomatic emergence. However, new diagnostic criteria for AD proposed by the National Institute on Aging and the Alzheimer’s Association, which include a preclinical, asymptomatic stage, are in accord with the notion that characteristic neuropathology may arise years or even decades before clinical presentation. This shift from a definitive to a probabilistic diagnostic approach is assisted by the use of novel biomarkers and imaging methodology, which may identify asymptomatic individuals and enable the timely implementation of intervention strategies aimed at curbing disease onset.9,10 Although intended primarily for research purposes, these criteria may be of immediate use to the clinician. As novel diagnostic tools are becoming increasingly available in general practice, accurate diagnosis facilitated by these emerging technologies may significantly optimize patient care.11 However, this approach has been critiqued because of the concept of disease being inexorably related to impairment, clinical difficulty in differentiating preclinical AD from other neurodegenerative diseases such as Fronto-Temporal Dementia and Progressive Supranuclear Palsy, a failure to acknowledge overlapping pathological changes underlying disease onset, and the need for further validation of suggested biomarkers for AD.12
Genetic determinants of AD risk
Recent genomic advances have greatly increased our understanding of the underlying pathogenesis of AD, which is known to involve a significant heritability component (~60%–80%) as is evident from epidemiological and twin studies.13 High-penetrance mutations in the amyloid precursor protein and presenilin 1 and presenilin 2 genes, which are inherited in a Mendelian pattern, result in excess cerebral accumulation of neurotoxic Aβ and are implicated in the development of early-onset AD, accounting for ~1% of cases. First-degree relatives of affected patients are also at a significantly increased risk of developing AD.
Although early-onset AD is relatively rare, late-onset AD has been linked to more than 600 genes, of which only a minority are supported by a sufficient level of evidence. This includes variation in the apolipoprotein E (APOE) gene that forms the focus of this review. The APOE gene is located on the long arm of chromosome 19 (q) at position 13.2 and consists of four exons and three introns totaling 3,597 base pairs. As a component of chylomicrons and intermediate-density lipoproteins, APOE plays a crucial regulatory role in lipid metabolism as well as the transport of cholesterol and fat-soluble compounds such as vitamins.14 The APOE gene is highly polymorphic, with the two most extensively studied single nucleotide polymorphisms resulting in an interchangeable arginine (Arg) to cysteine (Cys) substitution at amino acid positions 112 and 158, corresponding to the ε-2 (rs7412) and ε-4 (rs429358) alleles (and therefore E2 and E4 protein isoforms), respectively.15 Individuals with the APOE ε-4 allele are 15 times more likely to develop AD compared with noncarriers.
The APOE ε-4 allele is a major determinant of genetic predisposition for late-onset AD in different population groups and across boundaries for age and sex.16 This supports the notion that the amyloid cascade hypothesis plays a central pathogenic role in sporadic as well as monogenic forms of the condition.17 The APOE ε-4 allele affects nearly every pathological domain in AD, including Aβ aggregation, neurofibrillary tangle formation, regional and global cerebral atrophy, decreased synaptic plasticity, deranged lipid metabolism, and cholinergic dysfunction.18 Coinheritance of deleterious genetic variants implicated in these processes associated with oxidative stress and inflammation may lead to an earlier age of onset in AD, suggesting a combined detrimental effect on AD pathology.19,20 We hypothesized that the production of hydroxyl radicals contributing to lipotoxicity and oxidative stress in patients with AD needs to be counteracted by constantly providing the cells with antioxidants and other essential nutrients, and that a drug that would stimulate the regrowth of neurons may be beneficial.21
It is important to emphasize that the expression of low-penetrance polymorphic variants in genes such as APOE is highly context-dependent. The influence of modulating interactions with occupational exposures and other environmental risk factors (eg, smoking, low socioeconomic status, and poor dietary and lifestyle habits), together with associated medical comorbidities, may create a high-risk metabolic milieu conducive to disease development. Modifiable nongenetic factors are therefore an important focus for primary and secondary preventive strategies aimed at halting disease onset and progression.
Cholesterol metabolism, APOE genotype, and AD
The relationship between cholesterol and AD remains incompletely understood. Cholesterol plays a key role in neurodevelopment, maintains the structural integrity of neuronal cell membranes, and ensures physiological extracellular messenger activity as well as neuronal repair. It has been proposed that the use of cholesterol and apolipoproteins as biomarkers may facilitate prognostication and guide therapeutic decision making in a broad spectrum of psychiatric illnesses, including autistic spectrum disorders, major depressive disorder, schizophrenia, and AD, as reviewed by Woods et al.22 A reduction in neuronal cholesterol content promotes colocalization of beta-secretase 1 and amyloid precursor protein, disrupting plasmin generation and contributing to neurotoxic Aβ production and associated neurodegeneration.23,24 However, elevated cholesterol levels also promote Aβ production and aggregation, as well as tau hyperphosphorylation, neuritic plaque formation, and cerebrovascular ischemia.25,26 This apparent contradiction highlights the uncertainty regarding the molecular mechanisms implicated in cholesterol-mediated AD neuropathology.
Altered serum cholesterol levels may contribute to the pathogenesis of AD by modulating cerebral APOE mRNA expression.27 Limited amounts of cholesterol are able to cross the blood–brain barrier (BBB) incorporated as high-density lipoproteins. Large lipoproteins are prohibited,28 whereas oxidized derivatives of cholesterol (oxysterols), including 27-hydroxycholesterol, are able to cross the BBB. Oxysterol levels may be elevated in patients with AD, with these compounds being associated with increased cerebral Aβ production in vivo.29,30 These findings may partly explain the link between elevated circulating cholesterol levels and neuropathological findings in AD. Limiting oxidative stress by means of health-promoting lifestyle or dietary adaptations, including smoking cessation and ensuring adequate dietary antioxidant intake, might prove more beneficial as a preventive measure than decreasing endogenous cholesterol synthesis.80,82–84
Epidemiological studies investigating the role of cholesterol in AD have reported mixed results. Many have correlated elevated cholesterol levels with an increased risk of developing mild cognitive impairment (MCI) and AD later in life, as well as an increased rate of cognitive decline in those with established dementia, independent of APOE genotype. However, conflicting studies have either failed to replicate these findings or demonstrated an inverse relationship between cholesterol levels and AD risk. The majority of positive findings note an increase in AD risk associated with elevated cholesterol levels during midlife, whereas similar changes during later life have been correlated with a decreased risk for dementia.31–33 These conflicting findings may be explained by variation in the timing of cholesterol measurement with regard to age and the course of the underlying disease process.
In light of these findings, there is significant interest in the potential beneficial effects of lipid-lowering therapies, particularly statins, in mitigating the onset and progression of AD. Preclinical studies have clearly indicated that statins modulate the processing of amyloid precursor protein, correlating with a decrease in cerebral and cerebrospinal fluid levels of Aβ. Simvastatin in particular has been shown to improve cognitive functioning, protect against neurodegeneration in vivo, and promote cerebrovascular integrity in animal models.34,35 However, results from human observational studies have been mixed. Multiple retrospective case-control studies suggest statins may prevent the onset of MCI and AD, as well as alter the course of disease progression.36–40 A recent meta-analysis also concluded that statins may be beneficial in preventing AD.41 Certain studies have shown that statin use in individuals at an increased risk for AD improves verbal fluency and working memory. However, others have proposed a detrimental effect on certain cognitive abilities,42 and cohort studies have failed to associate statin use with a decreased risk of developing AD.43
Although an early randomized control trial (RCT) demonstrated significant improvement in cognitive performance and a decrease in serum Aβ levels in AD patients treated with statins,44 subsequent larger trials have failed to demonstrate that statins alter the course or progression of established AD or offer additional neuroprotective benefits.45,46 Another RCT also concluded that statin use does not improve cognition in established AD, and as such, is not currently recommended for AD prevention.47 Other lipid-lowering agents including niacin, cholestyramine, and fibrates are also not effective in protecting against AD.48 Various explanations have been put forth concerning such apparent negative findings evident for the majority of RCTs to date. Regarding study design, observational studies typically differ regarding the duration of statin use and period of observation, whereas discrepancy between the duration of statin use and the timing of clinical assessment is frequently noted between different studies. In addition, the distinction between preexisting and incident AD is often not apparent.26 Various forms of bias, including mortality, indication, and cessation bias, also play a role. The role of cardiometabolic risk factors, inflammation, and the presence of coexisting vascular dementia may also not be fully considered. The type and dosage of statins administered may vary, whereas statins also differ in their BBB permeability and lipophilicity. With the exception of their lipid-lowering properties, statins also improve BBB integrity; possess anti-inflammatory, antioxidant, and profibrinolytic properties, and improve endothelial dysfunction.49 To what extent these factors contribute to the aforementioned findings remains incompletely understood.
Future investigation is required to elucidate the effect of statin therapy in ε-4 allele carriers with AD who are normocholesterolemic.50 The beneficial role of statins in lowering dementia risk appears to be independent of APOE genotype, whereas the association between hyperlipidemia and increased dementia prevalence is only evident in patients without AD and in ε-4 allele noncarriers.51 Young et al developed a Gaussian predictive model integrating multimodal data gathered from volumetric magnetic resonance imaging, fludeoxyglucose positron emission tomography, determination of cerebrospinal fluid t-tau, p-tau, and aβ42 levels, and APOE genotyping, which correlated with conversion rates from MCI to AD.52 A higher frequency of the ε-4 allele observed in patients with primarily amnestic MCI further suggests an increased risk of conversion to AD in this subgroup.53 These findings are in accordance with results reported by Fagan et al, who showed that baseline fludeoxyglucose positron emission tomography measurements and episodic memory loss predicted the rate of conversion from MCI to AD, whereas the p-tau181p/Aβ1-42 ratio predicted further cognitive decline.54
Statin-induced adverse effects, including myalgia, which occurs in approximately 2% of patients;55 a benign elevation in liver enzyme levels,56 and an increased risk for new-onset diabetes57 are widely known. Cognitive effects such as memory loss and confusion have been associated with simvastatin in particular, tend to run a variable course, and significantly influence quality of life.58 These cognitive adverse effects have come under recent scrutiny by the US Food and Drug Administration. At least one study has suggested statins might adversely affect cognition in AD patients;59 however, a recent meta-analysis concludes that statins do not appear to adversely affect cognition in those with normal functioning at baseline.60
APOE genotype, cerebral vasculopathy, white matter pathology, and clinical heterogeneity in AD
A preponderance of evidence now suggests that AD, ischemic heart disease, and stroke share overlapping risk factors, including obesity, type 2 diabetes mellitus, hypertension, dyslipidemia, smoking, physical inactivity, oxidative stress, and hyperhomocysteinemia.61,62 A multidisciplinary therapeutic approach centered on lifestyle-based interventions targeting shared disease pathways may decrease disease risk across the diagnostic spectrum.63
In addition to its multifunctional role in AD, the APOE ε-4 allele is also associated with accelerated atherogenesis and greater risk for ischemic stroke. Multiple cerebral infarctions may lead to vascular dementia, which commonly coexists with AD.64,65 A recent meta-analysis clearly demonstrated a linear relationship between APOE genotype, serum low-density lipoprotein-cholesterol levels and carotid intimal media thickness, consistent with the view that ε-4 allele carriers are at an increased risk for ischemic stroke.66 It should, however, be emphasized that even homozygosity for the ε-4 allele only partially accounts for vascular risk.67
White matter pathology is frequently encountered in AD, being associated with focal symptomatology as well as global cognitive impairment. Aβ deposition in cortical and leptomeningeal arteries is strongly correlated with the APOE ε-4 allele, resulting in cerebral amyloid arteriopathy (CAA). This common (80%–90%) neuropathological characteristic of AD is closely related to structural white matter abnormalities, arising secondary to endothelial dysfunction, basement membrane thickening, lobar hemorrhages, and vessel stenosis.68,69 Vascular pathology underlying white matter dysfunction is observed in many forms of subcortical dementia, including extensive lacunar infarcts as well as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy.70 Hall et al71 evaluated the relationship between a biomarker panel reflecting cardiovascular risk and inflammation, as well as microvascular dysfunction and neuropsychiatric symptomatology in mild AD. The authors found a significant positive association between cholesterol and all four items on the neuropsychiatric inventory, which was restricted to male patients, reflecting the importance of sex as a determinant of clinical heterogeneity. The presence of CAA in APOE ε-4 allele carriers may further play a synergistic role in AD progression.69 However, other studies note that although white matter pathology originates secondary to vascular dysfunction, it is not directly related to APOE genotype, which rather influences the localization of CAA and thereby contributes to the emergence of focal cerebral pathology and corresponding clinical heterogeneity.72,73 It has been suggested that the presence of capillary CAA in APOE ε-4 allele carriers might constitute a specific subtype of AD.74 The localization of cerebral Aβ deposition and CAA may account for a degree of clinical heterogeneity observed in AD.75 Absence of the APOE ε-4 allele is further associated with an atypical clinical presentation in presenting at an earlier age, which merits further consideration of other risk factors for AD.76,77 Given the reciprocal influence of APOE genotype and CAA on parenchymal as opposed to vascular Aβ aggregation, further investigation of whether the combined presence of these factors influences clinical and therapeutic heterogeneity in AD is also warranted.
AD typically first presents with episodic memory loss. However, some patients may present at an earlier age with atypical symptoms. This clinical heterogeneity is partly a reflection of focal cerebral pathology, including atrophy and hypoperfusion; executive dysfunction and visuospatial symptoms are associated with synucleinopathy and microvascular pathology, focal temporal atrophy with an amnestic picture, and parietal pathology with disordered visuospatial processing. It is interesting to note that although the APOE ε-4 allele accelerates disease progression in AD, patients who present atypically or at an earlier age seldom carry the risk-associated allele.77,78 This suggests that the presence of such symptoms and associated pathology in ε-4 carriers might represent a distinct subtype of AD.
AD a paradigm shift from treatment to prevention
Health-promoting lifestyle changes are of proven benefit in ameliorating risk for cardiovascular disease and stroke. In line with a growing international health care focus on primary prevention of disease, dementia was identified as a major global health aim at the 2013 G8 summit. An international team of experts argued that public health policy should reflect current knowledge supporting the risk-modulating benefits of smoking cessation, regular physical exercise, a nutritional diet rich in fruits, vegetables, and fish; as well as adequate B vitamin intake in reducing the risk of AD and dementia.79 The implementation of national policies aimed at preventing cumulative risk for chronic noncommunicable diseases by means of lifestyle-based intervention strategies has proven beneficial in resource-limited settings in the developing world.80,81
There is particular interest in the Mediterranean diet as an effective strategy to decrease cumulative risk for AD and dementia-related mortality.82–84 Results from the Lyon Diet Heart Study showed an astonishing 70% reduction in mortality in patients randomly assigned to a Mediterranean-style diet compared with a low-cholesterol diet, despite only a 6% decrease in total cholesterol levels in the former group.85 Benefits attributable to the Mediterranean diet therefore appear to be largely independent of its cholesterol-lowering effects, but are rather accredited to a higher dietary intake of folate, as well as the protective benefits of flavonoids, omega-3 fatty acids, antioxidants, and moderate alcohol intake.86 We recently confirmed the cardiovascular health benefits of moderate polyphenol-rich red wine consumption in healthy volunteers participating in a 6-week crossover alcohol intervention study, where an increase in HDL cholesterol observed was less pronounced in APOE ε-4 carriers.87
Physical activity in middle age and later life may reduce the risk for progression from MCI to AD. However, results from epidemiological studies remain inconsistent, and no RCTs to date have been performed to definitely establish the value of lifestyle-based interventions aimed at improving neurocognitive performance in at-risk individuals.88 In line with a growing shift toward the prevention of AD, there is a need to better define the exact duration, type, and intensity of physical activity required to effectively reduce cumulative AD risk.89 The Exercise and Nutritional Interventions for Cognitive and Cardiovascular Health Enhancement (ENLIGHTEN) trial is set to establish the efficacy of physical exercise and the DASH (dietary approaches to stop hypertension) diet in patients at increased cardiometabolic risk, as well as assess the mechanisms whereby these interventions may improve neurocognitive performance.
Influence of APOE genotype on therapeutic decision making in AD
APOE genotyping, when performed as part of a comprehensive clinical risk assessment strategy considering environmental modulators of phenotypic expression, may facilitate the timely implementation of tailored lifestyle-centered treatment strategies and optimize pharmacotherapy in AD.69 Differentiating between hypercholesterolemic patients with the monogenic disorder, familial hypercholesterolemia (FH), and those carrying the APOE ε-4 allele is important, as treatment strategies would differ. A relatively high incidence of mild cognitive impairment has recently been reported in FH patients, possibly due to early exposure to elevated cholesterol or low-density lipoprotein dysfunction.90 The importance of the availability of a DNA test for diagnosis of FH as a high-risk cardiovascular subtype91 is emphasized by the clinical variability of this condition, which complicates clinical diagnosis.92,93 Although FH patients require lipid-lowering medication to reduce their risk of ischemic vascular disease, APOE ε-4 carriers are less responsive to statin therapy.94 APOE ε-4 carriers appear to be more vulnerable to the deleterious effects of excessive alcohol consumption, cigarette smoking, a sedentary lifestyle, and high dietary intake of saturated fat, suggesting interventions targeting these risks may be beneficial in decreasing risk aimed at AD prevention.95,96 Lowering of cumulative risk aimed at AD prevention is very important in APOE ε-4 allele carriers, as the benefits associated with many lifestyle-orientated interventions, including regular physical exercise and supplementation with omega-3 fatty acids, may be less pronounced or absent in AD patients carrying the APOE ε-4 allele (Table 1). This may be attributed to the attenuating effects of preclinical neuropathology, which can arise years or even decades before presentation. Furthermore, although a significant body of evidence implicates oxidative stress and chronic inflammation in the development and progression of AD, RCTs to date have failed to significantly associate antioxidant or omega-3 fatty acid supplementation with clinical improvement.97
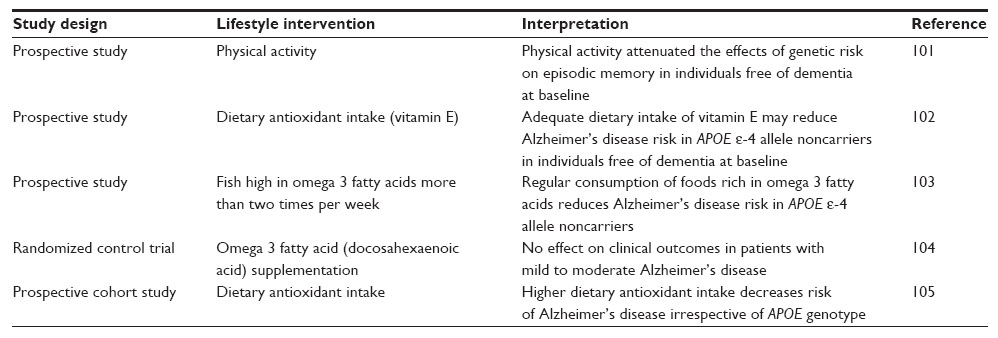 | Table 1 Influence of APOE genotype on the efficacy of lifestyle-based interventions in relation to Alzheimer’s disease |
Up to 80% of interpatient variation in the therapeutic response to medications commonly used to treat AD may be explained by genotype. Both APOE genotype and sex are important determinants of treatment response in AD.98 Multiple studies,99 have demonstrated that AD patients who carry the APOE ε-4 allele show a differential response to conventional pharmacotherapy (Table 2). Coinheritance of inactivating CYP2D6 alleles, conferring ultrarapid or poor metabolizer status, appear to have an additive effect.100
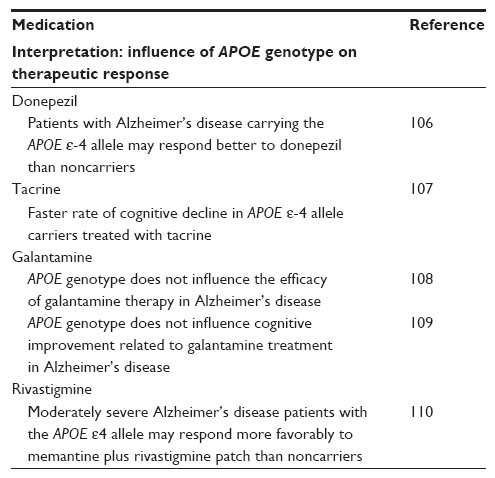 | Table 2 Influence of APOE genotype on the efficacy of acetylcholine-esterase inhibitors traditionally used in Alzheimer’s disease |
Conclusion
AD is commonly viewed as a hopeless diagnosis, relegating patients to a life marked by feelings of uncertainty and fear. Providing a caregiver, family member, or physician with additional genetic information may be viewed as contributing to this prevailing aura of futility. However, a growing appreciation that the phenotypic expression of risk-associated variants of low-penetrance in genes such as APOE ε-4, implicated in the development of AD, is largely dependent on a conducive metabolic milieu creates hope that timely health-promoting lifestyle-centered intervention strategies may overcome an apparently insurmountable genetic predisposition. Growing insight into the multifunctional role of the APOE ε-4 allele in explaining a certain degree of the marked interpatient etiological, clinical, and therapeutic heterogeneity noted for this condition gives hope that the identification of distinct treatable subtypes of AD may provide an opportunity for effective primary and secondary intervention. It may be viewed as the resilient duty of the physician to become increasingly knowledgeable about the therapeutic implications of personalized genotyping as a means to tailor treatment to the needs of the individual. A paradigm shift in thinking is required for genetic insights to be viewed as a beneficial avenue for intervention, rather than irrefutable confirmation of an incurable diagnosis.
A healthy lifestyle to prevent AD based on a similar approach applicable to cardiovascular risk reduction would be most beneficial when followed throughout life, rather than only after disease manifestation. This may be particularly important in individuals with a genetic predisposition to AD, to protect against disease development or reduce the risk of vascular disease and diabetes associated with this neurodegenerative condition.
Acknowledgments/Disclosure
This work is based on the research supported in part by the National Research Foundation (NRF) of South Africa (UID 83962). The Grantholder acknowledges that opinions, findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the authors, and that the NRF accepts no liability whatsoever in this regard. We also gratefully acknowledge the financial support from Winetech and the Technology for Human Resources and Industry Program (THRIP). MJK is a director and shareholder of Gknowmix (Pty) Ltd, which has developed a database tool for research translation under the auspices of the Innovation Centre of the South African Medical Research Council. As an extension of this initiative focused on capacity building, the Technology Innovation Agency (TIA) is acknowledged for an internship grant awarded to J Pretorius. The authors have no other conflicts of interest in this work.
References
James BD, Leurgans SE, Hebert LE, Scherr PA, Yaffe K, Bennett DA. Contribution of Alzheimer disease to mortality in the United States. Neurology. 2014;82(12):1045–1050. | |
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. | |
Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8). | |
Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9(1):63–75. | |
Kalaria RN, Maestre GE, Arizaga R, et al; World Federation of Neurology Dementia Research Group. Alzheimer’s disease and vascular dementia in developing countries: prevalence, management, and risk factors. Lancet Neurol. 2008;7(9):812–826. | |
Mavrodaris A, Powell J, Thorogood M. Prevalences of dementia and cognitive impairment among older people in sub-Saharan Africa: a systematic review. Bull World Health Organ. 2013;91(10):773–783. | |
Stefanacci RG. The costs of Alzheimer’s disease and the value of effective therapies. Am J Manag Care. 2011;17 Suppl 13:S356–S362. | |
Alzheimer’s Association, Thies W, Bleiler L. 2011 Alzheimer’s disease facts and figures. Alzheimers Dement. 2011;7(2):208–244. | |
Dubois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9(11): 1118–1127. | |
Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257–262. | |
Budson AE, Solomon PR. New diagnostic criteria for Alzheimer’s disease and mild cognitive impairment for the practical neurologist. Pract Neurol. 2012;12(2):88–96. | |
Giaccone G, Arzberger T, Alafuzoff I, et al; BrainNet Europe consortium. New lexicon and criteria for the diagnosis of Alzheimer’s disease. Lancet Neurol. 2011;10(4):298–299. | |
Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63(2):168–174. | |
Vance JE, Hayashi H. Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim Biophys Acta. 2010;1801(8):806–818. | |
Belbin O, Dunn JL, Ling Y, et al. Regulatory region single nucleotide polymorphisms of the apolipoprotein E gene and the rate of cognitive decline in Alzheimer’s disease. Hum Mol Genet. 2007;16(18):2199–2208. | |
Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997;278(16):1349–1356. | |
Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011;16(9):903–907. | |
Cedazo-Mínguez A. Apolipoprotein E and Alzheimer’s disease: molecular mechanisms and therapeutic opportunities. J Cell Mol Med. 2007;11(6):1227–1238. | |
van Rensburg SJ, Potocnik FC, De Villiers JN, Kotze MJ, Taljaard JJ. Earlier age of onset of Alzheimer’s disease in patients with both the transferrin C2 and apolipoprotein E-epsilon 4 alleles. Ann N Y Acad Sci. 2000;903:200–203. | |
Lehmann DJ, Schuur M, Warden DR, et al. Transferrin and HFE genes interact in Alzheimer’s disease risk: the Epistasis Project. Neurobiol Aging. 2012;33(1):202. e1–e13. | |
Van Rensburg SJ, Carstens ME, Potocnik FC, et al. Transferrin C2 and Alzheimer’s disease: another piece of the puzzle found? Med Hypotheses. 1995;44(4):268–272. | |
Woods AG, Sokolowska I, Taurines R, et al. Potential biomarkers in psychiatry: focus on the cholesterol system. J Cell Mol Med. 2012;16(6):1184–1195. | |
Abad-Rodriguez J, Ledesma MD, Craessaerts K, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol. 2004;167(5):953–960. | |
Stefani M, Liguri G. Cholesterol in Alzheimer’s disease: unresolved questions. Curr Alzheimer Res. 2009;6(1):15–29. | |
Sparks DL. Coronary artery disease, hypertension, ApoE, and cholesterol: a link to Alzheimer’s disease? Ann N Y Acad Sci. 1997;826:128–146. | |
Kandiah N, Feldman HH. Therapeutic potential of statins in Alzheimer’s disease. J Neurol Sci. 2009;283(1–2):230–234. | |
Petanceska SS, DeRosa S, Sharma A, et al. Changes in apolipoprotein E expression in response to dietary and pharmacological modulation of cholesterol. J Mol Neurosci. 2003;20(3):395–406. | |
Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12(5):284–296. | |
Björkhem I, Heverin M, Leoni V, Meaney S, Diczfalusy U. Oxysterols and Alzheimer’s disease. Acta Neurol Scand Suppl. 2001;85:43–49. | |
Ghribi O. Potential mechanisms linking cholesterol to Alzheimer’s disease-like pathology in rabbit brain, hippocampal organotypic slices, and skeletal muscle. J Alzheimers Dis. 2008;15(4):673–684. | |
Kivipelto M, Helkala EL, Laakso MP, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ. 2001;322(7300):1447–1451. | |
Mielke MM, Zandi PP, Sjögren M, et al. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005;64(10):1689–1695. | |
Solomon A, Kivipelto M, Wolozin B, Zhou J, Whitmer RA. Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement Geriatr Cogn Disord. 2009;28(1):75–80. | |
Ramirez C, Tercero I, Pineda A, Burgos JS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J Alzheimers Dis. 2012;4(1):161–174. | |
Tong XK, Lecrux C, Rosa-Neto P, Hamel E. Age-dependent rescue by simvastatin of Alzheimer’s disease cerebrovascular and memory deficits. J Neurosci. 2012;32(14):4705–4715. | |
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356(9242):1627–1631. | |
Miller LJ, Chacko R. The role of cholesterol and statins in Alzheimer’s disease. Ann Pharmacother. 2004;38(1):91–98. | |
Zamrini E, McGwin G, Roseman JM. Association between statin use and Alzheimer’s disease. Neuroepidemiology. 2004;23(1–2):94–98. | |
Wolozin B, Wang SW, Li NC, Lee A, Lee TA, Kazis LE. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. | |
Haag MD, Hofman A, Koudstaal PJ, Stricker BH, Breteler MM. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J Neurol Neurosurg Psychiatry. 2009;80(1):13–17. | |
Wong WB, Lin VW, Boudreau D, Devine EB. Statins in the prevention of dementia and Alzheimer’s disease: a meta-analysis of observational studies and an assessment of confounding. Pharmacoepidemiol Drug Saf. 2013;22(4):345–358. | |
Muldoon MF, Ryan CM, Sereika SM, Flory JD, Manuck SB. Randomized trial of the effects of simvastatin on cognitive functioning in hypercholesterolemic adults. Am J Med. 2004;117(11):823–829. | |
Rea TD, Breitner JC, Psaty BM, et al. Statin use and the risk of incident dementia: the Cardiovascular Health Study. Arch Neurol. 2005;62(7):1047–1051. | |
Friedhoff LT, Cullen EI, Geoghagen NS, Buxbaum JD. Treatment with controlled-release lovastatin decreases serum concentrations of human beta-amyloid (A beta) peptide. Int J Neuropsychopharmacol. 2001;4(2):127–130. | |
Rockwood K. Epidemiological and clinical trials evidence about a preventive role for statins in Alzheimer’s disease. Acta Neurol Scand Suppl. 2006;185:71–77. | |
Feldman HH, Doody RS, Kivipelto M, et al; LEADe Investigators. Randomized controlled trial of atorvastatin in mild to moderate Alzheimer disease: LEADe. Neurology. 2010;74(12):956–964. | |
Sano M, Bell KL, Galasko D, et al. A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology. 2011;77(6):556–563. | |
Shepardson NE, Shankar GM, Selkoe DJ. Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch Neurol. 2011;68(11):1385–1392. | |
Yang D, Knight RA, Han Y, et al. Statins Protect the Blood Brain Barrier Acutely after Experimental Intracerebral Hemorrhage. J Behav Brain Sci. 2013;3(1):100–106. | |
Caballero J, Nahata M. Do statins slow down Alzheimer’s disease? A review. J Clin Pharm Ther. 2004;29(3):209–213. | |
Dufouil C, Richard F, Fiévet N, et al. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology. 2005;64(9):1531–1538. | |
Young J, Modat M, Cardoso MJ, Mendelson A, Cash D, Ourselin S; Alzheimer’s Disease Neuroimaging Initiative. Accurate multimodal probabilistic prediction of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neuroimage Clin. 2013;2:735–745. | |
Sasaki M, Kodama C, Hidaka S, et al. Prevalence of four subtypes of mild cognitive impairment and APOE in a Japanese community. Int J Geriatr Psychiatry. 2009;24(10):1119–1126. | |
Fagan AM, Mintun MA, Shah AR, Holtzman DM, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer’s disease. EMBO Mol Med. 2009;1(8–9):371–380. | |
Opie LH. Exercise-induced myalgia may limit the cardiovascular benefits of statins. Cardiovasc Drugs Ther. 2013;27(6):569–572. | |
Bhardwaj SS, Chalasani N. Lipid-lowering agents that cause drug-induced hepatotoxicity. Clin Liver Dis. 2007;11(3):597–613. | |
Carter AA, Gomes T, Camacho X, Juurlink DN, Shah BR, Mamdani MM. Risk of incident diabetes among patients treated with statins: population based study. BMJ. 2013;346:f2610. | |
Evans MA, Golomb BA. Statin-associated adverse cognitive effects: survey results from 171 patients. Pharmacotherapy. 2009;29(7): 800–811. | |
Padala KP, Padala PR, McNeilly DP, Geske JA, Sullivan DH, Potter JF. The effect of HMG-CoA reductase inhibitors on cognition in patients with Alzheimer’s dementia: a prospective withdrawal and rechallenge pilot study. Am J Geriatr Pharmacother. 2012;10(5):296–302. | |
Swiger KJ, Manalac RJ, Blumenthal RS, Blaha MJ, Martin SS. Statins and cognition: a systematic review and meta-analysis of short- and long-term cognitive effects. Mayo Clin Proc. 2013;88(11):1213–1221. | |
Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62(10):1556–1560. | |
Rosendorff C, Beeri MS, Silverman JM. Cardiovascular risk factors for Alzheimer’s disease. Am J Geriatr Cardiol. 2007;16(3):143–149. | |
Kotze MJ, van Rensburg SJ. Pathology supported genetic testing and treatment of cardiovascular disease in middle age for prevention of Alzheimer’s disease. Metab Brain Dis. 2012;27(3):255–266. | |
Saidi S, Slamia LB, Ammou SB, Mahjoub T, Almawi WY. Association of apolipoprotein E gene polymorphism with ischemic stroke involving large-vessel disease and its relation to serum lipid levels. J Stroke Cerebrovasc Dis. 2007;16(4):160–166. | |
Zende PD, Bankar MP, Kamble PS, Momin AA. Apolipoprotein e gene polymorphism and its effect on plasma lipids in arteriosclerosis. J Clin Diagn Res. 2013;7(10):2149–2152. | |
Khan TA, Shah T, Prieto D, et al. Apolipoprotein E genotype, cardiovascular biomarkers and risk of stroke: systematic review and meta-analysis of 14,015 stroke cases and pooled analysis of primary biomarker data from up to 60,883 individuals. Int J Epidemiol. 2013;42(2):475–492. | |
Kofler BM, Miles EA, Curtis P, et al. Apolipoprotein E genotype and the cardiovascular disease risk phenotype: impact of sex and adiposity (the FINGEN study). Atherosclerosis. 2012;221(2):467–470. | |
McCarron MO, Nicoll JA. Apolipoprotein E genotype and cerebral amyloid angiopathy-related hemorrhage. Ann N Y Acad Sci. 2000;903: 176–179. | |
Love S. Contribution of cerebral amyloid angiopathy to Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2004;75(1):1–4. | |
Turner MA, Moran NF, Kopelman MD. Subcortical dementia. Br J Psychiatry. 2002;180:148–151. | |
Hall JR, Wiechmann AR, Johnson LA, et al. Biomarkers of vascular risk, systemic inflammation, and microvascular pathology and neuropsychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis. 2013;35(2):363–371. | |
Hirono N, Kitagaki H, Kazui H, Hashimoto M, Mori E. Impact of white matter changes on clinical manifestation of Alzheimer’s disease: A quantitative study. Stroke. 2000;31(9):2182–2188. | |
Kljajevic V, Meyer P, Holzmann C, et al; EDSD study group. The ε4 genotype of apolipoprotein E and white matter integrity in Alzheimer’s disease. Alzheimers Dement. 2014;10(3):401–404. | |
Thal DR, Papassotiropoulos A, Saido TC, et al. Capillary cerebral amyloid angiopathy identifies a distinct APOE epsilon4-associated subtype of sporadic Alzheimer’s disease. Acta Neuropathol. 2010;120(2): 169–183. | |
Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2): 106–118. | |
van der Flier WM, Pijnenburg YA, Fox NC, Scheltens P. Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ε4 allele. Lancet Neurol. 2011;10(3):280–288. | |
Lam B, Masellis M, Freedman M, Stuss DT, Black SE. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimers Res Ther. 2013;5(1):1. | |
Khachaturian AS, Corcoran CD, Mayer LS, Zandi PP, Breitner JC; Cache County Study Investigators. Apolipoprotein E epsilon4 count affects age at onset of Alzheimer disease, but not lifetime susceptibility: The Cache County Study. Arch Gen Psychiatry. 2004;61(5): 518–524. | |
Smith AD, Yaffe K. Dementia (including Alzheimer’s disease) can be prevented: statement supported by international experts. J Alzheimers Dis. 2014;38(4):699–703. | |
Sarrafzadegan N, Baghaei A, Sadri GH, et al. Isfahan healthy heart program: Evaluation of comprehensive, community-based interventions for non-communicable disease prevention. Prevention Control. 2006;2(2):73–84. | |
Harati H, Hadaegh F, Momenan AA, et al. Reduction in incidence of type 2 diabetes by lifestyle intervention in a middle eastern community. Am J Prev Med. 2010;38(6):628–636. | |
Scarmeas N, Stern Y, Tang MX, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006;59(6):912–921. | |
Scarmeas N, Luchsinger JA, Mayeux R, Stern Y. Mediterranean diet and Alzheimer disease mortality. Neurology. 2007;69(11):1084–1093. | |
Psaltopoulou T, Sergentanis TN, Panagiotakos DB, Sergentanis IN, Kosti R, Scarmeas N. Mediterranean diet, stroke, cognitive impairment, and depression: A meta-analysis. Ann Neurol. 2013;74(4): 580–591. | |
de Lorgeril M, Salen P, Martin JL, Monjaud I, Delaye J, Mamelle N. Mediterranean diet, traditional risk factors, and the rate of cardiovascular complications after myocardial infarction: final report of the Lyon Diet Heart Study. Circulation. 1999;99(6):779–785. | |
Sofi F, Macchi C, Abbate R, Gensini GF, Casini A. Effectiveness of the Mediterranean diet: can it help delay or prevent Alzheimer’s disease? J Alzheimers Dis. 2010;20(3):795–801. | |
Kotze MJ, Marnewick JL, Kidd M, Fisher LR, Velden DP. Assessment of the impact of hereditary factors on biochemical parameters of cardiovascular risk in relation to moderate alcohol consumption. Nutr Aging. 2014;2:189–195. | |
Pope SK, Shue VM, Beck C. Will a healthy lifestyle help prevent Alzheimer’s disease? Annu Rev Public Health. 2003;24:111–132. | |
Rolland Y, Abellan van Kan G, Vellas B. Physical activity and Alzheimer’s disease: from prevention to therapeutic perspectives. J Am Med Dir Assoc. 2008;9(6):390–405. | |
Zambón D, Quintana M, Mata P, et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am J Med. 2010;123(3):267–274. | |
Kotze MJ, Kriegshäuser G, Thiart R, et al. Simultaneous detection of multiple familial hypercholesterolemia mutations facilitates an improved diagnostic service in South african patients at high risk of cardiovascular disease. Mol Diagn. 2003;7(3–4):169–174. | |
Kotze MJ, Davis HJ, Bissbort S, Langenhoven E, Brusnicky J, Oosthuizen CJ. Intrafamilial variability in the clinical expression of familial hypercholesterolemia: importance of risk factor determination for genetic counselling. Clin Genet. 1993;43(6):295–299. | |
Kotze MJ, De Villiers WJ, Steyn K, et al. Phenotypic variation among familial hypercholesterolemics heterozygous for either one of two Afrikaner founder LDL receptor mutations. Arterioscler Thromb. 1993;13(10):1460–1468. | |
Baptista R, Rebelo M, Decq-Mota J, et al. Apolipoprotein E epsilon-4 polymorphism is associated with younger age at referral to a lipidology clinic and a poorer response to lipid-lowering therapy. Lipids Health Dis. 2011;10:48. | |
Kivipelto M, Rovio S, Ngandu T, et al. Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. J Cell Mol Med. 2008;12(6B):2762–2771. | |
Mangialasche F, Kivipelto M, Solomon A, Fratiglioni L. Dementia prevention: current epidemiological evidence and future perspective. Alzheimers Res Ther. 2012;4(1):6. | |
Persson T, Popescu BO, Cedazo-Minguez A. Oxidative stress in Alzheimer’s disease: why did antioxidant therapy fail? Oxid Med Cell Longev. 2014;2014:427318. | |
Farlow MR, Lahiri DK, Poirier J, Davignon J, Schneider L, Hui SL. Treatment outcome of tacrine therapy depends on apolipoprotein genotype and gender of the subjects with Alzheimer’s disease. Neurology. 1998;50(3):669–677. | |
Poirier J. Apolipoprotein E4, cholinergic integrity and the pharmacogenetics of Alzheimer’s disease. J Psychiatry Neurosci. 1999;24(2): 147–153. | |
Cacabelos R. Pharmacogenomics in Alzheimer’s disease. Methods Mol Biol. 2008;448:213–357. | |
Ferencz B, Jonsson Laukka E, Welmer AK, et al. The Benefits of Staying Active in Old Age: Physical Activity Counteracts the Negative Influence of PICALM, BIN1, and CLU Risk Alleles on Episodic Memory Functioning. Psychol Aging. Epub March 24, 2014. | |
Morris MC, Evans DA, Bienias JL, et al. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002;287(24):3230–3237. | |
Huang TL, Zandi PP, Tucker KL, et al. Benefits of fatty fish on dementia risk are stronger for those without APOE epsilon4. Neurology. 2005;65(9):1409–1414. | |
Quinn JF, Raman R, Thomas RG, et al. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304(17):1903–1911. | |
Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002;287(24): 3223–3229. | |
Choi SH, Kim SY, Na HR, et al. Effect of ApoE genotype on response to donepezil in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2008;25(5):445–450. | |
Sjögren M, Hesse C, Basun H, et al. Tacrine and rate of progression in Alzheimer’s disease – relation to ApoE allele genotype. J Neural Transm. 2001;108(4):451–458. | |
Aerssens J, Raeymaekers P, Lilienfeld S, Geerts H, Konings F, Parys W. APOE genotype: no influence on galantamine treatment efficacy nor on rate of decline in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12(2):69–77. | |
Suh GH, Jung HY, Lee CU, et al; Korean Galantamine Study Group. Effect of the apolipoprotein E epsilon4 allele on the efficacy and tolerability of galantamine in the treatment of Alzheimer’s disease. Dement Geriatr Cogn Disord. 2006;21(1):33–39. | |
Han HJ, Kim BC, Lee JY, et al. Response to rivastigmine transdermal patch or memantine plus rivastigmine patch is affected by apolipoprotein E genotype in Alzheimer patients. Dement Geriatr Cogn Disord. 2012;34(3–4):167–173. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.