")
Back to Journals » OncoTargets and Therapy » Volume 12
Apiosporamide, A 4-hydroxy-2-pyridone Alkaloid, Induces Apoptosis Via PI3K/Akt Signaling Pathway In Osteosarcoma Cells
Authors Zhang Y, Zhang Q, Bao J, Huang J, Zhang H
Received 7 June 2019
Accepted for publication 20 September 2019
Published 21 October 2019 Volume 2019:12 Pages 8611—8620
DOI https://doi.org/10.2147/OTT.S218692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yao Dai
Yuying Zhang,1 Qianqian Zhang,1 Jie Bao,1 Jintian Huang,2 Hua Zhang1
1Department of Biotechnology, School of Biological Science and Technology, University of Jinan, Jinan 250022, People’s Republic of China; 2Department of Orthopedics, Shandong Provincial Hospital Affiliated to Shandong University, Jinan 250021, People’s Republic of China
Correspondence: Hua Zhang
Department of Biotechnology, School of Biological Science and Technology, University of Jinan, 336 West Road of Nan Xinzhuang, Jinan 250022, People’s Republic of China
Tel +86 531 8973 6199
Email [email protected]
Background: Osteosarcoma (OS) is a common primary malignant bone tumour in children and young adults. Apiosporamide, a 4-hydroxy-2-pyridone alkaloid from a deep-sea-derived fungus, Arthrinium sp. UJNMF0008, showed anti-proliferative effects toward a panel of human cancer cell lines, and the molecular mechanism in MG63 cells was then investigated in the current work.
Methods: Cell viability was determined with MTT method. Cell proliferation was detected using colony-formation assay. Screening electron microscope was used for morphology observation. Cell cycle and apoptosis was analysed via flow cytometry. Real-time PCR was conducted to evaluate the mRNA expression related with cell apoptosis. The expression levels of proteins related to capase-mediated apoptotic pathway and PI3K/Akt signalling pathway were detected by Western blotting.
Results: Apiosporamide significantly decreased cell viability in cancer cells, and also exhibited excellent anti-proliferative effect. Apiosporamide caused cell cycle arrests at G0/G1 phase in MG63 cells. Moreover, apiosporamide induced apoptosis, activated caspase-3, caspase-8 and caspase-9, and regulated expression of Bax and Bcl-2 in MG63 cells. In addition, apiosporamide also attenuated PI3K/Akt signaling pathway.
Conclusion: Apiosporamide effectively suppressed MG63 cells proliferation by inducing apoptosis through PI3K/Akt and caspase-associated apoptotic pathway.
Keywords: apiosporamide, osteosarcoma, anti-proliferation, apoptosis, PI3K/Akt
Introduction
Osteosarcoma (OS) or osteogenic sarcoma is the most common type of cancer that starts in the bones. Specifically, it is highly aggressive malignant neoplasm.1 OS usually occurs in children, adolescents, and young adults,2 accounting for about 3–4% of all pediatric tumors and is second only to lymphoma.3 Although treatment for OS primarily involves surgical resection and chemotherapy, an average 5-year survival rate of localized OS is approximately 65%.4,5 However, the recurrence and metastasis of OS have poor prognoses, and most patients will die within 1 year after the definite diagnosis.6,7 Therefore, the development of novel therapeutic strategies for OS still remains a pressing task.
With the introduction of new sample collection techniques in recent years, more and more bioactive molecules have been discovered from marine organisms, such as microbes, phytoplankton, sponges, algae, cnidarians, bryozoans, molluscs, tunicates, echinoderms, and mangroves.8–13 Most notably, the isolated compounds from marine resources have been attracted much attention for their anti-cancer effect and fewer side effects.14,15 Apiosporamide (Figure 1), a 4-hydroxy-2-pyridone alkaloid, was first isolated and identified as an antifungal agent from the coprophilous fungus Apiospora montagnei in 1994, and its absolute configuration had been confirmed by total synthesis of its enantiomer.16,17 During our investigation into the metabolites of a marine-derived fungus Arthrinium sp. UJNMF0008, apiosporamide was re-obtained and its cytotoxicity against human OS cell lines (MG63 and U2OS) was reported for the first time.18 In the present study, apiosporamide was further tested for its cytotoxic activity against several cancer cell lines (MDA-MB-231, A549 and MCF-7), which revealed that the best cytostatic effect of apiosporamide was still toward MG63 cell line. Therefore, to explore the underling mechanism and potential as a new therapeutic agent of apiosporamide, we primarily evaluated its effect on MG63 cells in the current work.
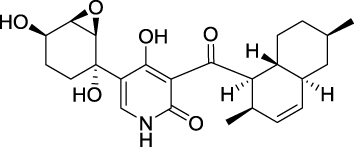 |
Figure 1 Chemical structure of apiosporamide. |
Materials And Methods
Cell Culture
Cell lines U2OS, MG63, A549, MCF-7 and MDA-MB-231 were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, People’s Republic of China). All cells were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (Thermo, Waltham, MA, USA) with 10% fetal bovine serum (Gibco Invitrogen, Carlsbad, CA, USA) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. Apiosporamide was initially dissolved in DMSO (dimethylsulfoxide) to make a 20 mM stock solution and diluted with culture medium to the final test concentrations, which contained no more than 0.05% DMSO.
MTT Assay
The cells were cultured in 96-well plates, and then seeded at a density of 1×104 cells per well. The cells were treated with apiosporamide (>98% purity)18 at various concentrations for indicated times (24, 48, 72 hrs). The viability of cells was measured by the MTT (5 mg/L, Sigma-Aldrich, St. Louis, MO, USA) assay.19 In addition, to study the effects of PI3K on the anti-proliferative action of apiosporamide, MG-63 cells were treated with apiosporamide in the presence of 5 μM LY294002 (>98% purity, Beyotime Biotechnology) or 10 μM AK323727 (>98% purity, Beyotime Biotechnology) for 24 hrs. The survival rate was calculated according to the following formula: Survival rate= absorbance of treatment/absorbance of control×100%. IC50 was calculated based on nonlinear regression analysis of survival curves (SPSS software, version 13.0, Chicago, IL, USA). All data were collected from three independent experiments.19
Colony-Formation Assay
MG63 cells were plated at a density of 1×103 cells/well in 6-well plates. The medium was changed with fresh medium containing apiosporamide (3 and 9 μM) in the drug treatment group.
After 14 days, colonies were fixed in 4% paraformaldehyde for 15 mins and stained by 0.2% crystal violet at room temperature. Colonies containing >50 cells were scored using a light microscope. Each experiment was repeated 3 times in duplicate.
Cell Cycle Analysis
MG63 cells were incubated with the apiosporamide for 24 hrs, and then fixed in cold 70% ethanol overnight at 4°C. The cells were suspended in a staining buffer (10 μg/mL propidium iodide (PI), 0.5% Tween 20, 0.1% RNase in PBS) for 30 mins at room temperature. Cell cycle analysis was performed on the flow cytometer (Becton-Dickinson).
Electron Microscope Analysis
MG63 cells were treated with apiosporamide for 24 hrs, and fixed with 4% glutaraldehyde for 2 hrs at room temperature. Cells were subsequently carried out in 1% OsO4 in 0.15 M phosphate buffer for 1 hr, followed by a rapid wash in the same buffer. The cells were dehydrated in increasing grades of ethanol (50%, 70%, 95%, 100%, 15–20 mins each) and then critical point drying. Observation under conventional scanning electron microscope (JSM-4800, Japan) was generally performed.19
Apoptosis Analysis
Cell apoptosis assays were evaluated with an Annexin V-FITC apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA) as described previously.19 MG63 cells were treated with apiosporamide (0, 9.0, 12, 15 μM) for 24 hrs. The data were analyzed using Flowjo 7.0.
Quantitative Real-Time PCR
MG63 cells were treated with or without apiosporamide for 24 hrs. Total RNA was extracted using TRIzol reagent (Invitrogen, Waltham, MA, USA), and complementary DNA was synthesized using a RT reaction kit (Promega, Madison, WI, USA). qRT-PCR assays were performed as described previously.19 Gene expression levels were calculated relative to the housekeeping β-actin and all the reactions were repeated at least three times. Primer sequences were synthesized as shown in Table 1.
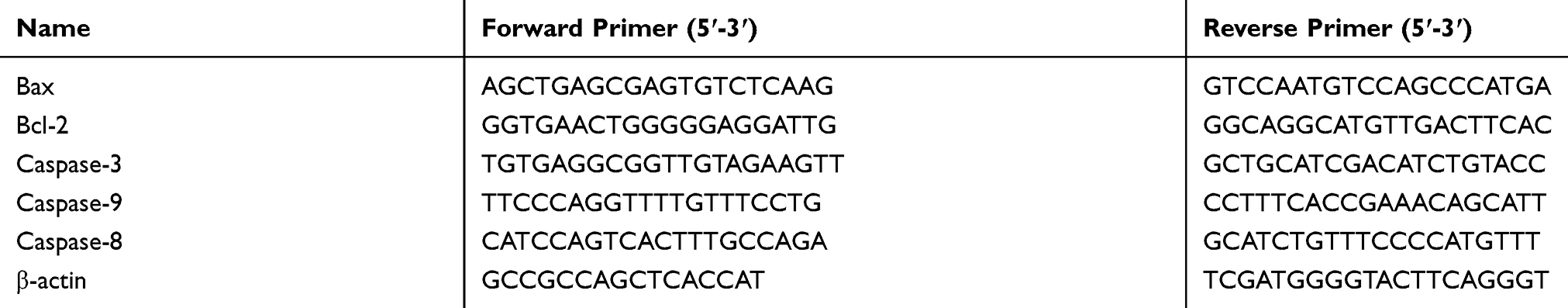 |
Table 1 The Primers Of Real-Time PCR |
Western Blot Analysis
The MG63 cells were prepared using 1× RIPA buffer containing protease inhibitor cocktails (Roche, South San Francisco, CA, USA). Protein concentrations were determined using BCA protein assay kit (Beyotime Institute of Biotechnology, Jiangsu, People’s Republic of China). The membranes were blocked with 3% BSA solution at room temperature for 60 mins. A 1:1000 dilution of the primary antibodies (Bax, Bcl-2, caspase-3, caspase-8, caspase-9, Akt, p-Akt, PI3K, PDK1, CDK6, cyclinE1, cyclinD1, p21, p-Rb and β-actin) were incubated overnight at 4°C. After being washed three times for 5 mins with TBST buffer (0.05% Tris-buffered saline and Tween 20), the membranes were incubated with the appropriate secondary antibody (dilution of 1:5000) for 2 hrs at room temperature. Finally, the membranes were washed three times for 5 mins with TBST buffer. Finally, the bands were visualized with enhanced chemiluminescence reagent (Merck Millipore, Darmstadt, Germany) using imaging system (Chemi-Doc XRS imager, Bio-Rad, Hercules, CA, USA).
Statistical Analysis
All data presented in this study were expressed as mean±SD of at least three independent experiments. SPSS 18.0 (SPSS Inc, Chicago, IL, USA) was used to perform all statistical analyses. The statistical significance of the differences between groups was evaluated by Student’s t-test. p<0.05 was considered to indicate a statistically significant difference. *p<0.05, **p<0.01 and ***p<0.001, respectively.
Results
Effect Of Apiosporamide On Cell Viability
To investigate the effect of apiosporamide on the proliferation of cancer cells, we used MG63, U2OS, MDA-MB-231, MCF-7 and A549 cell lines, all of which were assessed for viability using a MTT assay following exposure to increasing concentrations of apiosporamide for 24, 48 and 72 hrs. The results are summarized in Table 2. Apiosporamide showed obvious proliferation inhibitory effects on all cell lines, and MG63 cells were most sensitive to the treatment. Therefore, the anti-proliferative effect of apiosporamide on MG63 cells was then examined by colony-formation assay. As shown in Figure 2, apiosporamide treatment significantly reduced the number of colonies in a dose-dependent manner when compared with the untreated cells. These results indicated that apiosporamide treatment effectively inhibited the proliferation of MG63 cells.
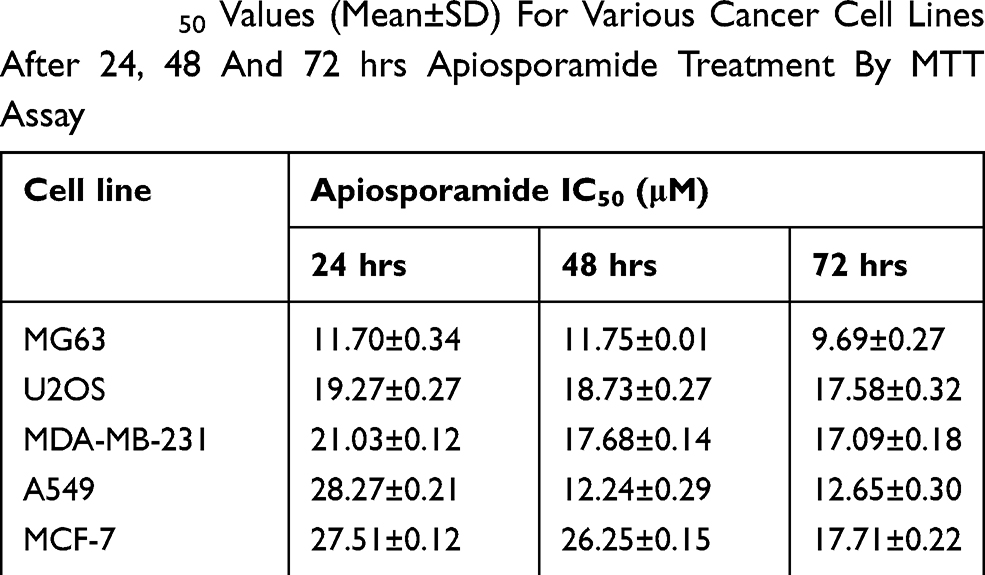 |
Table 2 IC50 Values (Mean±SD) For Various Cancer Cell Lines After 24, 48 And 72 hrs Apiosporamide Treatment By MTT Assay |
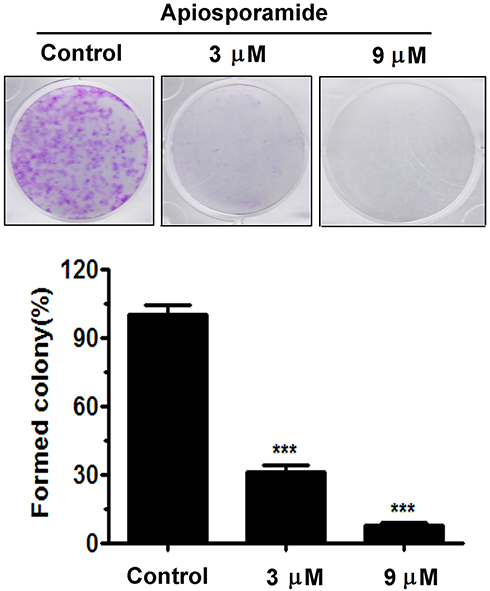 |
Figure 2 Colony-formation assay was performed in MG63 cells. The numbers of cell colonies were counted. n=3; ***P<0.001. |
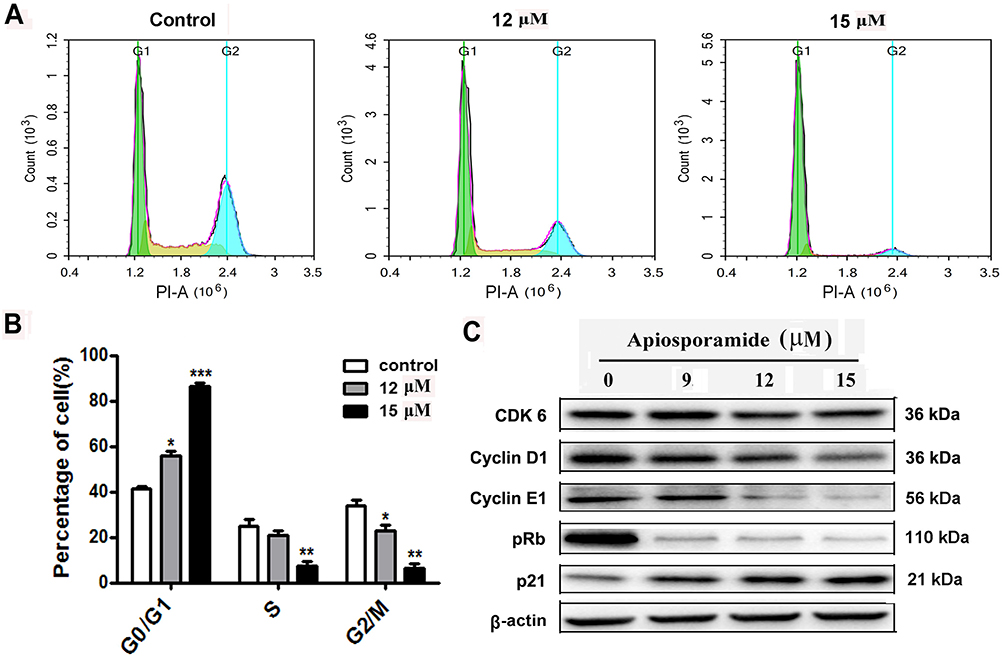 |
Figure 3 Apiosporamide-induced cell cycle arrest at G0/G1 phase. Notes: (A and B) MG63 cells were treated with apiosporamide for 24 hrs. The distribution of cell cycle was assessed by flow cytometry. The percentage of cells in each phase was shown as mean± SD (n = 3). (C) MG63 cells were exposed to apiosporamide for 24 hrs. The level of cell cycle-regulated protein was analyzed by Western blotting. *P<0.05, **P<0.01 and ***P<0.001 vs vehicle control. Abbreviation: Rb, retinoblastoma tumor suppressor protein. |
Apiosporamide Caused Cycle Arrest At G0/G1 Phase In OS Cells
The treatment effect of apiosporamide on OS cell cycle phases was examined by flow cytometer analysis. After treatment of MG63 cells with different concentrations of apiosporamide for 24 hrs, the number of cells in the G0/G1 phase increased with dose-dependent manner from a percentage of 41.25% in no treated cells to 86.38% for 15 μM of apiosporamide (Figure 3A and B). In addition, we also explored the effect of apiosporamide on cell cycle in U2OS cells, results indicated that apiosporamide caused cell cycle arrests at G0/G1 phase, too (Figure S1). Then, a Western blot of G1 phase-associated proteins was conducted in order to further confirm the results. As shown in Figure 3C, apiosporamide decreased the expression levels of cyclin D1, cyclin E1 and CDK6; at the same time the expression of CDK inhibitors, p21 was upregulated. The phosphor-Rb proteins decreased gradually with the increase of apiosporamide treatment concentration (Figure 3C). All these observations demonstrated that apiosporamide triggered G0/G1 phase arrest by regulating cell cycle-related proteins.
Apiosporamide Induces Apoptosis Of OS Cells
We further explored whether the inhibition of cell viability by apiosporamide treatment was due to the induction of apoptosis. SEM (screening electron microscope) was used to estimate apiosporamide-dependent changes of MG63 cell morphology. As shown in Figure 4A, the results demonstrated the occurrence of typical cell apoptosis, after treatment with apiosporamide for 24 hrs. To further quantify apoptosis induced by apiosporamide, flow cytometric analysis via Annexin V-FITC and PI was performed. We found that a significant increase of the number of apoptotic cells was observed in MG63 cells in a dose-dependent manner after exposure to apiosporamide for 24 hrs (Figure 4B). The ratio of apoptotic cells was 10.68% for control (0.2% DMSO) whereas 21.73% for 15 μM apiosporamide-treated group (Figure 4C). Furthermore, flow cytometric analysis was performed on U2OS cell. An increase of the number of apoptotic cells was observed after exposed to apiosporamide in a dose-dependent way (Figure S1). These results were consistent with the cell viability assay, suggesting that higher cell growth inhibition resulting from apiosporamide could be, at least partly, due to the induction of more apoptosis in OS cells.
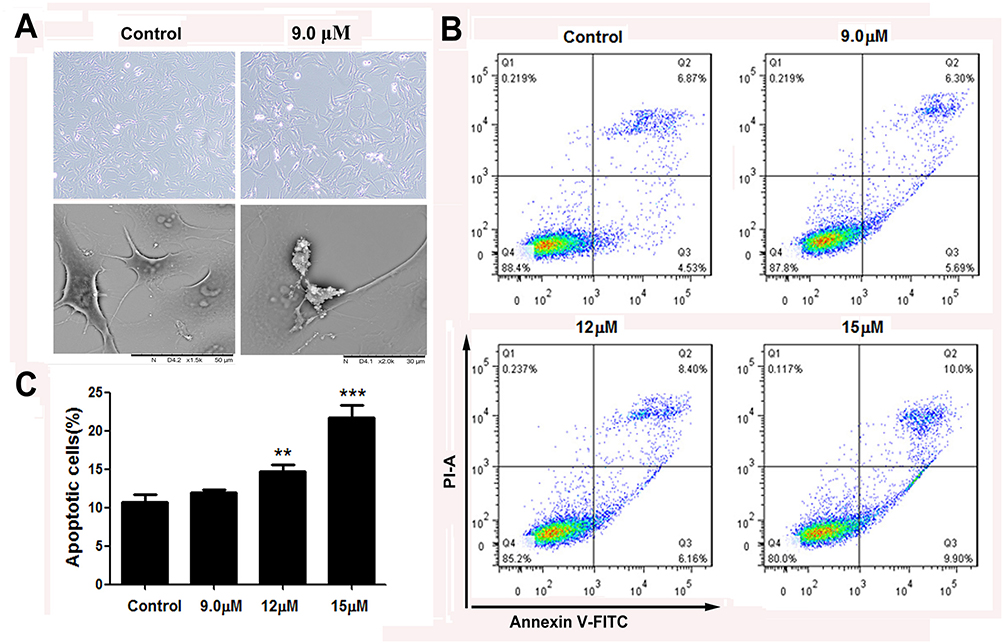 |
Figure 4 Apoptosis of MG63 cells following treatment with apiosporamide. Notes: (A) SEM micrographs of apiosporamide-treated MG63 cells for 24 hrs. (B) Apoptosis was determined by an Annexin V-FITC/PI apoptosis detection kit, and representative results from flow cytometry were shown. (C) Quantitative analysis of apoptosis of MG63 cells as shown in (B). Values were presented as mean±SD (n=3). **P<0.01 and ***P<0.001 vs normal control. Abbreviations: SEM, screening electron microscope; Annexin V-FITC, annexin V fluorescein isothiocyanate; PI, propidium iodide. |
Apiosporamide Increase Activation Of Caspase-3, -8 And -9 In MG63 Cells
In order to investigate the molecular mechanism of apiosporamide-induced cell apoptosis, Western-blot and real-time PCR were performed to determine the protein and mRNA expression related to cell apoptosis. Data showed that the mRNA levels of caspase-3, -8 and -9 upon application of apiosporamide were much higher than the control (Figure 5A), apiosporamide treatment strongly triggered the cleavage caspase-3, -8, and -9 at 24 hrs (Figure 5B). Moreover, apiosporamide treatment also downregulated the expression of anti-apoptotic protein Bcl-2 and upregulated pro-apoptotic protein Bax (Figure 5C and D). These results indicated that apiosporamide induced apoptosis by regulating apoptosis-associated regulators.
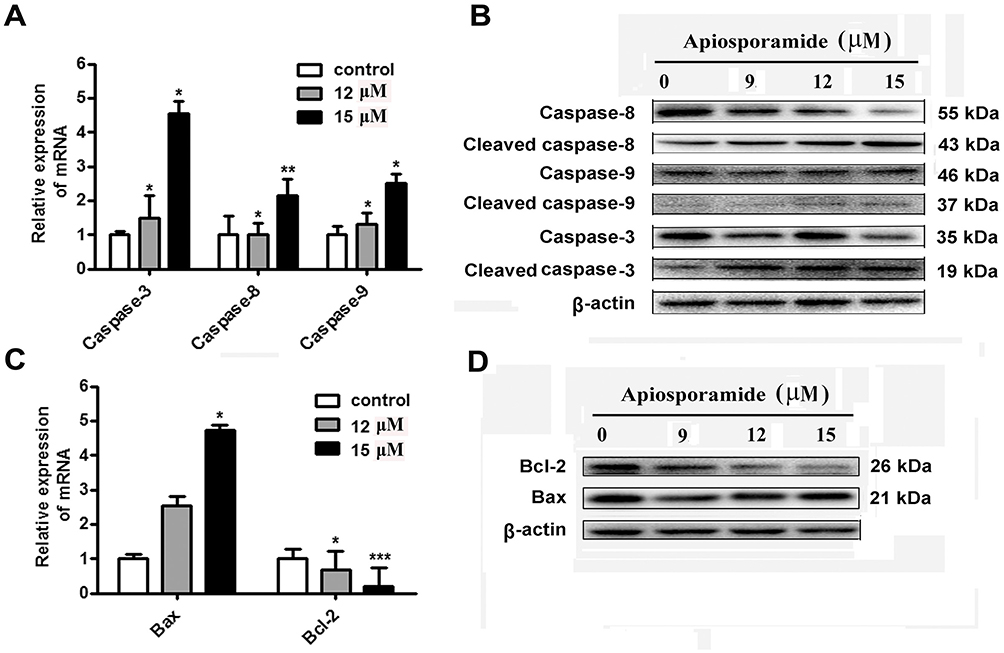 |
Figure 5 Effect of apiosporamide on apoptosis regulators. Notes: (A and C) Real-time PCR analyzed the mRNA expression of caspase-3, caspase-8, caspase-9, Bax and Bcl-2 in apiosporamide-treated cells. Data were presented as mean±SD (n=3), *P<0.05, **P<0.01, ***P<0.001 compared to control cells. (B and D) MG63 cells were treated with indicated concentrations of apiosporamide for 24 hrs and protein levels of Bcl-2, Bax, caspases-3, -8 and -9 were analyzed using Western blotting. |
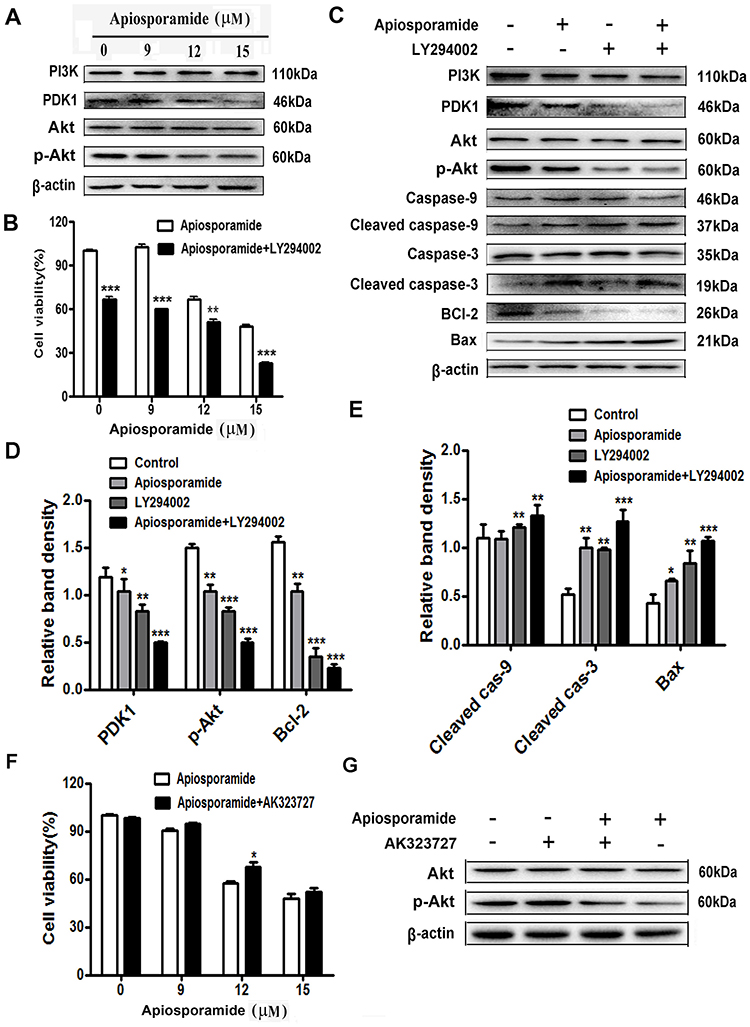 |
Figure 6 PI3K/Akt pathway mediated apisoporamide-induced apoptosis of MG63 cells. Notes: (A) Cells were treated with various concentrations of apisoporamide for 24 hrs. The expressions of PI3K, PDK1, p-Akt and Akt were analyzed by Western blotting. (B) MG63 cells were incubated with LY294002 (5 μM) and apisoporamide (0, 9, 12, 15 μM) for 24 hrs. Cell viability was measured by MTT assay. Data were presented as mean±SD (n=3). (C) Western blotting results showed a significant change in the relative protein expression after MG63 cells were treated with apisoporamide (12 μM) and/or LY294002 (5 μM) for 24 hrs. (D and E) The immunoblot signals were quantified by densitometry, and mean data from independent experiments were normalized to the untreated cells. The bars are the means±SD (n = 3). (F) MG63 cells were co-treated with AK323727 (10 μM) and apisoporamide (0, 9, 12, 15 μM) for 24 hrs. Cell viability was measured by MTT assay. Data were presented as mean±SD (n=3). (G) Western blotting analysis of Akt, p-Akt and β-actin expression. MG63 cells were treated with apisoporamide (15 μM) and/or AK323727 (10 μM) for 24 hrs. *P<0.05, **P<0.01 and ***P<0.001 vs vehicle control. Abbreviations: PI3K, phosphatidylinositol 3 kinase; PDK1, phosphoinositide-dependent kinase-1; Akt, protein kinase B. |
Apiosporamide Induces Apoptosis Of MG63 Cells By Inhibiting PI3K/Akt Pathway
PI3K/Akt pathway is a vital controller in tumorigenesis, proliferation, apoptosis, metastasis, etc.20,21 Thus, we assessed whether apiosporamide induced apoptosis via PI3K/Akt pathway in MG63 cells, the protein expression of PI3K, PDK1, Akt and its phosphorylated form were examined by Western blot. As shown in Figure 6A, no obvious difference was observed in the expression of PI3K and Akt between the apisoporamide-treated cells and the control group. Interestingly, the levels of p-Akt and PDK1 in apisoporamide-treated cells were significantly decreased compared with the control. These results suggested that apisoporamide exhibited inhibitory effect on PI3K/Akt pathway.
To further confirm the role of the PI3K/Akt pathway in the anti-proliferative effect exerted by apisoporamide, we used PI3K inhibitor LY294002 to interfere with the action of apisoporamide. Notably, MTT analysis showed that the proliferation of MG63 cells treated with a combination of apisoporamide and LY294002 was significantly lower than that only treated with apisoporamide (Figure 6B). Furthermore, Western blotting analysis showed that co-treatment with apisoporamide and LY294002 induced apoptosis more obviously compared with apisoporamide alone treated group. As seen in Figure 6C and 6E, co-treatment with apisoporamide and LY294002 markedly increased the levels of cleaved caspase-3, caspase-9 and Bax, while it also significantly lowered the expression levels of PDK1, p-Akt and Bcl-2 (Figure 6C and 6D). Additionally, MG63 cells were treated with apisoporamide and 10 μM AK323727, an Akt agonist, to test if the activation of Akt signaling could reverse apisoporamide’s inhibitory effect on MG63 cells, as demonstrated in Figure 6F and 6G. Results of cell proliferation suggested that, when compared to the apisoporamide-treated group, the apisoporamide and AK323727 co-treated group increased the cell viability. WB results showed that AK323727 increased p-Akt levels in apisoporamide-treated MG63 cells. Taken together, these results revealed that apisoporamide induced MG63 cell apoptosis via the PI3K/Akt signaling pathway.
Discussion
Apiosporamide was first isolated from the fungus Apiospora montagnei Saccardo (Amphisphaeriaceae) in 1994 by Gloer.16 This unique 4-hydroxy-2-pyridinone alkaloid exhibited potent antifungal activity against the coprophilous fungus Ascobolus furfuraceus and antibacterial activity against Bacillus subtilis and Staphylococcus aureus.17 In addition, studies revealed that YM-215343, a compound closely related to apiosporamide, showed antifungal activity against the pathogenic fungi, Candida albicans, Cryptococcus neoformans and Aspergillus fumigatus, and also exhibited cytotoxicity against HeLa S3 cells with an IC50 of 3.4 μg/mL.22 In our previous work, we have also observed that apiosporamide displayed anti-proliferative effects toward two OScell lines.18 In the present study, the cytotoxicity of apiosporamide was examined on different kinds of human cancer cell lines by MTT assay, and the results showed that IC50 values of apisosporamide ranged from 9.69±0.27 μM to 28.27±0.21 μM in several tested cell lines. Interestingly, the results above showed that the inhibitory effects of apisoporamide against tumor cells were cell type-dependent as well as dose-dependent. These data indicated that MG63 cells were more sensitive to apisoporamide treatment.
Cell cycle arrest in cancer cells has become a major indicator of anticancer effects. Anticancer agents may result in an arrest of cells in various phases of the cell cycle, thereby reducing the growth and proliferation of cancerous cells.23 Our present work demonstrated that apiosporamide inhibited OS cells growth via the G0/G1 arrest of the cell cycle. Furthermore, we analyzed the alteration of the protein expression level of cyclin D1, CDK6 and p21. These data suggested that the accumulation of p21, which is one of the cyclin-dependent kinase inhibitors, is strongly associated with the G0/G1 arrest.24 Moreover, apiosporamide treatment leads to downregulation of cyclin D1 protein. The reduction in cyclin D1 results in the release of p21, which binds to and inhibits cyclin D1/CDK6, thus preventing cell cycle progression from the G1 to the S phase. In addition, the retinoblastoma tumor suppressor protein (Rb) might contribute to regulating the cell cycle arrest in preventing tumor.25 Therefore, the phosphorylation level of Rb (p-Rb) was also tested in apiosporamide-treated MG63 cells and was demonstrated to decrease significantly.
Apoptosis is a process programmed by the genome of the cell and is intimately linked to the development of tumor. Current cancer therapy such as chemotherapy has been made to develop strategies that trigger apoptosis in malignant cancer cells, and a large number of molecules and pathways are involved in this process. It is well known that caspase signaling is closely associated with apoptosis. The present study found that apiosporamide insignificantly induced generation of apoptotic cells through the activation of caspase-3, -8 and -9. Consistent with the FACS results, our assay revealed that apiosporamide promoted the apoptotic process at the late stages as determined by Annexin V/PI-labelled cells. In addition, previous studies reported that many signals for cellular life and death were regulated by the Bcl-2 family proteins. The Bcl-2 family includes both pro-life (e.g., Bcl-2) and pro-death (e.g., Bax) proteins. It was thought that high level of anti-apoptotic Bcl-2 and/or a decreased expression of pro-apoptotic Bax is a balance, which could control the sensitivity of cells to apoptotic stresses.26 Our results showed significant increases of Bax but decrease of anti-apoptosis protein Bcl-2 in MG63 cells with treated apiosporamide compared with controls. These data indicated that apiosporamide induces cell apoptosis by modulating the activity of proteins associated with apoptosis.
The PI3K/Akt signaling pathway has been investigated commonly for its crucial role in various types of cancer and is considered to get involved in tumorigenesis by simultaneously promoting proliferation and inhibiting apoptosis.27–29 Accumulating studies display that activation of the PI3K/Akt pathway is linked to OS cells proliferation and apoptosis.30–32 Therefore, inhibition of PI3K/Akt signaling can serve as a potentially useful strategy for the treatment of OS. Normally, activation of PI3K activates downstream protein kinases, PDK1, both PDK1 and Akt have a PH domain showing selectivity for phosphoinositides phosphorylated in position three produced by PI3K at the plasma membrane.33,34 PDK1-mediated phosphorylation is a key event in Akt activation.35,36 Subsequently, p-Akt could regulate the expression of many molecules to affect cell proliferation and apoptosis.37 The present study found that with the significant decrease of the expression level of PDK1, the p-Akt expression level significantly decreased in MG63 cells exposed to apiosporamide. Since Akt is a downstream target of PI3K, the observed inhibition of p-Akt suggests that apiosporamide also inhibits PI3K. Accumulating evidence indicates that apiosporamide has an important role for the PI3K signaling pathway in OS.
To further determine whether the PI3K/Akt signaling pathway is important in the apoptosis of OS cells, MG63 cells were treated with LY294002 which is a PI3K inhibitor.38 The results showed that cells were treated with 5 μM LY294002 in combination with various concentrations of apiosporamide, the rate of cell viability decreased significantly compared with apiosporamide alone. Following treatment with LY294002, the phospho-Akt expression level in MG63 cell lines decreased significantly, suggesting that the apiosporamide is highly potent in inhibiting the PI3K/Akt signaling pathway. In addition, PI3K activity is essential for retaining Bax in the cytoplasm and Akt is capable of suppressing Bax translocation to mitochondria.39 Our results further confirmed that LY294002 combined with apiosporamide could significantly induce higher expression level of Bax and decrease the expression of Bcl-2 in MG63 cells than the effects of LY294002 or apiosporamide alone; however, it also prominently increases in cleaved caspase-3 and caspase-9 expression when compared with apiosporamide treatment alone. And more importantly, co-treatment with Akt activator, AK323727, effectively blocked apiosporamide-inhibited MG63 cell proliferation. These results suggested that apiosporamide induces apoptosis partly through inhibiting the PI3K/Akt pathway in MG63 cells, predominantly through downregulation of anti-apoptotic factors and upregulation of apoptotic factors.
In conclusion, the present study revealed that apiosporamide inhibited the proliferation, caused cell cycle arrest in the G0/G1 phase and induced the apoptosis of OS cells. In addition, its role in promoting tumor cell apoptosis was associated with the inhibition of the PI3K/Akt signaling pathway and activation caspase-mediated apoptotic pathway. The mechanism of apiosporamide-induced apoptosis in OS cells requires further verification. Furthermore, in vivo experiments are required to understand the anti-tumor effects of apiosporamide.
Acknowledgments
This research work was financially supported by the National Natural Science Foundation of China (no. 31501104), the Natural Science Foundation of Shandong Province (no. JQ201721), the Innovation Team Project of Jinan Science & Technology Bureau (no. 2018GXRC003), the Young Taishan Scholars Program (no. tsqn20161037) and the Shandong Talents Team Cultivation Plan of University Preponderant Discipline (no. 10027).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Luetke A, Meyers PA, Lewis A, Juergens H. Osteosarcoma treatment—where do we stand? A state of the art review. Cancer Treat Rev. 2014;40(4):523–532. doi:10.1016/j.ctrv.2013.11.006
2. Moore DD, Luu HH. Osteosarcoma. Cancer Treat Res. 2014;162:65–92.
3. Wycislo KL, Fan TM. The immunotherapy of canine osteosarcoma: a historical and systematic review. J Vet Intern Med. 2015;29(3):759–769. doi:10.1111/jvim.12542
4. Jaffe N. Osteosarcoma: review of the past, impact on the future. The American experience. Cancer Treat Res. 2009;152:239–262.
5. Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. 2018;18(1):39–50. doi:10.1080/14737140.2018.1413939
6. Meyers PA. Muramyl tripeptide (mifamurtide) for the treatment of osteosarcoma. Expert Rev Anticancer Ther. 2009;9(8):1035–1049. doi:10.1586/era.09.69
7. He JP, Hao Y, Wang XL, et al. Review of themolecular pathogenesis of osteosarcoma. Asian Pac J Cancer Prev. 2014;15(15):5967–5976. doi:10.7314/APJCP.2014.15.15.5967
8. Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2012;29(2):144–222. doi:10.1039/c2np00090c
9. Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2013;30:237–323.
10. Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2014;31(2):160–258. doi:10.1039/c3np70117d
11. Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2015;32(2):116–121. doi:10.1039/c4np00144c
12. Blunt JW, Copp BR, Keyzers RA, Munro MH, Prinsep MR. Marine natural products. Nat Prod Rep. 2016;33(3):382–431. doi:10.1039/c5np00156k
13. Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR. Marine natural products. Nat Prod Rep. 2017;34(3):235–294. doi:10.1039/c6np00124f
14. Mioso R, Marante FJ, Bezerra RS, Borges FV, Santos BV, Laguna IH. Cytotoxic compounds derived from marine sponges: a review (2010–2012). Molecules. 2017;22(2):208. doi:10.3390/molecules22020208
15. Gammone MA, Riccioni G, Galvano F, D’Orazio N. Novel therapeutic strategies against cancer: marine-derived drugs may be the answer? Anticancer Agents Med Chem. 2016;16(12):1549–1557.
16. Alfatafta AA, Gloer JB, Scott JA, Malloch D. Apiosporamide, a new antifungal agent from the coprophilous fungus Apiospora montagnei. J Nat Prod. 1994;57(12):1696–1702. doi:10.1021/np50114a012
17. Williams DR, Kammler DC, Donnell AF, Goundry WR. Total synthesis of (+)-apiosporamide: assignment of relative and absolute configuration. Angew Chem Int Ed Engl. 2005;44(41):6715–6718. doi:10.1002/anie.200502015
18. Bao J, Zhai H, Zhu K, et al. Bioactive pyridone alkaloids from a deep-sea-derived fungus Arthrinium sp. UJNMF0008. Mar Drugs. 2018;16(5):
19. Zhang YY, Zhang QQ, Song JL, Zhang L, Jiang CS, Zhang H. Design, synthesis, and antiproliferative evaluation of novel coumarin/2-cyanoacryloyl hybrids as apoptosis inducing agents by activation of caspase-dependent pathway. Molecules. 2018;23(8):
20. Polivka J
21. Zhu M, Ying J, Lin C, et al. b-Escin inhibits the proliferation of osteosarcoma cells via blocking the PI3K/Akt pathway. RSC Adv. 2018;8:29637–29644. doi:10.1039/C8RA03578D
22. Shibazaki M, Taniguchi M, Yokoi T, et al. YM-215343, a novel antifungal compound from Phoma sp. QN04621. J Antibiot (Tokyo). 2004;57(6):379–382. doi:10.7164/antibiotics.57.379
23. Shah MA, Schwartz GK. Cyclin-dependent kinases as targets for cancer therapy. Cancer Chemother Biol Response Modif. 2005;22:135–162.
24. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. doi:10.1038/nrc2657
25. Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi:10.1038/nrc2399
26. Volkmann N, Marassi FM, Newmeyer DD, Hanein D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ. 2014;21:206–215. doi:10.1038/cdd.2014.119
27. Xie X, Tang B, Zhou J, Gao Q, Zhang P. Inhibition of the PI3K/Akt pathway increases the chemosensitivity of gastric cancer to vincristine. Oncol Rep. 2013;30(2):773–782. doi:10.3892/or.2013.2520
28. Chang F, Lee JT, Navolanic PM, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. 2003;17(3):590–603. doi:10.1038/sj.leu.2402824
29. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi:10.1038/nrd1902
30. Zhao J, Zhang ZR, Zhao N, Ma BA, Fan QY. VEGF silencing inhibits human osteosarcoma angiogenesis and promotes cell apoptosis via PI3K/AKT signaling pathway. Cell Biochem Biophys. 2015;73(2):519–525. doi:10.1007/s12013-015-0692-7
31. Wang T, Gong X, Jiang R, Li H, Du W, Kuang G. Ferulic acid inhibits proliferation and promotes apoptosis via blockage of PI3K/Akt pathway in osteosarcoma cell. Am J Transl Res. 2016;8(2):968–980.
32. Chen ZZ. Berberine induced apoptosis of human osteosarcoma cells by inhibiting phosphoinositide 3 kinase/protein kinase B (PI3K/Akt) signal pathway activation. Iran J Public Health. 2016;45(5):578–585.
33. Alessi DR, James SR, Downes CP, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase. Balpha Curr Biol. 1997;7(4):261–269. doi:10.1016/S0960-9822(06)00122-9
34. James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, Alessi DR. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J. 1996;315:709–713. doi:10.1042/bj3150709
35. Gagliardi PA, Puliafito A, Primo L. PDK1: at the crossroad of cancer signaling pathways. Semin Cancer Biol. 2018;48:27–35. doi:10.1016/j.semcancer.2017.04.014
36. Wang G, Liu X, Xie J, Meng J, Ni X. PDK-1 mediated Hippo-YAP-IRS2 signaling pathway and involved in the apoptosis of non-small cell lung cancer cells. Biosci Rep. 2019;39(5):
37. Sussman M. “AKT”ing lessons for stem cells: regulation of cardiac myocyte and progenitor cell proliferation. Trends Cardiovasc Med. 2007;17(7):235–240. doi:10.1016/j.tcm.2007.08.003
38. Wu D, Tao J, Xu B, et al. Phosphatidylinositol 3-kinase inhibitor LY294002 suppresses proliferation and sensitizes doxorubicin chemotherapy in bladder cancer cells. Urol Int. 2011;87:105–113. doi:10.1159/000322849
39. Tsuruta F, Masuyama N, Gotoh Y. The phosphatidylinositol 3-kinase (PI3K)-Akt pathway suppresses Bax translocation to mitochondria. J Biol Chem. 2002;277(16):14040–14047. doi:10.1074/jbc.M108975200
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.