")
Back to Journals » Cancer Management and Research » Volume 13
Apatinib Promotes Ferroptosis in Colorectal Cancer Cells by Targeting ELOVL6/ACSL4 Signaling
Received 15 August 2020
Accepted for publication 13 December 2020
Published 11 February 2021 Volume 2021:13 Pages 1333—1342
DOI https://doi.org/10.2147/CMAR.S274631
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Chien-Feng Li
Xiangyang Tian,1 Shuyuan Li,2 Guoyan Ge2
1Department of Oncology, Peace Hospital, Changzhi Medical College, Changzhi, Shanxi Province, 046000, People’s Republic of China; 2Department of Tumor Spleen and Stomach, Hospital of Traditional Chinese Medicine of Changzhi City, Changzhi, Shanxi Province, 046013, People’s Republic of China
Correspondence: Xiangyang Tian
Peace Hospital, Changzhi Medical College, No. 110 Yan’an South Road, Luzhou District, Changzhi, Shanxi Province, 046000, People’s Republic of China
Email [email protected]
Background: Colorectal cancer (CRC) is a common digestive system malignancy. Ferroptosis, a new form of regulated cell death, plays a vital role in the pathogenesis and therapy of cancers.
Objective: We aimed to study the role of apatinib in ferroptosis of CRC cells and its potential mechanisms.
Materials and Methods: Human CRC HCT116 cells were exposed to apatinib. Cell viability was examined using a CCK-8 kit. The concentrations of intracellular iron and reactive oxygen species (ROS) were detected using kits. Additionally, Western blot analysis was used to determine the expression of ferroptosis-related proteins. Elongation of very long-chain fatty acids family member 6 (ELOVL6) was one of the targets of apatinib predicted by SwissTargetPrediction. Therefore, ELOVL6 expression was evaluated after treatment with apatinib. Subsequently, the effects of ELOVL6 overexpression on ferroptosis of HCT116 cells were investigated. Finally, STRING database was applied to predict the potential proteins interacting with ELOVL6, and co-immunoprecipitation (co-IP) assay was applied for confirmation.
Results: Results indicated that apatinib decreased cell viability and increased the contents of intracellular iron ROS. Moreover, significantly upregulated ACSL4 expression was observed, accompanied by notable downregulation of GPx4 and FTH1 expression after apatinib exposure. Furthermore, ELOVL6 expression was remarkably enhanced in HCT116 cells, which was dramatically inhibited under apatinib intervention. ELOVL6 overexpression reversed the effects of apatinib on cell viability and ferroptosis of HCT116 cells. Moreover, ACSL4, a vital regulator of ferroptosis, could interact with ELOVL6 directly, which was confirmed by the result of co-IP.
Conclusion: These findings demonstrated that apatinib promoted ferroptosis in CRC cells by targeting ELOVL6/ACSL4, providing a new mechanism support for apatinib application in the clinical treatment of CRC.
Keywords: colorectal cancer, apatinib, ferroptosis, reactive oxygen species, ELOVL6
Introduction
Colorectal cancer (CRC) is a common and adverse malignancy in digestive system and a major cause for cancer-related mortality worldwide.1,2 Recent statistics have revealed that 1.8 million CRC cases are diagnosed annually and that each year, 881,000 CRC-related deaths occur worldwide.3 Although considerable progress has been made in the treatment of CRC recently, the prognosis of patients with CRC still remains dismal.4 Therefore, it is urgent to obtain a clearer understanding about the mechanisms underlying the anti-cancer effects of drugs, thereby developing more effective preventive strategies for CRC therapy.
Ferroptosis was recently identified as a new form of regulated cell death by Brent R. Stockwell’s laboratory in 2012.5 It is characterized by an overwhelming and iron-dependent accumulation of lipid peroxide and reactive oxygen species (ROS) upon a distinct set of genes, thus evoking a different method of death from apoptosis and necrosis.6,7 For excessive growth, tumor cells present a higher demand for iron than non-tumor cells, which enables tumor cells more susceptible to iron-catalyzed necrosis.8 Ferroptosis plays a vital role in the pathogenesis and cancer therapy.9 A growing body of literature has proved that ferroptosis, as a new form to regulate cell death, is a novel method for the destruction of cancer cells.10 Therefore, triggering ferroptosis might offer a novel therapeutic strategy for treating CRC.
Apatinib, also known as YN968D1, is a new oral small-molecule agent with antiangiogenic effect for the treatment of multiple solid tumors, including gastric cancer, colorectal cancer and hepatocellular carcinoma.11,12 It has been well reported that apatinib severs as an optional treatment in metastatic CRC.13 Apatinib, used as third-line treatment, effectively improves the prognosis of patients with heavily treated metastatic CRC.14 Besides, apatinib could induce apoptosis and protective autophagy in CRC cells by endoplasmic reticulum stress.15 Additionally, the SwissTargetPrediction website (http://www.swisstargetprediction.ch/) predicts that elongation of very long-chain fatty acids family member 6 (ELOVL6) ELOVL6 is one target of apatinib. Accumulating evidence has demonstrated that ELOVL6 is high-expressed and serves as a negative clinical predictor in a plenty of carcinomas.16,17 However, whether the anti-CRC activity of apatinib acts by regulating ferroptosis of CRC cells through ELOVL6 remains to be elucidated.
In the present study, human colorectal cancer HCT116 cells were exposed to apatinib for investigating its effects on viability and ferroptosis. Afterwards, ELOVL6 was overexpressed to explore the underlying regulatory mechanisms of apatinib in ferroptosis of CRC cells. Our study is of great significance since it provides a new mechanism support for apatinib application in the clinical treatment of colorectal cancer.
Materials and Methods
Cell Culture and Treatment
Human colorectal cancer HCT116 cells and human normal intestinal epithelial cell line (HIEC) were procured from the Cell Bank of Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. Cells were maintained in Dulbecco’s modified Eagle medium (DMEM; Invitrogen, Carlsbad, USA) supplemented with 10% heat-inactivated FBS (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in fully humidified air of 95% air and 5% CO2. HCT116 cells were, respectively, exposed to apatinib (Hengrui Medicine Co. Ltd., Jiangsu, China) of different concentrations at 5 μM, 10 μM and 20 μM for 24 h. Untreated HCT116 cells were used as the control cells.
Cell Viability Assay
The cytotoxicity of apatinib on CRC cells was determined using a Cell Counting Kit-8 (CCK-8) assay kit (Shanghai Yi Sheng Biotechnology Co. Ltd., China). Briefly, HCT116 cells in the logarithmic growth period were collected and dispensed into 96-well tissue culture plates (4000 cells/well), followed by treatment with the indicated concentrations of apatinib. Following incubation for 24, 48 or 72 h, ten microliters of CCK-8 solution was added to each hole. After incubation at 37°C for further 2 h, the optical density (OD) at 450 nm of cells was detected utilizing a microplate reader (Bio-Rad Laboratories, Richmond, CA, USA).
Cell Transfection
Prior to cell transfection, HCT116 cells were plated into a 6-well plate and cultured until 80% confluence at 37°C in a humidified environment of 5% CO2. ELOVL6-overexpressing plasmid (Oe-ELOVL6) or the empty vector plasmid (Oe-NC), provided by Shanghai GenePharma co., ltd (Shanghai, China), was transfected into HCT116 cells. Transfection was performed by Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) as per the manufacturer’s protocol. At 24 h after transfection, cells were harvested and the transfection efficiency was evaluated by applying reverse transcription-quantitative polymerase chain reaction (RT-qPCR).
Measurement of Total Iron Content and Ferrous Iron (Fe2+) Concentration
An iron assay kit (Baiaolaibo, Beijing, China) was employed to measure total iron concentration in cell lysates in accordance with the manufacturer’s guidelines. The detection of relative intracellular Fe2+ level was performed with the iron assay kit (Abcam) following manufacturer’s recommendations. The experiments were independently repeated at least three times.
Detection of Intracellular Reactive Oxygen Species (ROS)
The accumulation of intracellular ROS was examined by a ROS assay kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) with usage of 2, 7-dichlorofluorescein diacetate (DCFH-DA, Invitrogen, USA) as a fluorescence probe. After appropriate treatment, the HCT116 cells were collected and washed with ice-cold phosphate buffered saline (PBS) thrice, followed by incubation in DMEM supplemented with 10 μM DCFH-DA for 20 min at 37°C in the dark. After centrifugation at 800 g, 4°C for 5 min, the fluorescence was evaluated by a fluorimeter excited at 488 nm and emitted at 525 nm. The images were photographed by a confocal microscope.
Test for the Markers of Oxidative Stress
Cells were collected and centrifuged at 3000 x g for 10 min at 4°C, and the supernatant was collected for subsequent examinations. The concentrations of malondialdehyde (MDA) and glutathione (GSH) as well as the activity of glutathione peroxidase (GPx) in HCT116 cells were detected using commercial kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) according to the colorimetric methods.
RT-qPCR Analysis
Total RNA in cells was extracted using TRIzol reagent (Invitrogen). The complementary DNA (cDNA) was synthesized by a PrimeScript RT Reagent Kit (Takara, Japan). PCR then was performed with 2 μg cDNA as the templet using Power SYBR Master Mix (Applied Biosystems, Foster, USA) on the ABI 7500 PCR system (Applied Biosystems). All primers used in the present study were synthesized by Shanghai GenePharma co., ltd (Shanghai, China). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was the internal reference gene. Relative expression was calculated using the 2−ΔΔCt method.
Western Blot Analysis
Total proteins were extracted from cells by using RIPA Lysis Buffer (Beyotime, Shanghai, China). A bicinchoninic acid protein assay kit (Beyotime, Shanghai, China) was applied to detect the protein concentration. A total of 40 μg protein samples in cell lysates were loaded on SDS-PAGE, followed by transferring onto PVDF membranes. Membranes were subsequently immersed into 5% nonfat milk, and then incubated with primary antibodies. All blots were incubated with HRP-conjugated secondary antibody (A0216, Beyotime, Shanghai, China) for 1 h and next visualized using Odyssey Infrared Imaging Scanner (LI-COR Biosciences). GAPDH was used as the loading control. The intensities of bands were calculated using Image-J software (National Institutes of Health).
Co-Immunoprecipitation (IP) Assay
For co-IP assay, HCT116 cells were washed in 2 mL Phosphate Buffer Saline (PBS; Beyotime, Shanghai, China) and centrifuged at 850 x g at room temperature for 5 min to collect the cells. Then, cells were lysed in lysis Buffer (Beyotime, Shanghai, China). Lysates were incubated with specific antibody and control lgG plus Protein A/G beads (Santa Cruz Biotechnology, USA) at 4°C for 2 h. After washing the beads with PBS three times, immunoprecipitates were analyzed by Western blot analysis.
Statistical Analysis
All data were represented as mean values ± standard deviation. Statistical analysis was performed with GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). Differences between groups were evaluated by Student’s t-test. Comparisons involving multiple samples were analyzed by one-way analysis of variance (ANOVA) followed by Turkey’s post hoc test. P values less than 0.05 were considered statistically significant.
Results
Apatinib Inhibits Cell Viability and Promotes Ferroptosis in HCT116 Cells
The chemical structural formula of apatinib was presented in Figure 1A. The viability of HCT116 cells after treatment with apatinib at different concentrations was determined using a CCK-8 assay kit. As exhibited in Figure 1B, apatinib dose-dependently decreased cell viability compared with the untreated control group. Apatinib at concentration of 5, 10 or 20 μM to treat HCT116 cells was selected for the following experiments, which was consistent with the previous study.15 Iron accumulation and lipid ROS production are two critical signaling events in ferroptosis.18 Result of Figure 1C and D indicated that the levels of total iron and Fe2+ were dramatically reduced after apatinib intervention relative to the control group. Additionally, the ROS accumulation was significantly elevated in a dose-dependent manner of apatinib (Figure 1E). Moreover, notably increased concentration of MDA while decreased contents of GSH and GPx were observed under the influence of apatinib (Figure 1F–H). Summing up, these data suggest that apatinib suppresses cell viability and promotes ferroptosis in HCT116 cells.
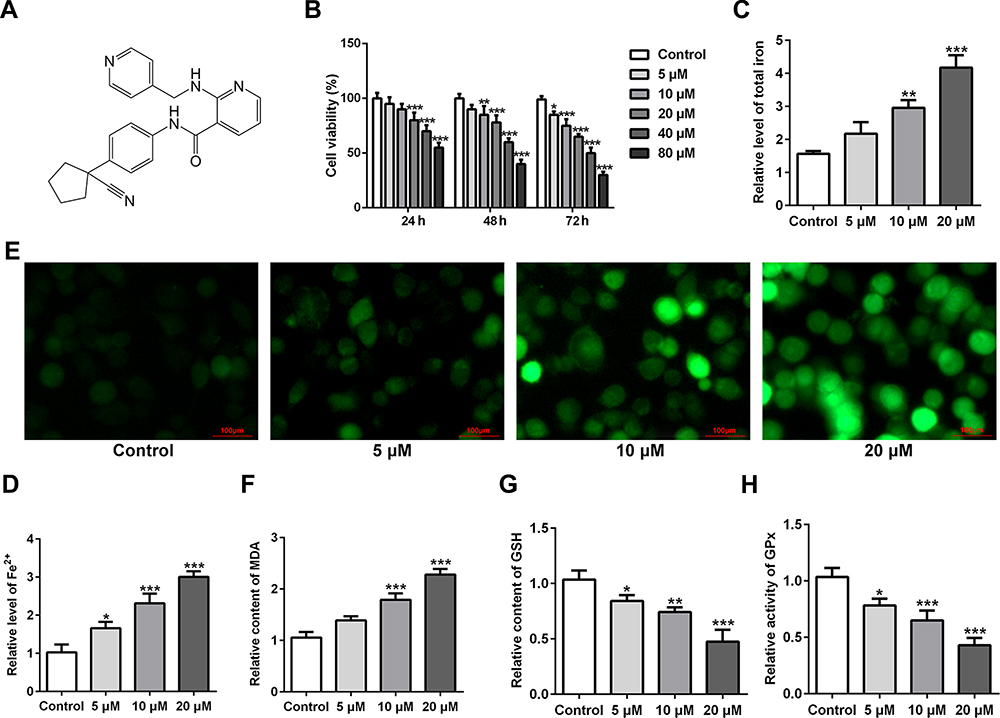 |
Figure 1 Apatinib inhibits cell viability and promotes ferroptosis in HCT116 cells. (A) The chemical structural formula of Apatinib. (B) Cell viability of HCT116 cells after treatment with apatinib (5, 10 and 20 μM) was determined using a cell counting kit-8 assay kit. The relative concentrations of (C) total iron and (D) Fe2+ were measured using commercially available kits, respectively. (E) The level of ROS was detected by a ROS assay kit using 2, 7-dichlorofluorescein diacetate (DCFH-DA, Invitrogen, USA). The levels of (F) MDA, (G) GSH and (H) GPx were evaluated using commercial kits. *P<0.05, **P<0.01 and ***P<0.001 vs control. Abbreviations: ROS, reactive oxygen species; MDA, malondialdehyde; GSH, glutathione; GPx, glutathione peroxidase. |
Apatinib Regulates the Expression of Ferroptosis-Associated Proteins in HCT116 Cells
Subsequently, the expression of ferroptosis-associated proteins was separately examined via applying Western blot analysis. It is observable from Figure 2 that apatinib remarkably upregulated the expression of long-chain acyl-CoA synthetases 4 (ACSL4) whilst downregulated the levels of glutathione peroxidase 4 (GPx4) and ferritin heavy chain (FTH1) compared with the control group. These data show evidence that apatinib can regulate the expression of ferroptosis-associated proteins in HCT116 cells.
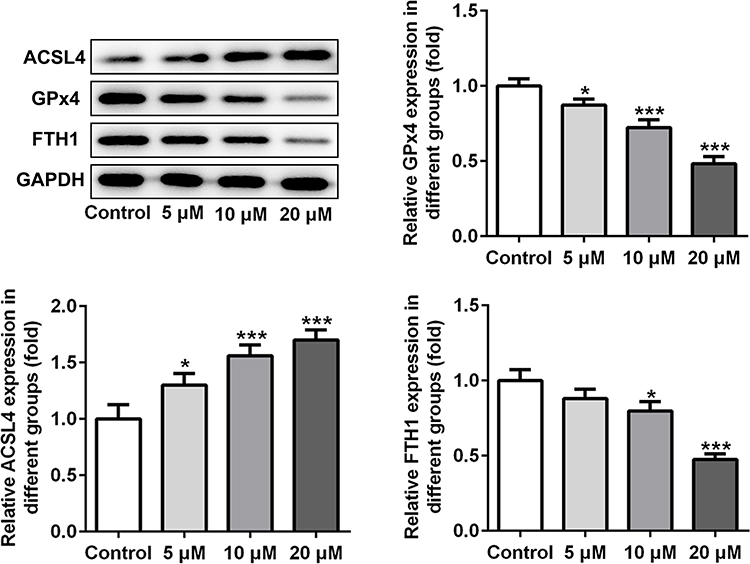 |
Figure 2 Apatinib regulates the expression of ferroptosis-associated proteins in HCT116 cells. The expression of ACSL4, GPx4 and FTH1 was determined using Western blot analysis. *P<0.05 ***P<0.001 vs control. Abbreviations: ACSL4, long-chain acyl-CoA synthetases 4; GPx4, glutathione peroxidase 4; FTH1, ferritin heavy chain. |
Apatinib Inhibits the ELOVL6 Expression in HCT116 Cells
To study the underlying mechanisms of apatinib in modulating ferroptosis of HCT116 cells, the SwissTargetPrediction website (http://www.swisstargetprediction.ch/) was employed. ELOVL6 was noticed as a target of apatinib. The GEPIA database (http://gepia.cancer-pku.cn/) displayed the high expression of ELOVL6 in CRC tissues compared with the normal tissues (Figure 3A). Then, the expression of ELOVL6 in HCT116 cells was determined using RT-qPCR and Western blot analyses. As shown in Figure 3B and C, ELOVL6 was heightened in HCT116 cells in comparison with the HIEC cells. Moreover, apatinib exposure remarkably downregulated ELOVL6 at the transcriptional and protein levels in a dose-dependent manner (Figure 3D and E). These observations reveal that apatinib possesses an inhibitory effect on the ELOVL6 expression in HCT116 cells.
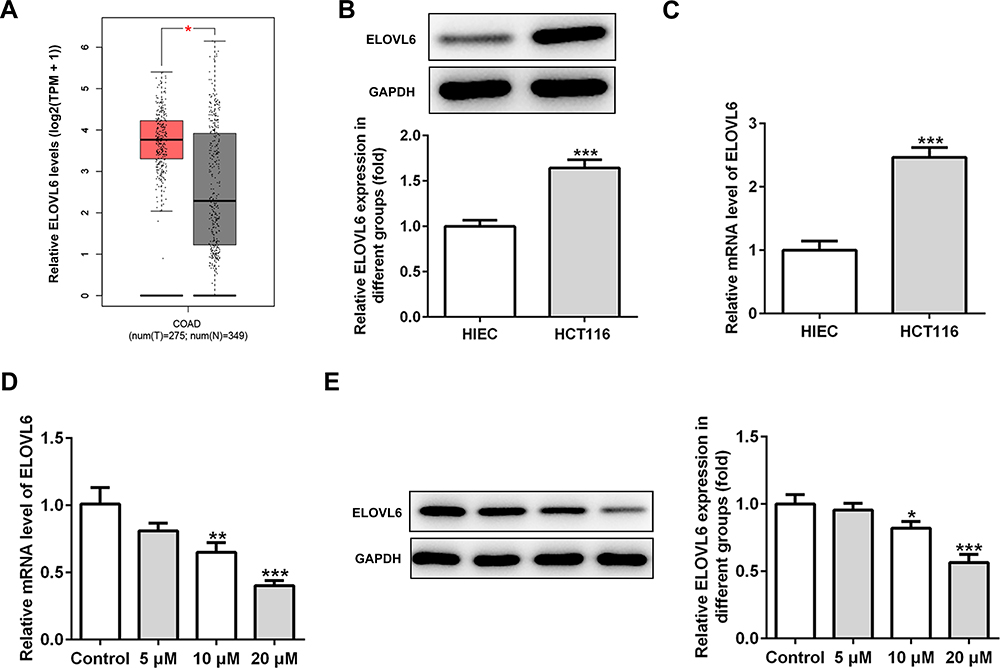 |
Figure 3 Apatinib inhibits the ELOVL6 expression in HCT116 cells. (A) The GEPIA database (http://gepia.cancer-pku.cn/) displayed that ELOVL6 is highly expressed in CRC tissues compared with the normal tissues. The expression of ELOVL6 in HCT116 and HIEC cells was detected using (B) Western blot analysis and (C) RT-qPCR, respectively. ***P<0.001 vs HIEC. (D) RT-qPCR and (E) Western blot analyses (40 μg protein sample per lane) were employed to evaluate the level of ELOVL6 after treatment of apatinib. *P<0.05, **P<0.01 and ***P<0.001 vs control. Abbreviations: ELOVL6, elongation of very long-chain fatty acids family member 6; CRC, colorectal cancer. |
ELOVL6 Overexpression Reverses the Effects of Apatinib on Cell Viability and Ferroptosis
To further investigate the specific role of ELOVL6 in CRC cells, ELOVL6 expression was overexpressed by transfecting overexpressing plasmid. After transfection, significant upregulation of ELOVL6 mRNA and protein expression are observed in Figure 4A and B. Afterwards, cell viability was tested using CCK-8 assay. In Figure 4C, ELOVL6 upregulation notably elevated the viability of HCT116 cells as compared to the 20 μM apatinib+Oe-NC group. As displayed in Figure 4D, the expression of ELOVL6 protein was conspicuously upregulated after ELOVL6 overexpression in apatinib-treated HCT116 cells. Additionally, the levels of intracellular total iron and Fe2+ were examined to analyze the function of ELOVL6 overexpression in ferroptosis. Results of Figure 4E and F elucidated that the suppressive effects of apatinib on both total iron and Fe2+ in HCT116 cells were reversed with ELOVL6 overexpression. Moreover, the production of ROS was significantly suppressed in apatinib-treated and ELOVL6-upregulated HCT116 cells relative to the apatinib-challenged HCT116 cells (Figure 4G). Consistently, in comparison with the 20 μM apatinib+Oe-NC group, ELOVL6 overexpression notably reduced the content of MDA while enhanced the levels of GSH and GPx (Figure 4H–J). Collectively, these results implicate that ELOVL6 overexpression neutralizes the effects of apatinib on cell viability and ferroptosis.
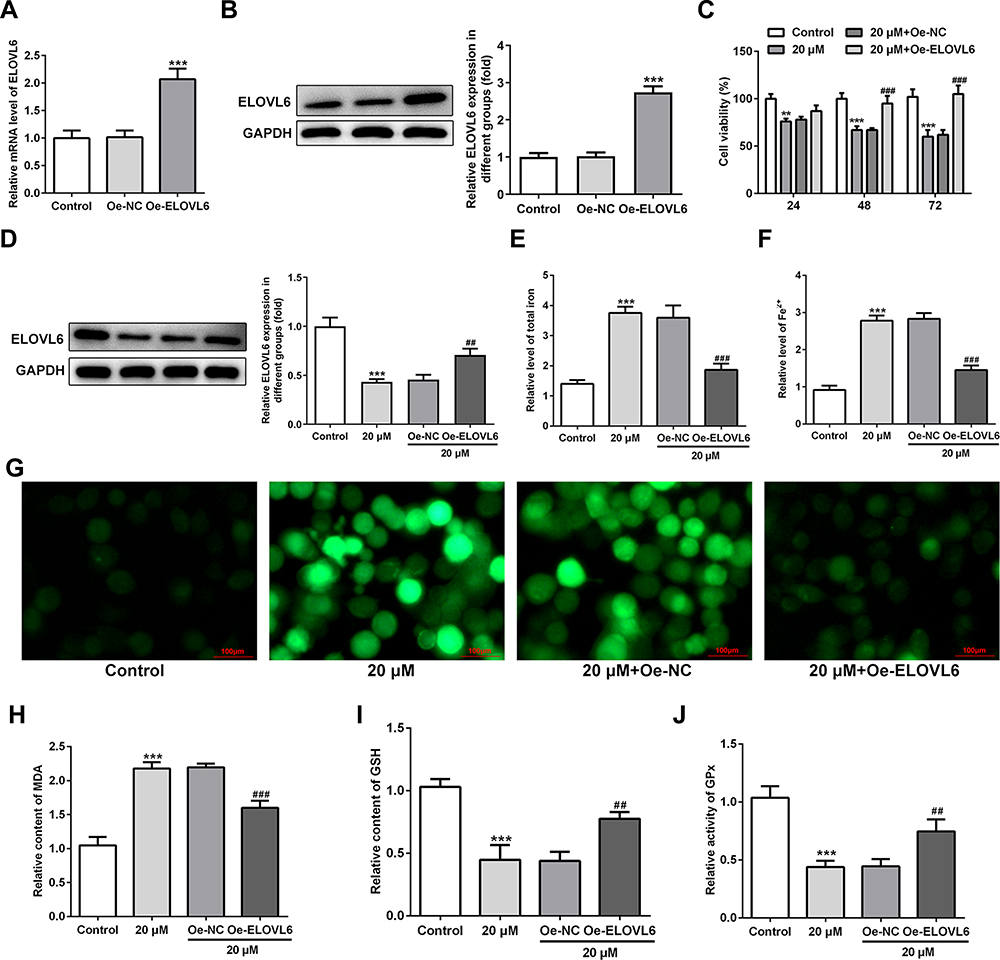 |
Figure 4 ELOVL6 upregulation reverses the effects of apatinib on cell viability and ferroptosis. (A and B) ELOVL6 overexpression was examined, respectively, using RT-qPCR and Western blot analysis after transfection. ***P<0.001 vs Oe-NC. (C) Cell viability of HCT116 cells was determined using a cell counting kit-8 assay kit. (D) The protein level of ELOVL6 after apatinib treatment upon transfection was examined with Western blotting. The relative concentrations of (E) total iron and (F) Fe2+ were detected using respective kits. (G) ROS level was detected by a ROS assay kit with 2, 7-dichlorofluorescein diacetate (DCFH-DA, Invitrogen, USA). The levels of (H) MDA, (I) GSH and (J) GPx were evaluated using commercial kits. **P<0.01 and ***P<0.001 vs control; ##P<0.01, ###P<0.001 vs Oe-NC. Abbreviations: ELOVL6, elongation of very long-chain fatty acids family member 6; ROS, reactive oxygen species; MDA, malondialdehyde; GSH, glutathione; GPx, glutathione peroxidase. |
ELOVL6 Overexpression Restores the Regulatory Effects of Apatinib on the Expression of Ferroptosis-Associated Proteins via Regulating ACSL4 Expression
Then, the role of ELOVL6 overexpression in modulating the levels of ferroptosis-associated proteins was determined through Western blot analysis. As displayed in Figure 5A–D, the increase in ACSL4 expression as well as the decrease in GPx4 and FTH1 expression resulted from apatinib treatment was reversed by ELOVL6 upregulation. Subsequently, STRING database was applied to predict the potential proteins interacting with ELOVL6, which illustrated that ACSL4, a vital regulator of ferroptosis, could interact directly with ELOVL6 (Figure 5E). Further, Co-IP assay explored their binding affinity, proving the strong interaction between ELOVL6 and ACSL4 (Figure 5F). Through the above findings, we conclude that ELOVL6 elevation abolishes the regulatory impacts of apatinib on the expression of ferroptosis-associated proteins via ACSL4.
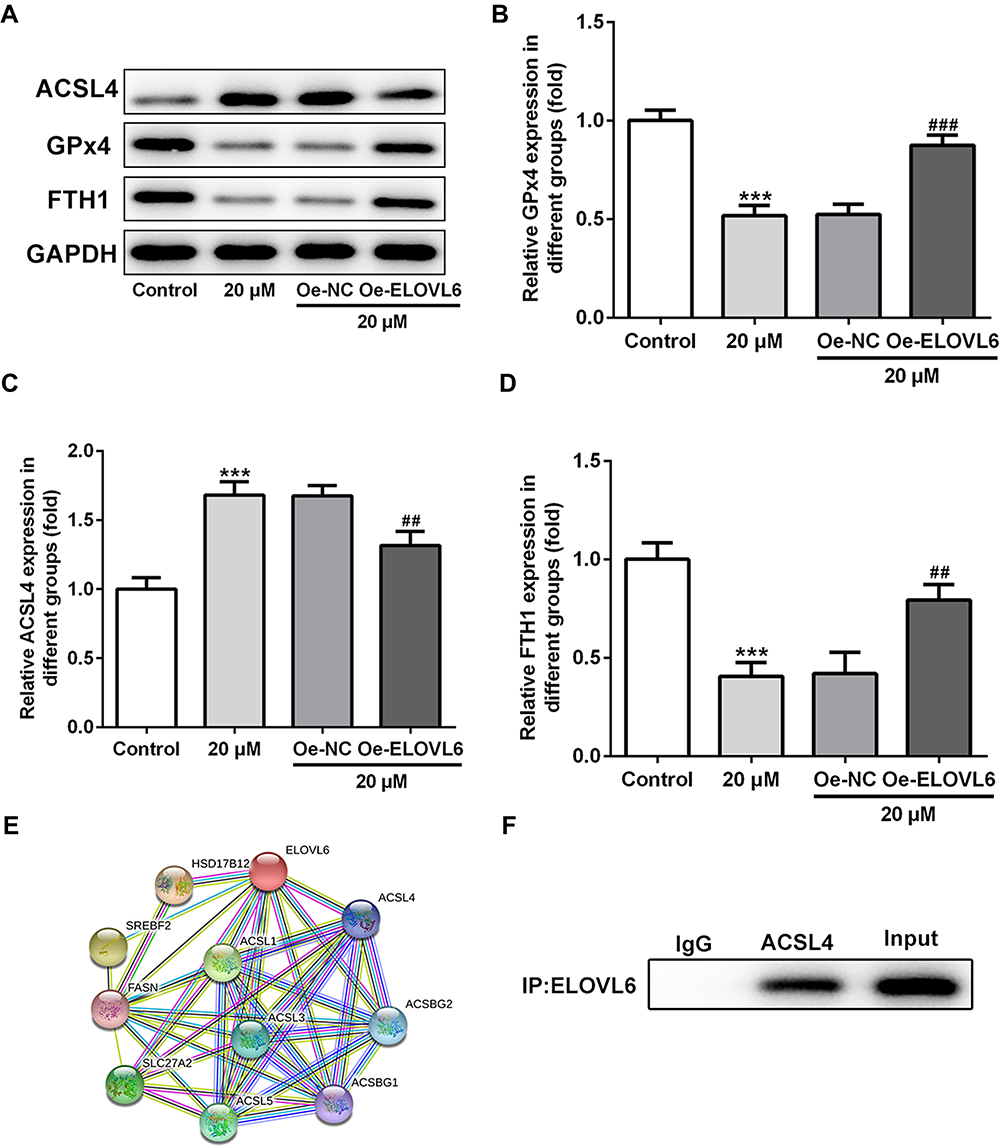 |
Figure 5 ELOVL6 overexpression restores the regulatory effects of apatinib on the expression of ferroptosis-associated proteins via ACSL4. (A–D) The expression of ACSL4, GPx4 and FTH1 was determined using Western blot analysis. ***P<0.001 vs control; ##P<0.01, ###P<0.001 vs Oe-NC. (E) The protein–protein interaction network for ELOVL6. (F) Immunoprecipitation was performed with indicated antibodies in HCT116 cells. The immunocomplexes were subjected to Western blot assay. Abbreviations: ELOVL6, elongation of very long-chain fatty acids family member 6; ACSL4, long-chain acyl-CoA synthetases 4; GPx4, glutathione peroxidase 4; FTH1, ferritin heavy chain. |
Discussion
Existing studies have indicated that apatinib inhibits the progression of CRC via promoting apoptosis. However, the detailed mechanisms underlying apatinib-induced cell death require further exploration. Among the mechanisms accounting for cell death, ferroptosis is a newly identified and unique category of regulated cell death.19 Ferroptosis is featured with iron dependence and lipid peroxidation production, which makes it distinguished from other kinds of cell death such as apoptosis and necrosis.20 Numerous studies have confirmed that ferroptosis plays a crucial role in killing tumor cells and inhibiting tumor growth in multiple cancers. For instance, ferroptosis activation suppresses CRC cell growth and tumorigenesis.21 In the present study, we demonstrated that apatinib treatment led to a decrease in cell viability and an increase in ferroptosis at least partly via targeting ELOVL6/ACSL4 signaling in HCT116 cells.
Iron accumulation and lipid ROS production are two critical events in ferroptosis, which is characterized by high intracellular Fe2+ concentration and ROS level.18 The decrease in total iron, Fe2+ and ROS levels has been widely reported in diverse cancers.22,23 Significant increases in intracellular iron content, ferrous iron concentration and ROS level were revealed in HCT116 cells after apatinib intervention. As is known to us all, ferroptosis is linked to a fatal accumulation of lipid peroxidation, evoking membrane rupture and cell content release.24 MDA, a representative end-product of lipid peroxidation, contributes to cell membrane injury and then ferroptosis.6 Moreover, the GSH antioxidant system is recognized as a major cell-protection system, preventing ferroptoticcell death.25 Furthermore, GPx is a unique member of glutathione peroxidases that regulates the ferroptosis to protect cells against detrimental lipid peroxidation.26 The present study unveiled that apatinib notably enhanced the levels of ROS and MDA, accompanied by decreases in GSH content and GPx activity, suggesting the activation of ferroptosis after apatinib exposure in HCT116 cells. It has been documented that excessive iron involves in lipid peroxidation through multiple mechanisms.27 ACSL4, a limiting enzyme for catalyzing the conversion of long-chain fatty acids to acyl-coenzyme, plays a crucial role in the process of iron-dependent oxidative stress.28,29 GPx4, a significant antioxidant enzyme in mammals, has been proved to perform a suppressive role in lipid ROS production during ferroptoticcell death.30 Besides, the inhibitory effect of GSH on ferroptosis is mainly attributed to the activation of GPx4.26 Moreover, FTH1 is an antioxidant that can cause the resistance to ferroptosis.31 In the current study, significant upregulation of ACSL4 and downregulation of GPx4 and FTH1 were viewed in apatinib-treated HCT116 cells, affirming the regulatory effects of apatinib on ferroptosis-related proteins.
To fully investigate the underlying mechanisms of apatinib in ferroptosis of HCT116 cells, the SwissTargetPrediction website (http://www.swisstargetprediction.ch/) was employed, the result of which delineated that ELOVL6 was targeted by apatinib. ELOVL6 was reported to be a tumor suppressor gene or an oncogene in various tumors. For instance, in hepatocellular carcinoma, low level of ELOVL6 indicates poor prognosis.32 Inhibition of ELOVL6 potentiates upregulation of genes related to breast cancer development and metastasis in vivo.33 Additionally, in a study investigating pediatric primary central nervous system germ cell tumors, the expression of ELOVL6 was observed to be abundant in germinoma.34 However, the GEPIA database (http://gepia.cancer-pku.cn/) displayed that ELOVL6 is expressed at high level in CRC tissues. Meanwhile, in the current study, the expression of ELOVL6 in mRNA and protein levels in HCT116 cells presented the same result. Therefore, whether apatinib targeted ELOVL6 to promote ferroptosis of HCT116 cells was explored. Results demonstrated that apatinib inhibited the expression of ELOVL6, and ELOVL6 overexpression reversed the effects of apatinib on ferroptosis of HCT116 cells. Additionally, to further excavate the molecular mechanisms underlying ELOVL6-mediated ferroptosis, STRING database was applied to search for the potential proteins interplaying with ELOVL6. Among the gained proteins, ACSL4, a vital regulator of ferroptosis, was identified as a biomarker and contributor of ferroptosis.28 It have been reported that ACSL4 was downregulated in human glioma tissues and cells, and it could suppress glioma cells proliferation via activating ferroptosis.35 Further, the interplay between ELOVL6 and ACSL4 was confirmed by co-IP assay. Thus, these findings suggest that apatinib inhibits cell viability and promotes ferroptosis in HCT116 cells at least partly via targeting ELOVL6/ACSL4 signaling.
Conclusion
In summary, the present research first discovered the effect of apatinib on the ferroptosis of CRC HCT116 cells. Mechanically, we demonstrated that apatinib boosted ferroptosis in CRC cells by targeting ELOVL6 and subsequently regulating ACSL4 expression, which provides a new mechanism support for apatinib application in the clinical treatment of patients suffering from colorectal cancer. However, the usage of single CRC cell line is a limitation of this study. Therefore, a comprehensive analysis is required in the future.
Data Sharing Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Disclosure
The authors declare that they have no competing interests.
References
1. Meng S, Jian Z, Yan X, Li J, Zhang R. LncRNA SNHG6 inhibits cell proliferation and metastasis by targeting ETS1 via the PI3K/AKT/mTOR pathway in colorectal cancer. Mol Med Rep. 2019;20(3):2541–2548. doi:10.3892/mmr.2019.10510
2. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–386. doi:10.1002/ijc.29210
3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
4. van der Werf A, Arthey K, Hiesmayr M, et al. The determinants of reduced dietary intake in hospitalised colorectal cancer patients. Support Care Cancer. 2018;26(6):2039–2047. doi:10.1007/s00520-018-4044-1
5. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
6. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
7. Nguyen THP, Mahalakshmi B, Velmurugan BK. Functional role of ferroptosis on cancers, activation and deactivation by various therapeutic candidates-an update. Chem Biol Interact. 2020;317:108930. doi:10.1016/j.cbi.2019.108930
8. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–849. doi:10.1016/j.ccell.2019.04.002
9. Shen LD, Qi WH, Bai JJ, et al. Resibufogenin inhibited colorectal cancer cell growth and tumorigenesis through triggering ferroptosis and ROS production mediated by GPX4 inactivation. Anat Rec. 10.
10. Chen P, Li X, Zhang R, et al. Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics. 2020;10(11):5107–5119. doi:10.7150/thno.44705
11. Scott AJ, Messersmith WA, Jimeno A. Apatinib: a promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs Today (Barc). 2015;51(4):223–229. doi:10.1358/dot.2015.51.4.2320599
12. Ma JL, Zhang T, Suo FZ, et al. Lysine-specific demethylase 1 activation by vitamin B2 attenuates efficacy of apatinib for proliferation and migration of gastric cancer cell MGC-803. J Cell Biochem. 2018;119(6):4957–4966. doi:10.1002/jcb.26741
13. Li A, Wang K, Xu A, et al. Apatinib as an optional treatment in metastatic colorectal cancer. Medicine (Baltimore). 2019;98(35):e16919. doi:10.1097/MD.0000000000016919
14. Liang L, Wang L, Zhu P, et al. A pilot study of apatinib as third-line treatment in patients with heavily treated metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17(3):e443–e449. doi:10.1016/j.clcc.2018.02.011
15. Cheng X, Feng H, Wu H, et al. Targeting autophagy enhances apatinib-induced apoptosis via endoplasmic reticulum stress for human colorectal cancer. Cancer Lett. 2018;431:105–114. doi:10.1016/j.canlet.2018.05.046
16. Su YC, Feng YH, Wu HT, et al. Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Sci Rep. 2018;8(1):6586. doi:10.1038/s41598-018-24633-3
17. Feng YH, Chen WY, Kuo YH, et al. Elovl6 is a poor prognostic predictor in breast cancer. Oncol Lett. 2016;12(1):207–212. doi:10.3892/ol.2016.4587
18. Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–465. doi:10.1002/hep.29117
19. Chen Y, Li N, Wang H, et al. Amentoflavone suppresses cell proliferation and induces cell death through triggering autophagy-dependent ferroptosis in human glioma. Life Sci. 2020;247:117425. doi:10.1016/j.lfs.2020.117425
20. Sang M, Luo R, Bai Y, et al. Mitochondrial membrane anchored photosensitive nano-device for lipid hydroperoxides burst and inducing ferroptosis to surmount therapy-resistant cancer. Theranostics. 2019;9(21):6209–6223. doi:10.7150/thno.36283
21. Sui XB, Zhang RN, Liu SP, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. 2018;9:8. doi:10.3389/fphar.2018.01371
22. Bao WE, Liu XW, Lv YL, et al. Nanolongan with multiple on-demand conversions for ferroptosis-apoptosis combined anticancer therapy. ACS Nano. 2019;13(1):260–273. doi:10.1021/acsnano.8b05602
23. Chen Y, Fan ZM, Yang Y, Gu CY. Iron metabolism and its contribution to cancer (Review). Int J Oncol. 2019;54(4):1143–1154. doi:10.3892/ijo.2019.4720
24. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
25. Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523(4):966–971. doi:10.1016/j.bbrc.2020.01.066
26. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
27. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–4975. doi:10.1073/pnas.1603244113
28. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. doi:10.1016/j.bbrc.2016.08.124
29. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
30. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170. doi:10.1007/82_2016_508
31. Bai T, Lei PX, Zhou H, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med. 2019;23(11):7349–7359. doi:10.1111/jcmm.14594
32. Li H, Wang XL, Tang J, Zhao HB, Duan M. Decreased expression levels of ELOVL6 indicate poor prognosis in hepatocellular carcinoma. Oncol Lett. 2019;18(6):6214–6220. doi:10.3892/ol.2019.10974
33. Zakharova GS, Poloznikov AA, Astakhova LA, et al. The effect of ELOVL6 fatty acid elongase inhibition on the expression of genes associated with the metastasis of breast cancer. Russ Chem Bull. 2018;67(12):2307–2315. doi:10.1007/s11172-018-2374-2
34. Wong JM, Chi SN, Marcus KJ, et al. Germinoma with malignant transformation to nongerminomatous germ cell tumor. J Neurosurg Pediatr. 2010;6(3):295–298. doi:10.3171/2010.6.PEDS09541
35. Cheng J, Fan YQ, Liu BH, Zhou H, Wang JM, Chen QX. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. 2020;43(1):147–158. doi:10.3892/or.2019.7419
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.