")
Back to Journals » OncoTargets and Therapy » Volume 11
Apatinib-loaded lipid nanobubbles combined with ultrasound-targeted nanobubble destruction for synergistic treatment of HepG2 cells in vitro
Authors Tian Y, Liu Z , Zhang L, Zhang J, Han X, Wang Q, Cheng W
Received 11 April 2018
Accepted for publication 25 May 2018
Published 10 August 2018 Volume 2018:11 Pages 4785—4795
DOI https://doi.org/10.2147/OTT.S170786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Yuhang Tian,1,* Zhao Liu,1,* Lei Zhang,1 Jia Zhang,2 Xue Han,1 Qiucheng Wang,1 Wen Cheng1
1Department of Ultrasound, Harbin Medical University Cancer Hospital, Harbin 150080, People’s Republic of China; 2Department of Microsystems and Microstructure Manufacturing, Ministry of Education, Harbin Institute of Technology, Harbin 150080, People’s Republic of China
*These authors contributed equally to this work
Purpose: Apatinib, an oral small-molecule antiangiogenetic medicine, is used to treat patients with advanced hepatocellular carcinoma. However, its systemic toxic side effects cannot be ignored. The ultrasound (US)-targeted nanobubble destruction technology can minimize systemic drug exposure and maximize therapeutic efficacy. The aim of this study was to develop novel GPC3-targeted and drug-loaded nanobubbles (NBs) and further assess the associated therapeutic effects on hepatocellular carcinoma cells in vitro.
Materials and methods: Apatinib-loaded NBs were prepared by a mechanical vibration method. GPC3, a liver tumor homing peptide, was coated onto the surface of apatinib-loaded NBs through biotin–avidin interactions to target liver cancer HepG2 cells. The effects of different treatment groups on cell proliferation, cell cycle, and apoptosis of HepG2 cells were tested.
Results: The NBs could achieve 68% of optimal drug encapsulation. In addition, ligand binding assays demonstrated that attachment of targeted NBs to human HepG2 liver cancer cells was highly efficient. Furthermore, cell proliferation assays indicated that the antiproliferative activities of GPC3-targeted and apatinib-loaded NBs in combination with US (1 MHz, 1 W/cm2, 30 s) were, respectively, 44.11%±2.84%, 57.09%±6.38%, and 67.51%±2.89% after 24, 48, and 72 h of treatment. Treatment with GPC3-targeted and apatinib-loaded NBs also resulted in a higher proportion of cells in the G1 phase compared with other treatment groups such as apatinib only and nontargeted apatinib-loaded NBs when US was utilized.
Conclusion: US-targeted and drug-loaded nanobubble destruction successfully achieved selective growth inhibition and apoptosis in HepG2 cells in vitro. Therefore, GPC3-targeted and apatinib-loaded NBs can be considered a novel chemotherapeutic approach for treating liver cancer in combination with US.
Keywords: ultrasound, apatinib, lipid nanobubble, liver cancer, GPC3, targeted delivery
Introduction
Hepatocellular carcinoma (HCC), one of the most common malignant tumors, ranks fourth in incidence and is the third leading cause of cancer death in People’s Republic of China.1 Women aged 50 or older are at high risk of suffering from HCC.2 Early-stage HCC is eligible for hepatectomy, which can improve liver function and the patients’ quality of life, but is also limited to Barcelona Clinic Liver Cancer (BCLC) A stage.3 Due to the lack of representative early symptoms and effective early-stage diagnostic methods, most patients present advanced liver cancer at first diagnosis and are ineligible for hepatectomy.
Chemotherapy is one of the most effective approaches for treating HCC patients. However, traditional chemotherapeutics require further assessment to maximize drug toxicity in killing cancer cells, while minimizing side effects such as asthenia, nausea, hypersensitive reactions, peripheral pain, and vomiting.4–8 Thus, a novel targeted drug delivery system is imminently required, which can minimize systemic drug exposure and maximize therapeutic efficacy.
In the past decades, a lot of efforts have been made in developing new drug delivery and release systems, including water-soluble prodrugs, microemulsions, liposomes, and nanoparticles.9–15 The ultrasound (US)-targeted nanobubble destruction (UTND) method has become a new trend for drug delivery to solid tumors.16–19 Compared with other drug delivery systems, UTND has multiple significant advantages. First of all, nanobubbles (NBs) are easily prepared by modified emulsification processes20 and used as US contrast agents to visualize tumors. In addition, NBs in combination with US can induce acoustic cavitation, stimulating cell membrane permeabilization and improving drug uptake by tumor cells.21–26 Previous studies particularly paid attention to nontargeted NBs that are easily accumulated in the reticuloendothelial system, resulting in lower drug concentration at the tumor site. To increase therapeutic efficacy and reduce systemic toxicity, it is essential to construct targeted and drug-loaded NBs, carrying tumor-specific ligands such as antibodies and peptides.
Thus, we hypothesized that GPC3-targeted and drug-loaded NBs used in combination with UTND might provide a new approach for targeted chemotherapy. In this study, we coupled the Anti-GPC3 (liver cancer homing peptide) antibody with apatinib-loaded NBs to test the hypothesis that GPC3-coated and drug-loaded NBs can enhance antitumor efficacy via UTND.
Materials and methods
Ethics statement
Approval from institutional research ethics committee of Harbin Medical University Cancer Hospital was obtained prior to the use of the HepG2 cells for research purposes.
Cell lines and culture
Human hepatocellular carcinoma HepG2 cells were a generous gift from the Institute of Cancer Research affiliated to Harbin Medical University (Harbin, People’s Republic of China). The cells were grown in HyClone minimum Eagle’s medium (MEM) (Thermo Fisher Scientific, Shanghai, People’s Republic of China) at 37°C in a humidified incubator containing 5% CO2, supplemented with 10% fetal bovine serum (FBS; GIBCO, Carlsbad, CA, USA), 100 μg/mL streptomycin, and 100 U/mL penicillin (GIBCO). Exponentially growing cells were used in all experiments.
Preparation of apatinib-loaded NBs
DSPC and DSPE-PEG2000 were purchased from Avanti Polar Lipids (Alabaster, AL, USA). NALNBs were produced by a modified emulsification process.16,27 An appropriate amount of lipid mixture (DSPC and DSPE-PEG2000 at a molar ratio of 9:1) and a given amount of apatinib (Hengrui Medicine Co., Ltd., Jiangsu, People’s Republic of China) were added into lipid components. The phospholipids were dissolved in chloroform and methanol (2:1, v/v). The resulting solution was moved to a round bottom flask, with the solvent removed by rotary vacuum evaporation at 55°C. The dried phospholipid samples were hydrated at 40°C with 4 mL PBS (pH 7.4). Next, the vial was degassed, and perfluoropropane (C3F8; Research Institute of Physical and Chemical Engineering of Nuclear Industry, Tianjin, People’s Republic of China) was added. The compounds were then mechanically vibrated for 45 s in a dental amalgamator (YJT Medical Apparatuses and Instruments, Shanghai, People’s Republic of China). The mixture was resuspended in 4 mL sterile PBS, and centrifuged at 800 rpm for 3 min. Finally, the supernatant was collected as nontargeted and apatinib-loaded NBs (NALNBs). Unloaded NBs (nondrug loaded) were similarly prepared but without apatinib addition.
Preparation of GPC3-targeted and apatinib-loaded NBs (TALNBs)
TALNBs were also produced by a modified emulsification process.20 There were some dissimilarities between TALNBs and NALNBs. The production process included the following 3 steps. First, biotinylated apatinib-loaded nanobubbles (BALNBs) were prepared as described above just replacing DSPE-PEG2000 with DSPE-PEG2000-biotin (Avanti Polar Lipids). The obtained BALNBs were subsequently washed with PBS and centrifuged at 800 rpm for 3 min to remove excess unbound lipids. To ensure there were no unbound lipids, an additional centrifugation was performed. Then, 50 μg of streptavidin (SA; Beijing Biosynthesis Biotechnology Co., Ltd., Beijing, People’s Republic of China) per 108 NBs was added to the washed dispersion.28 The resulting NBs were then incubated at 4°C for 30 min, washed 3 times by centrifugation as described earlier, to remove unbound streptavidin and obtain BSALNBs. Next, 20 μg of biotinylated Anti-GPC3 antibody (Beijing Biosynthesis Biotechnology Co., Ltd.) per 108 NBs was added to the NBs and incubated at 4°C for another 30 min. The mixture was then washed with PBS to remove unbound antibodies and obtain TALNBs. NBs were quantified on a hemocytometer (Bürker-Türk, Wertheim, Germany), with the size determined by a Zetasizer Nano ZS90 analyzer (Malvern Instruments Ltd., Worcestershire, UK).
Detection of streptavidin-conjugated with biotinylated and apatinib-loaded nanobubbles (BSALNBs)
After BSALNB production, 0.05 μg of FITC-labeled streptavidin (Beijing Biosynthesis Biotechnology Co., Ltd.) per 108 NBs was added for incubation at room temperature with gentle shaking for 30 min.29 Next, the mixture was washed with PBS 3 times to remove unbound FITC-labeled streptavidin and obtain BSALNBs. The fluorescence intensity of FITC was detected on a flow cytometer (Becton Dickinson Co., Ltd., Franklin Lakes, NJ, USA). The mean fluorescence intensity of FITC in BSALNBs reflected the relative levels of streptavidin incorporated into BALNBs.
Measurement of Anti-GPC3 antibody concentration on the surface of TALNBs
Relative levels of the Anti-GPC3 antibody bound to TALNBs were detected by measuring the mean fluorescence intensity of ALEXA488. Results were compared among NALNBs, BALNBs, and BSALNBs. The biotinylated Anti-GPC3 antibody was incubated, respectively, with NALNBs, BALNBs, and BSALNBs at 4°C for 30 min, and washed with PBS 3 times to remove the unbound antibody. Then, NALNBs, BALNBs, and BSALNBs were incubated with ALEXA-labeled goat anti-rabbit IgG (Beijing Biosynthesis Biotech Co., Ltd.) at 4°C for 1 h and washed with PBS 3 times to remove unbound IgG. Attention was paid to avoid light during incubation with ALEXA-labeled IgG. Relative levels of the Anti-GPC3 antibody bound to NBs were detected on a flow cytometer (Becton Dickinson Co., Ltd.).
Targeted binding of TALNBs to HepG2 cells in vitro
The binding capacity of TALNBs for GPC3-positive hepatocellular carcinoma cells was assessed. HepG2 cells (5×104/mL) were cultured in Costar 6-well plates as monolayers in 90% MEM containing 10% FBS for 24 h. The medium was supplemented with 1% penicillin/streptomycin and maintained in a humidified atmosphere containing 5% CO2 at 37°C. Static binding of TALNBs was performed. Briefly, TALNBs and NALNBs (1×108 NBs/mL) were added to 6-well plates, respectively, and incubated with HepG2 cells for 1 h; analysis was performed by scanning electron microscopy (Hitachi, Tokyo, Japan).
Apatinib encapsulation efficiency (EE) of drug-loaded NBs
The content of apatinib loaded was determined by ultraviolet (UV) measurements (GBC Scientific Equipment Inc., Victoria, Australia). A freshly prepared solution of apatinib was used to construct standard curve. Samples with 0.5, 1, 1.5, and 2 mg of apatinib, respectively, were added to the lipid mixture to produce drug-loaded NBs. After shaking, the NB suspensions were centrifuged at 800 rpm for 3 min. Next, solutions containing free apatinib were collected, and apatinib concentrations were determined on a UV–visible spectrometer. The amounts of uncombined apatinib were obtained by absorption measurements using a standard curve. EE (%) was calculated using the following formula:
|
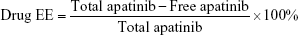
US destructibility of TALNBs in vitro
UTND was assessed by a previously developed method.30 Briefly, NBs were exposed to US at a frequency of 1 MHz and an output intensity of 3.5 W/cm2 using a 20 mm probe (Institute of Ultrasound Imaging, Second Affiliated Hospital of Chongqing Medical University, Chongqing, People’s Republic of China). Each US cycle included 4 frames of 30 s at a duty cycle of 50%. After US exposure, NB concentrations were determined on a hemocytometer (Bürker-Türk). The NBs were divided into 4 groups: blank (no sonication), unloaded NBs, NALNBs, and TALNBs.
US-mediated drug delivery in vitro
HepG2 cells (3×105 cells/well) were seeded in 6-well plates for overnight growth to allow cell attachment and cultured in MEM containing 10% FBS at 37°C. Afterward, the cells were equally divided into the following treatment groups: (a) culture medium only (negative control); (b) apatinib only; (c) NALNBs only; (d) TALNBs only; (e) TALNBs + US. In treatment groups (b)–(e), the apatinib dose was 10 μmol/L. In treatment groups (c) and (d), NALNBs (1×108 NBs/mL) and TALNBs (1×108 NBs/mL) were incubated with HepG2 cells for 1 h, and free NBs that did not bind to the cells were washed with PBS 3 times. In treatment group (e), after incubation with TALNBs and washing with PBS, the cells were exposed to a 20 mm diameter transducer (Institute of Ultrasound Imaging, Second Affiliated Hospital of Chongqing Medical University), which was immersed into the cell culture medium, 2 mm above the cell suspension. US parameters were: frequency, 1 MHz; intensity, 1.0 W/cm2; irradiation, 30 s/well.
Cell viability assay
After exposure to the US pulse, the cells were harvested and counted. Then, 8×103 cells/well were seeded into 96-well plates, and cultured for 24, 48, and 72 h, respectively. After 3 PBS washes, 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt (cell counting kit-8 [CCK-8]; Beyotime Institute of Biotechnology, Jiangsu, People’s Republic of China) was added to the samples. Absorbance of formazan at 450 nm was determined on a microplate reader (BioTek, Winooski, VT, USA).
Cell cycle analysis
HepG2 cells treated in different groups for 24 h were digested with trypsin-EDTA and washed twice with PBS by centrifugation at 1,000 rpm for 5 min. Next, the cells were fixed with 5 mL of 75% ethanol and incubated at 4°C overnight. Afterward, the cells were incubated with 0.25 mg/mL ribonuclease solution at 37°C for 30 min, followed by staining with 50 μL/mL propidium iodide in the dark for 30 min. In all groups, cell cycle stages were analyzed by flow cytometry (Becton Dickinson Co., Ltd.).
Cell apoptosis analysis
HepG2 cells treated in different groups for 24 h were stained with an apoptosis kit (Hangzhou Longji Biological Technology Co., Ltd, Hangzhou, People’s Republic of China) following the manufacturer’s instructions. Propidium iodide at a concentration of 0.5 μg/mL was incubated with the cells, and apoptosis was analyzed on a flow cytometer (Becton Dickinson Co., Ltd.).
Statistical analysis
The SPSS 13.0 software package (SPSS Inc., Chicago, IL, USA) was used for statistical analysis. Data are mean ± SD. The various groups were compared by one-way analysis of variance with a Dunnett method. Two-sided P<0.05 was considered statistically significant.
Results
Preparation and characterization of TALNBs
As schematically shown in Figure 1A, NBs loaded with apatinib were initially prepared; apatinib, mixed with other lipid components, forms the outer lipid shell of NBs. Subsequently, TALNBs were fabricated by conjugating apatinib-loaded NBs with biotinylated Anti-GPC3 antibody based on the biotin–streptavidin–biotin linkage.28 Due to specific binding of the Anti-GPC3 antibody, the ligand overexpressed on the surface of HCC,31–33 TALNBs can bind to tumor cells. After NB irradiation with an adequate intensity US pulse, drug can be released into tumor site (Figure 1B).
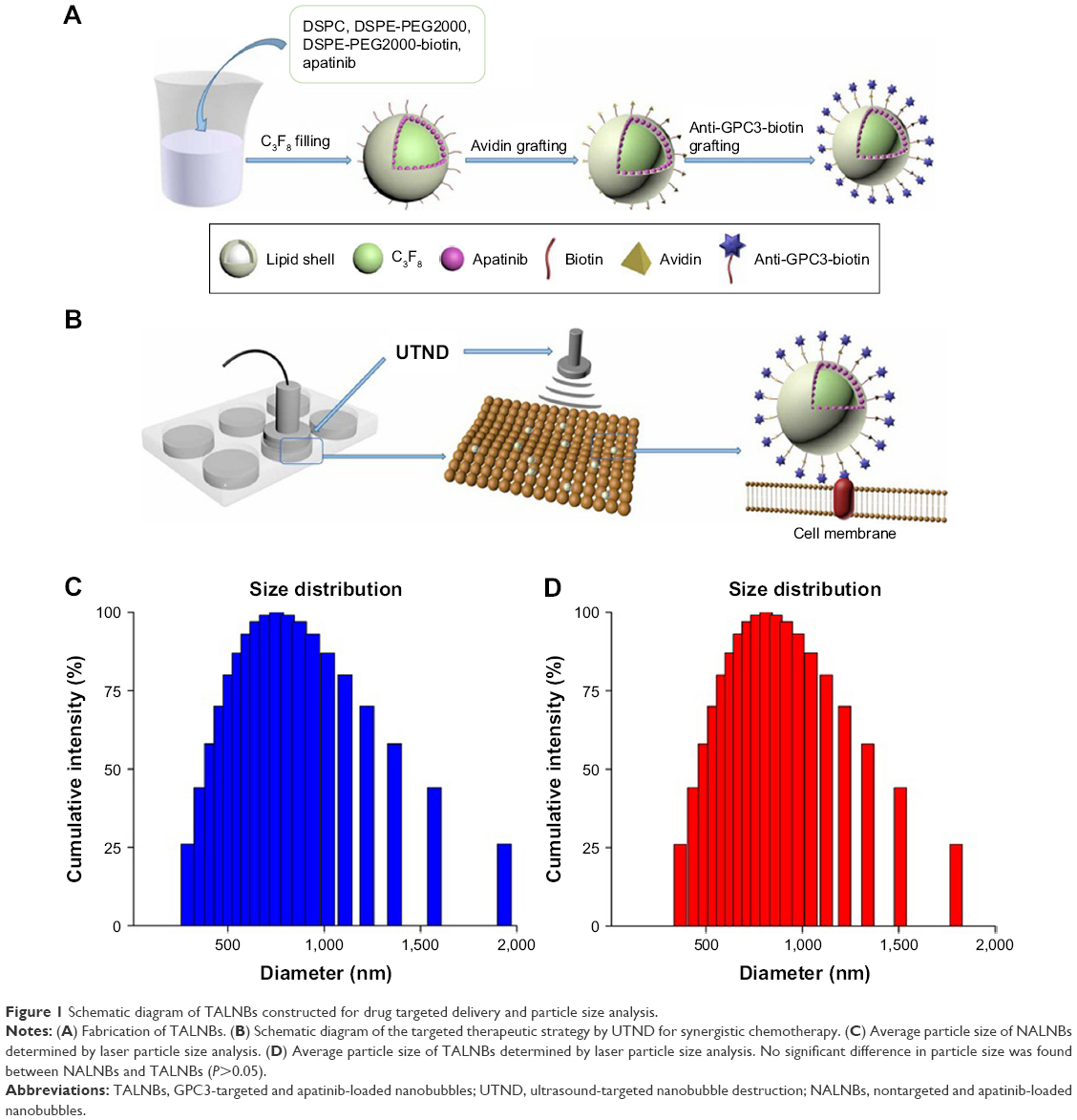 | Figure 1 Schematic diagram of TALNBs constructed for drug targeted delivery and particle size analysis. |
The synthesized NALNBs had a size distribution of 752.7±16.3 nm and a polydispersity index of 0.21±0.03. Meanwhile, TALNBs had a size distribution of 811.6±45.8 nm and a polydispersity index of 0.18±0.04. No significant particle size difference was observed between TALNBs and NALNBs (Figure 1C and D).
Conjugation of Anti-GPC3 polyclonal antibodies with drug-loaded NBs
Conjugation of Anti-GPC3 polyclonal antibodies with apatinib-loaded NBs was assessed by flow cytometry (Figure 2). First, NALNBs without FITC were used as blank control. Fluorescence intensities of BALNBs after incubation with FITC-labeled streptavidin showed that about 91.8%±4.1% of BALNBs were successfully conjugated with FITC-labeled streptavidin (Figure 2A). Next, Figure 2B shows the fluorescence intensities of BALNBs and BSALNBs after incubation with biotinylated Anti-GPC3 polyclonal antibodies and ALEXA488-labeled goat anti-rabbit IgG (secondary antibody for Anti-GPC3), with NALNBs used as blank control, similarly. The results showed that about 74.3%±3.9% of BSALNBs were successfully conjugated with Anti-GPC3 polyclonal antibodies and ALEXA488-labeled goat anti-rabbit IgG. Meanwhile, the fluorescence intensity of BALNBs was only 49.5%±5.5%. These findings indicated that the fluorescence intensity of BSALNBs was significantly higher than that of BALNBs (P<0.05), suggesting successful conjugation of Anti-GPC3 polyclonal antibodies.
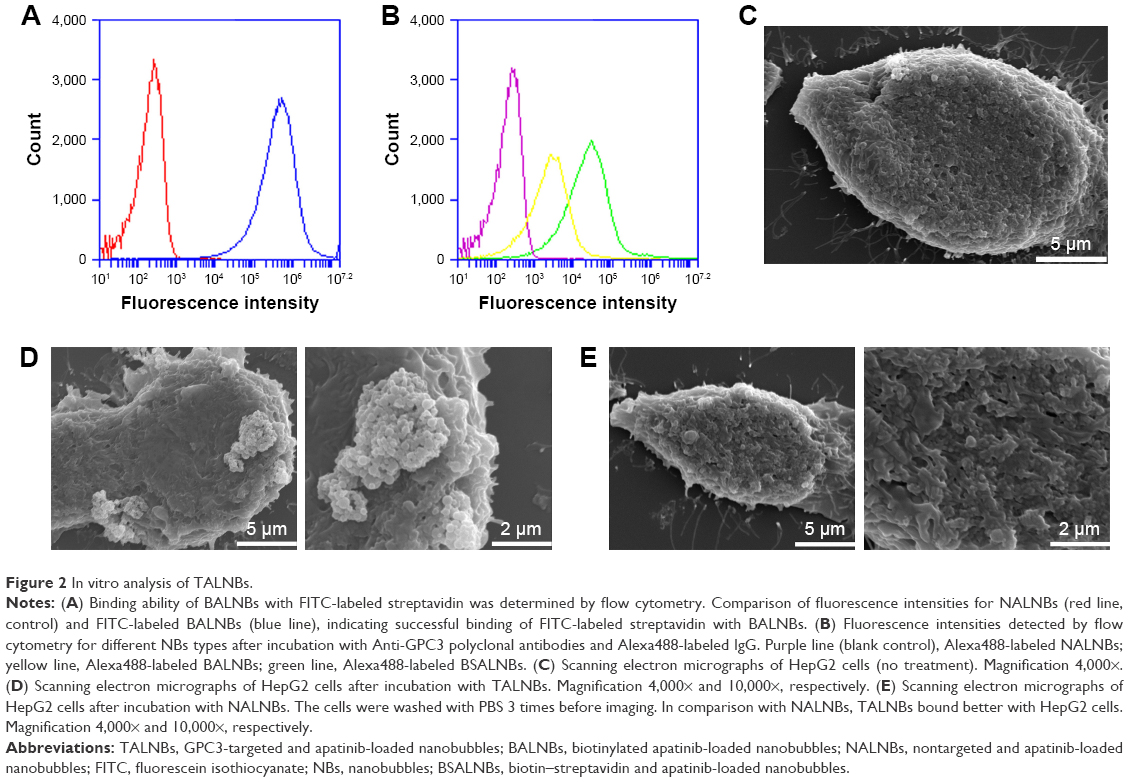 | Figure 2 In vitro analysis of TALNBs. |
Binding affinity of TALNBs for HepG2 cells
HepG2 cells were incubated with TALNBs and NALNBs, respectively. Scanning electron micrographs showed that there was no binding of NALNBs to HepG2 cells (Figure 2E, 4,000× and 10,000×, respectively). In contrast, large amounts of TALNBs were targeted to the surface of HepG2 cells successfully (Figure 2D, 4,000× and 10,000×, respectively). HepG2 cells (no treatment) were used as the control (Figure 2C, 4,000×).
Apatinib EE of drug-loaded NBs
The encapsulated amounts of apatinib were determined by centrifuging unbound apatinib from the NBs. Table 1 shows that about 65% of apatinib was encapsulated into NBs when loading 0.5 mg of apatinib. This resulted in a drug concentration of 16 μg/108 NBs, with an NB concentration of 4.91×108 NBs/mL. With increasing amounts of loaded apatinib, EE increased first before decreasing, while the concentration decreased gradually. After loading 2 mg of apatinib, EE was only about 40%, resulting in a drug concentration of 93 μg/108 NBs, with an NB concentration of 2.12×108 NBs/mL. Considering that 1 mg of apatinib loaded may result in optimal EE (about 68% EE), this amount of apatinib was used to prepare TALNBs.
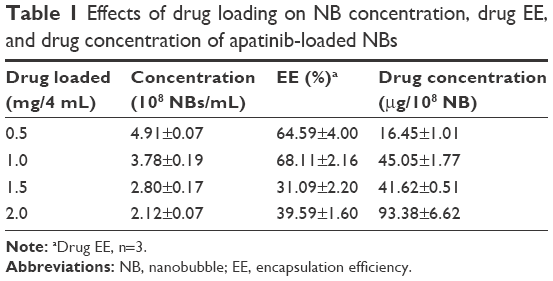 | Table 1 Effects of drug loading on NB concentration, drug EE, and drug concentration of apatinib-loaded NBs |
In vitro US destructibility of TALNBs
US destructibility of TALNBs was assessed for NALNBs, unloaded NBs, and blank (no sonication). The corresponding fold line diagram of NB concentration versus sonication time is shown in Figure 3. Attenuation half-life (t1/2) values for unloaded NBs, NALNBs, and TALNBs were 65, 50, and 45 s, respectively, with no significant difference between the 3 types NBs (P>0.05).
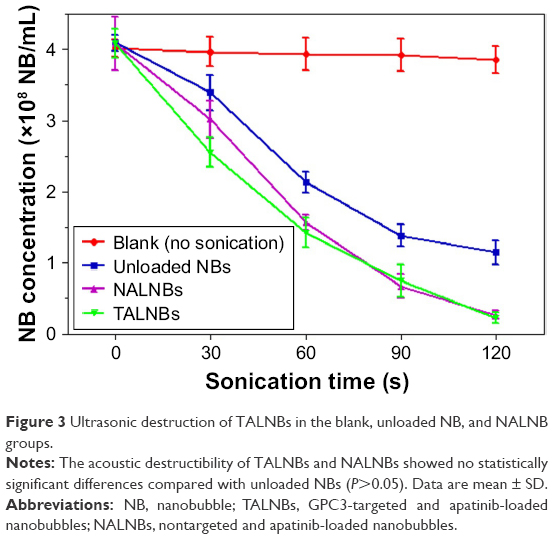 | Figure 3 Ultrasonic destruction of TALNBs in the blank, unloaded NB, and NALNB groups. |
Cell viability after US irradiation
Cell proliferation in different treatment groups was evaluated by CCK-8 assay, with untreated cells used as negative control. Figure 4 shows the cell proliferation inhibitory rates at 24, 48, and 72 h, respectively, for different treatment groups. The cell proliferation inhibitory rates were 44.11%±2.84%, 57.09%±6.38%, and 67.51%±2.89% at 24, 48, and 72 h for treatment group (e), respectively. Compared with other treatment groups, group (e) showed significantly higher rates (P<0.05), suggesting that US-mediated TALNB destruction effectively inhibited cell proliferation.
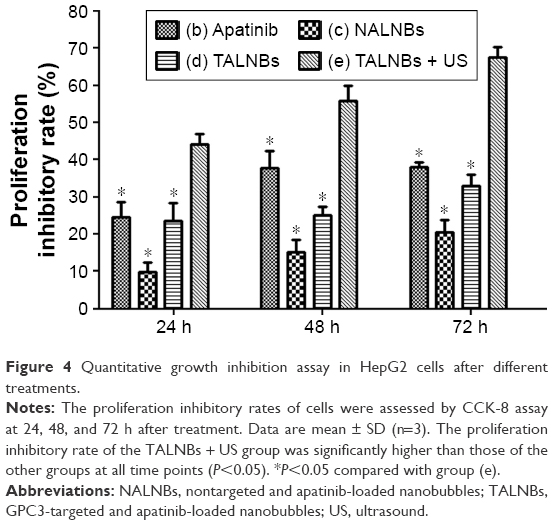 | Figure 4 Quantitative growth inhibition assay in HepG2 cells after different treatments. |
Cell cycle arrest after US irradiation
Cell cycle distribution in different treatment groups was analyzed by flow cytometry. Figure 5 compares cell cycle stages in the control and treatment groups. Accordingly, cell proportions for treatment group (e) in the G0/G1, S, and G2/M phases were 67.69%±4.65%, 19.83%±2.41%, and 12.48%±2.43%, respectively. In comparison with the other groups, group (e) showed a significantly higher proportion of cells in the G0/G1 phase (P<0.05) and markedly reduced amounts of cells in the S phase (P<0.05), indicating that US-mediated delivery of TALNBs induced a significant G1 phase arrest and a concomitant reduction of the S phase.
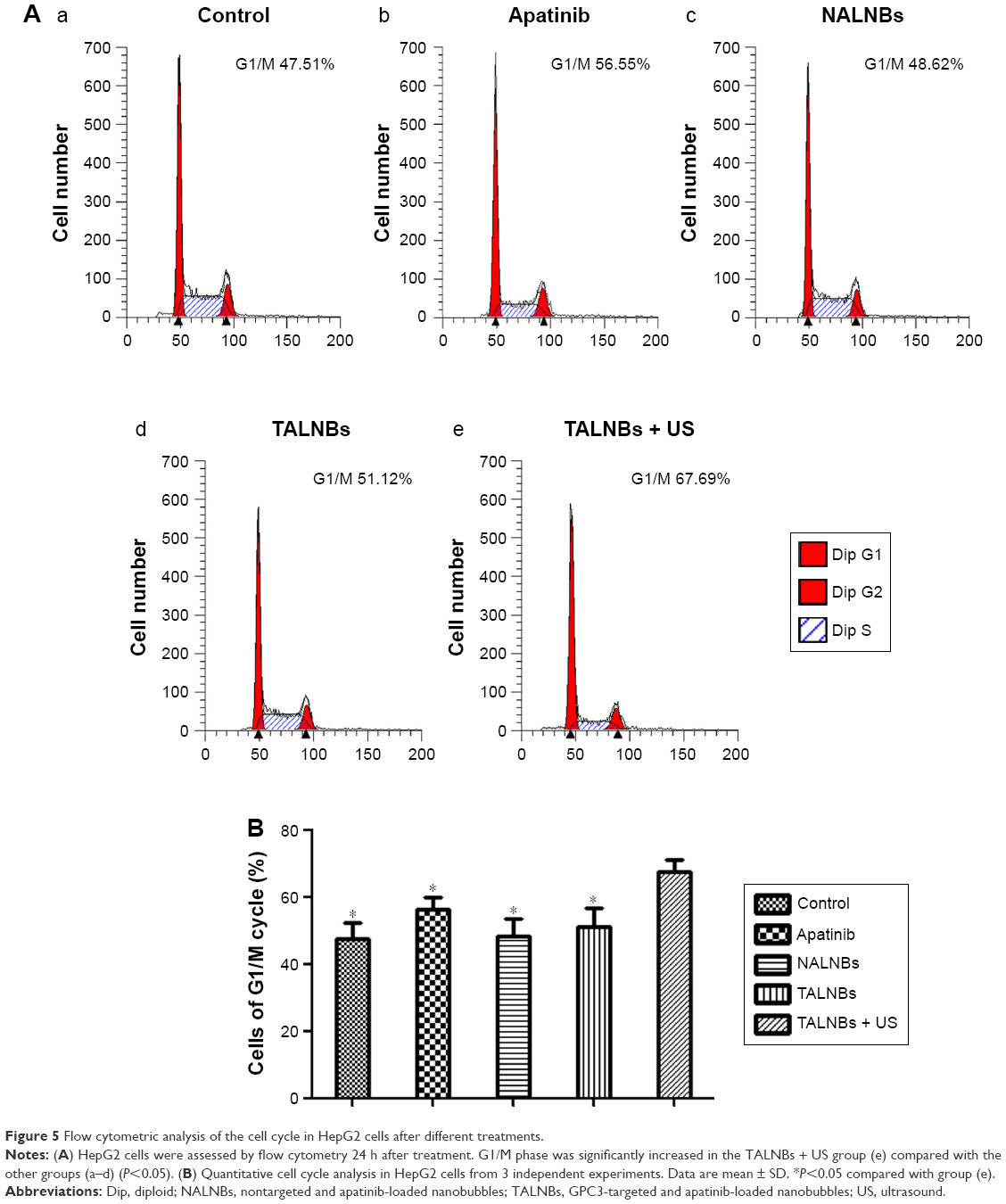 | Figure 5 Flow cytometric analysis of the cell cycle in HepG2 cells after different treatments. |
Cell apoptosis after US irradiation
Apoptotic rates in different treatment groups were determined by flow cytometry (Figure 6). Accordingly, the apoptotic rate in the TALNBs + US treatment group was 53.6%±7.9%. In comparison with other treatment groups, administration of TALNBs + US led to a significantly increased apoptotic rate (P<0.05), indicating a significant increase of cell apoptosis efficiency by ultrasonic destruction of TALNBs.
 | Figure 6 Flow cytometric analysis of cell apoptosis in HepG2 cells. |
Discussion
Many drugs for HCC treatment have passed clinical trials. Especially, apatinib, a novel and selective inhibitor of VEGF receptor 2, has emerged as a focal point for effectively improving treatment compliance and prolonging the survival period of patients with advanced gastric cancer34,35 and HCC.36 Apatinib has a wide range of tumor inhibitory effects both in vivo and in vitro, via competitive binding of the VEGFR-2 site.37,38 Furthermore, clinical studies of apatinib in HCC have shown promising clinical efficacy.36,39
The UTND technology is considered a novel approach for controlling drug delivery and contrast-enhanced US imaging.40–42 The apatinib API (Active Pharmaceutical Ingredient) used by us is lipophilic, but poor soluble in water. NBs are composed of amphiphilic phospholipids. Loading apatinib into the shell of NBs could increase the solubility of apatinib in PBS, simultaneously achieving satisfactory drug EE. In addition, apatinib encapsulation could prevent nonspecific killing of normal cells. Multiple studies have indicated that the affinity of binding to tumor cells could be enhanced by conjugating targeted antibodies or peptides with NBs.20,29,43 Therefore, NBs may be actively targeted to HCC cells via GPC3 receptors overexpressed on their surface.
GPC3 was initially found to encode a protein of the human glypican family.44 It is anchored to the cell membrane via a glycosylphosphatidylinositol (GPI) anchor.45,46 In addition, studies have indicated that GPC3 might serve as a specific target, which is highly expressed in HCC but not in normal liver cells and nonneoplastic liver lesions.47–51 By carrying the Anti-GPC3 antibody, NBs could be held in place before ultrasonic damage. Meanwhile, the drug is released directly to cancer cells rather than going through systemic circulation, thereby increasing treatment efficacy.
In this study, TALNBs were synthesized by combining an Anti-GPC3 antibody to apatinib-loaded NBs through biotin–avidin interactions. Although this binding method was successful in vitro, its clinical feasibility might be hampered by many factors. First, not all HCC tumors always express GPC3.52 Besides, GPC3 can be secreted into the blood in some GPC3-positive HCC patients.52 TALNBs might bind to free GPC3 in the blood, resulting in increasing blood concentration of apatinib, thus causing a side effect. Also, NBs, as foreign bodies, may stimulate in vivo the immune system unnecessarily. Furthermore, various in vivo factors, such as tumor vessels, blood flow rate, and wall shear stress, might alter the binding affinity of TALNBs for tumors. Each avidin consists of 4 subunits, combining with 4 biotin molecules. In the process of fabricating TALNBs, the biotin–avidin interactions were used, thus leading to larger size distribution than NALNBs. TALNBs were able to yield a drug EE of 68%, corroborating previous findings.29 The amounts of NBs decreased as more drug was loaded, which could be explained by the disruption of the whole NB structure during apatinib loading. By scanning electron microscopy, we found that Anti-GPC3 antibody conjugation with apatinib-loaded NBs significantly increased their attachment to HepG2 cells. We also found that the half-life of TALNBs was shorter compared with that of NALNBs, which might be directly related to differences in their surface static charges. Avidin is rich in charged amino acids.53 Previous studies showed that inclusion of charged species in the NB surface can disrupt the stability of the NB monolayer shell.54,55 In this study, US parameters were: frequency, 1 MHz; intensity, 1.0 W/cm2; irradiation, 30 s/well. Applying this level of US pulses alone may not induce significant adverse effects on cell viability, as shown previously.56 CCK-8 analysis carried out at 24, 48, and 72 h after treatment revealed that the cell proliferation inhibitory rate for TALNBs was improved significantly after US application. Use of TALNBs + US also led to a higher proportion of cells in the G1 phase, in accordance with a previous report.57 The mechanism behind US mediated delivery of drug-loaded NBs might be associated with the following influencing factors. First, physical interactions between the Anti-GPC3 antibody and GPC3 receptors contribute to the adhesion of TALNBs on the surface of HepG2 cells, improve local drug release, and shorten the time required for drug delivery into HepG2 cells. Second, applying US to NBs can lead to NB oscillation; after exposure to US, the drug-loaded NBs can be destroyed for enhanced delivery and release of the encapsulated drugs.58,59 Third, US exposure of NBs creates a critical transmembrane shear force, which leads to increased permeability and drug release through the cell membrane.60–62 Although, the above results indicated that tumor cells could be inhibited to a certain extent in the apatinib only, NALNBs only, TALNBs with or without US groups, the inhibitory effects of TALNBs combined with US were the most significant, which may be due to many factors, including sonoporation, cavitation effects, and the anticancer activity of apatinib. In vitro, the TALNBs + US group showed higher apoptosis efficiency compared with the other treatment groups. In order to avoid nonspecific destruction of NBs in blood circulation, US is applied to the tumor site rather than entire liver in vivo. However, the whole liver cannot be uniformly sonicated unlike cultured cells. We did not explicitly examine the dose of TALNBs and US’s pulse energy for TALNB application in vivo experiments. Therefore, a great deal of in vivo preclinical studies is required to evaluate the clinical applicability of the above approach.
Conclusion
Overall, we produced TALNBs for US-mediated delivery of drugs to HCC cells. This study demonstrated that US-targeted and drug-loaded nanobubble destruction successfully achieves selective growth inhibition and apoptosis in HepG2 cells in vitro. Therefore, TALNBs mediated by US may represent a novel and effective therapeutic approach for HCC.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant numbers 81571682, 2016), Hai Yan Science Foundation of Harbin Medical University Cancer Hospital (grant numbers JJQN2018-18).
Disclosure
The authors report no conflicts of interest in this work.
References
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Tsoulfas G, Agorastou P, Tooulias A, Marakis GN. Current and future challenges in the surgical treatment of hepatocellular carcinoma: a review. Int Surg. 2014;99(6):779–786. | ||
Roayaie S, Jibara G, Tabrizian P, et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology. 2015;62(2):440–451. | ||
Sisignano M, Angioni C, Park CK, et al. Targeting CYP2J to reduce paclitaxel-induced peripheral neuropathic pain. Proc Natl Acad Sci U S A. 2016;113(44):12544–12549. | ||
Jordan K, Gralla R, Jahn F, Molassiotis A. International antiemetic guidelines on chemotherapy induced nausea and vomiting (CINV): content and implementation in daily routine practice. Eur J Pharmacol. 2014;722(1433):197–202. | ||
Tricco AC, Blondal E, Veroniki AA, et al. Comparative safety and effectiveness of serotonin receptor antagonists in patients undergoing chemotherapy: a systematic review and network meta-analysis. BMC Med. 2016;14(1):216. | ||
Sakamoto J, Matsui T, Kodera Y. Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancer. 2009;12(2):69–78. | ||
Yong WP, Soo RA, Yong WP, Innocenti F. Clinical pharmacology and pharmacogenetics of gemcitabine. Drug Metab Rev. 2009;41(2):77–88. | ||
Gund M, Khanna A, Dubash N, Damre A, Singh KS, Satyam A. Water-soluble prodrugs of paclitaxel containing self-immolative disulfide linkers. Bioorg Med Chem Lett. 2015;25(1):122–127. | ||
Min HS, You DG, Son S, et al. Echogenic glycol chitosan nanoparticles for ultrasound-triggered cancer theranostics. Theranostics. 2015;5(12):1402–1418. | ||
Liu F, Feng L, Zhang L, Zhang X, Zhang N. Synthesis, characterization and antitumor evaluation of CMCS-DTX conjugates as novel delivery platform for docetaxel. Int J Pharm. 2013;451(1–2):41–49. | ||
Swet JH, Pacheco HJ, Iannitti DA, El-Ghanam A, Mckillop IH. A silica-calcium-phosphate nanocomposite drug delivery system for the treatment of hepatocellular carcinoma: in vivo study. J Biomed Mater Res Part B Appl Biomater. 2014;102(1):190–202. | ||
Mishra N, Yadav NP, Rai VK, et al. Efficient hepatic delivery of drugs: novel strategies and their significance. Biomed Res Int. 2013;2013:382184. | ||
Tam K. The roles of doxorubicin in hepatocellular carcinoma. ADMET DMPK. 2013;1(3):29–44. | ||
Wang L, Li M, Zhang N. Folate-targeted docetaxel-lipid-based-nanosuspensions for active-targeted cancer therapy. Int J Nanomedicine. 2012;7(7):3281–3294. | ||
Zhang Y, Chang R, Li M, Zhao K, Zheng H, Zhou X. Docetaxel-loaded lipid microbubbles combined with ultrasound-triggered microbubble destruction for targeted tumor therapy in MHCC-H cells. Onco Targets Ther. 2016;9(1):4763–4771. | ||
Cool SK, Geers B, Roels S, et al. Coupling of drug containing liposomes to microbubbles improves ultrasound triggered drug delivery in mice. J Control Release. 2013;172(3):885–893. | ||
De Cock I, Zagato E, Braeckmans K, et al. Ultrasound and microbubble mediated drug delivery: acoustic pressure as determinant for uptake via membrane pores or endocytosis. J Control Release. 2015;197:20–28. | ||
Yan F, Li L, Deng Z, et al. Paclitaxel-liposome-microbubble complexes as ultrasound-triggered therapeutic drug delivery carriers. J Control Release. 2013;166(3):246–255. | ||
Yan F, Li X, Jin Q, et al. Therapeutic ultrasonic microbubbles carrying paclitaxel and LyP-1 peptide: preparation, characterization and application to ultrasound-assisted chemotherapy in breast cancer cells. Ultrasound Med Biol. 2011;37(5):768–779. | ||
Hamano N, Negishi Y, Omata D, et al. Bubble liposomes and ultrasound enhance the antitumor effects of AG73 liposomes encapsulating antitumor agents. Mol Pharm. 2013;10(2):774–779. | ||
Florinas S, Nam HY, Kim SW. Enhanced siRNA delivery using a combination of an arginine-grafted bioreducible polymer, ultrasound, and microbubbles in cancer cells. Mol Pharm. 2013;10(5):2021–2030. | ||
Escoffre JM, Piron J, Novell A, Bouakaz A. Doxorubicin delivery into tumor cells with ultrasound and microbubbles. Mol Pharm. 2011;8(3):799–806. | ||
Lentacker I, De Cock I, Deckers R, De Smedt SC, Moonen CTW. Understanding ultrasound induced sonoporation: Definitions and underlying mechanisms. Adv Drug Deliv Rev. 2014;72:49–64. | ||
Mo S, Coussios CC, Seymour L, Carlisle R. Ultrasound-enhanced drug delivery for cancer. Expert Opin Drug Deliv. 2012;9(12):1525–1538. | ||
Li Y, Wang P, Chen X, et al. Activation of microbubbles by low-intensity pulsed ultrasound enhances the cytotoxicity of curcumin involving apoptosis induction and cell motility inhibition in human breast cancer MDA-MB-231 cells. Ultrason Sonochem. 2016;33:26–36. | ||
Kang J, Wu X, Wang Z, et al. Antitumor effect of docetaxel-loaded lipid microbubbles combined with ultrasound-targeted microbubble activation on VX2 rabbit liver tumors. J Ultrasound Med. 2010;29(1):61–70. | ||
Stieger SM, Dayton PA, Borden MA, et al. Imaging of angiogenesis using Cadence™ contrast pulse sequencing and targeted contrast agents. Contrast Media Mol Imaging. 2008;3(1):9–18. | ||
Liu H, Chang S, Sun J, et al. Ultrasound-mediated destruction of LHRHa-targeted and paclitaxel-loaded lipid microbubbles induces proliferation inhibition and apoptosis in ovarian cancer cells. Mol Pharm. 2014;11(1):40–48. | ||
Tinkov S, Winter G, Coester C, Bekeredjian R. New doxorubicin-loaded phospholipid microbubbles for targeted tumor therapy: Part I – formulation development and in-vitro characterization. J Control Release. 2010;143(1):143–150. | ||
Baumhoer D, Tornillo L, Stadlmann S, Roncalli M, Diamantis EK, Terracciano LM. Glypican 3 expression in human nonneoplastic, preneoplastic, and neoplastic tissues: a tissue microarray analysis of 4,387 tissue samples. Am J Clin Pathol. 2008;129(6):899–906. | ||
Llovet JM, Chen Y, Wurmbach E, et al. A molecular signature to discriminate dysplastic nodules from early hepatocellular carcinoma in HCV cirrhosis. Gastroenterology. 2016;131(6):1758–1767. | ||
Zhu ZW, Friess H, Wang L, et al. Enhanced glypican-3 expression differentiates the majority of hepatocellular carcinomas from benign hepatic disorders. Gut. 2001;48(4):558–564. | ||
Li J, Qin S, Xu J, et al. Apatinib for chemotherapy-refractory advanced metastatic gastric cancer: results from a randomized, placebo-controlled, parallel-arm, phase II trial. J Clin Oncol. 2013;31(26):3219–3225. | ||
Li J, Qin S, Xu J, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34(13):1448–1454. | ||
Qin S. Apatinib in Chinese patients with advanced hepatocellular carcinoma: a phase II randomized, open-label trial. J Clin Oncol. 2014;32(Suppl 15):abstr 4019. | ||
Tian S, Quan H, Xie C, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102(7):1374–1380. | ||
Scott AJ, Messersmith WA, Jimeno A. Apatinib: a promising oral antiangiogenic agent in the treatment of multiple solid tumors. Drugs Today. 2015;51(4):223–229. | ||
Kou P, Zhang Y, Shao W, et al. Significant efficacy and well safety of apatinib in an advanced liver cancer patient: a case report and literature review. Oncotarget. 2017;8(12):20510–20515. | ||
Escoffre JM, Mannaris C, Geers B, et al. Doxorubicin liposome-loaded microbubbles for contrast imaging and ultrasound-triggered drug delivery. IEEE Trans Ultrason Ferroelectr Freq Control. 2013;60(1):78–87. | ||
Li P, Zheng Y, Ran H, et al. Ultrasound triggered drug release from 10-hydroxycamptothecin-loaded phospholipid microbubbles for targeted tumor therapy in mice. J Control Release. 2012;162(2):349–354. | ||
Liao AH, Li YK, Lee WJ, et al. Estimating the delivery efficiency of drug-loaded microbubbles in cancer cells with ultrasound and bioluminescence imaging. Ultrasound Med Biol. 2012;38(11):1938–1948. | ||
Chang S, Guo J, Sun J, et al. Targeted microbubbles for ultrasound mediated gene transfection and apoptosis induction in ovarian cancer cells. Ultrason Sonochem. 2013;20(1):171–179. | ||
Filmus J, Church JG, Buick RN. Isolation of a cDNA corresponding to a developmentally regulated transcript in rat intestine. Mol Cell Biol. 1988;8(10):4243–4249. | ||
Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9(5):224. | ||
Filmus J, Capurro M. Glypican-3: a marker and a therapeutic target in hepatocellular carcinoma. FEBS J. 2013;280(10):2471–2476. | ||
Liu JW, Zuo XL, Wang S. Diagnosis accuracy of serum Glypican-3 level in patients with hepatocellular carcinoma and liver cirrhosis: a meta-analysis. Eur Rev Med Pharmacol Sci. 2015;19(19):3655–3673. | ||
Sham JG, Kievit FM, Grierson JR, et al. Glypican-3-targeted 89Zr PET imaging of hepatocellular carcinoma. J Nucl Med. 2014;55(5):799–804. | ||
Feng M, Ho M. Glypican-3 antibodies: a new therapeutic target for liver cancer. FEBS Lett. 2014;588(2):377–382. | ||
Gao W, Tang Z, Zhang YF, et al. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun. 2015;6:6536. | ||
Gao H, Li K, Tu H, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–6428. | ||
Nakatsura T, Yoshitake Y, Senju S, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. 2003;306(1):16–25. | ||
Yao Z, Zhang M, Sakahara H, et al. The relationship of glycosylation and isoelectric point with tumor accumulation of avidin. J Nucl Med. 1999;40(3):479–483. | ||
Klibanov AL. Targeted delivery of gas-filled microspheres, contrast agents for ultrasound imaging. Adv Drug Deliv Rev. 1999;37(1–3):139–157. | ||
Borden MA, Longo ML. Dissolution behavior of lipid monolayer-coated, air-filled microbubbles: effect of lipid hydrophobic chain length. Langmuir. 2002;18(24):9225–9233. | ||
Wang JF, Wang JB, Chen H, et al. Ultrasound-mediated microbubble destruction enhances gene transfection in pancreatic cancer cells. Adv Ther. 2008;25(5):412–421. | ||
Lu W, Ke H, Ding Q, et al. Apatinib has anti-tumor effects and induces autophagy in colon cancer cells. Iran J Basic Med Sci. 2017;20(9):990–995. | ||
Escoffre JM, Novell A, Piron J, Zeghimi A, Doinikov A, Bouakaz A. Microbubble attenuation and destruction: are they involved in sonoporation efficiency? IEEE Trans Ultrason Ferroelectr Freq Control. 2013;60(1):46–52. | ||
Luan Y, Faez T, Gelderblom E, et al. Acoustical properties of individual liposome-loaded microbubbles. Ultrasound Med Biol. 2012;38(12):2174–2185. | ||
Wu J. Theoretical study on shear stress generated by microstreaming surrounding contrast agents attached to living cells. Ultrasound Med Biol. 2002;28(1):125–129. | ||
Wu J, Ross JP, Chiu JF. Reparable sonoporation generated by microstreaming. J Acoust Soc Am. 2002;111(3):1460–1464. | ||
Wu J, Nyborg WL. Ultrasound, cavitation bubbles and their interaction with cells. Adv Drug Deliv Rev. 2008;60(10):1103–1116. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.