Back to Journals » International Journal of Nanomedicine » Volume 16
Antiviral Drug Delivery System for Enhanced Bioactivity, Better Metabolism and Pharmacokinetic Characteristics
Authors Chen R, Wang T ![]() , Song J, Pu D, He D, Li J, Yang J
, Song J, Pu D, He D, Li J, Yang J ![]() , Li K, Zhong C
, Li K, Zhong C ![]() , Zhang J
, Zhang J ![]()
Received 14 April 2021
Accepted for publication 11 July 2021
Published 22 July 2021 Volume 2021:16 Pages 4959—4984
DOI https://doi.org/10.2147/IJN.S315705
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Ran Chen,1,* Tingting Wang,2,* Jie Song,1,* Daojun Pu,3,* Dan He,1 Jianjun Li,1 Jie Yang,1 Kailing Li,1 Cailing Zhong,1 Jingqing Zhang1
1Chongqing Research Center for Pharmaceutical Engineering, School of Pharmacy, Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 2Biochemistry and Molecular Biology Laboratory, Experimental Teaching and Management Center, Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 3Pharmaceutical Institute, Southwest Pharmaceutical Limited Company, Chongqing, 400038, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingqing Zhang Email [email protected]
Abstract: Antiviral drugs (AvDs) are the primary resource in the global battle against viruses, including the recent fight against corona virus disease 2019 (COVID-19). Most AvDs require multiple medications, and their use frequently leads to drug resistance, since they have poor oral bioavailability and low efficacy due to their low solubility/low permeability. Characterizing the in vivo metabolism and pharmacokinetic characteristics of AvDs may help to solve the problems associated with AvDs and enhance their efficacy. In this review of AvDs, we systematically investigated their structure-based metabolic reactions and related enzymes, their cellular pharmacology, and the effects of metabolism on AvD pharmacodynamics and pharmacokinetics. We further assessed how delivery systems achieve better metabolism and pharmacology of AvDs. This review suggests that suitable nanosystems may help to achieve better pharmacological activity and pharmacokinetic behavior of AvDs by altering drug metabolism through the utilization of advanced nanotechnology and appropriate administration routes. Notably, such AvDs as ribavirin, remdesivir, favipiravir, chloroquine, lopinavir and ritonavir have been confirmed to bind to the severe acute respiratory syndrome-like coronavirus (SARS-CoV-2) receptor and thus may represent anti-COVID-19 treatments. Elucidating the metabolic and pharmacokinetic characteristics of AvDs may help pharmacologists to identify new formulations with high bioavailability and efficacy and help physicians to better treat virus-related diseases, including COVID-19.
Keywords: antiviral drug, delivery systems, metabolism, pharmacokinetics, pharmacodynamics
Graphical Abstract:

Introduction
Many infectious diseases have rapid propagation and high infection rates with potential for human pandemics. Emerging infectious diseases caused by such viruses as severe acute respiratory syndrome (SARS), Middle East respiratory syndrome (MERS) and Ebola virus, present major threats to public health.1 The current outbreak of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to spread.2 COVID-19 may cause severe cytokine storm and aggravate immunopathological damage, which make antiviral treatment more complicated. The related cytokines include but not limited to TNF-α, IL-1 and IL-6.3–5 With the explosive growth of confirmed cases, the World Health Organization (WHO) declared the outbreak to be a public health emergency of international concern on January 30, 2020.6 At present, COVID-19 has spread to more than 200 countries and regions around the world, and the outbreak of COVID-19 has caused serious damage worldwide. As of 14:00 on July 19, 2021, there were a total of 191,229,635 diagnosed cases of COVID-19 diagnosed, and 4,105,799 deaths globally. Compared with SARS and MERS, the spread rate of COVID-19 is more rapid, probably due to the increased globalization and virus adaptability to various environments.7
At present, drugs employed to fight viral infections include natural medicines8–11 (eg, liquorice, Scutellaria baicalensis and forsythia), chemical drugs12 (eg, ribavirin, remdesivir, favipiravir) and biotechnology-derived drugs3 (eg, IFN-α, IFN-β and peptide). Natural antiviral medicines have multiple targets and moderate effects, and they usually contain complex ingredients but exhibit only limited efficacy.13 Biotechnology-derived AvDs have high curative effects and induce low drug resistance, but they usually need to be administered by injection due to their poor stability and bioavailability, and they are very easily inactivated in vivo.14 Chemical AvDs inhibit viruses quickly and strongly, and as most are administered orally, their use and storage are convenient. Chemical AvDs are the main treatment employed in antiviral therapy.15 The physicochemical properties of these drugs, as well as their metabolism, affect their efficacy to varying degrees.16–18 To improve the antiviral efficacy of AvDs, we need to comprehensively understand the characteristics of AvDs. In this review, we systematically investigated the structural, physicochemical, metabolic, kinetic and bioactive characteristics and pharmacological effects of popular nucleoside analogs (NA-AvDs) and non-nucleoside analogs of AvDs (NN-AvDs) currently on the market (Figure 1).
 |
Figure 1 The schematic diagram for antiviral drug delivery systems to improve bioactivity, increase metabolism and pharmacokinetic characteristics. (A) AvDs have poor absorption due to low solubility/low permeability. (B) drug delivery systems are used to enhance absorption and bioavailability of AvDs. (C) AvDs are metabolized by hepatic microsomal enzymes. (D) The pharmacodynamics activities of AvDs are enhanced. The upward arrows (↑↑↑): refers to an increase in the absorption/bioavailability of the drug. |
The scientific community is urged to explore and develop novel potent antiviral agents. Considering that vaccine development is time-consuming and that viruses mutate quickly, antivirals remain the main treatment for viral infections. To date, there are α-interferon, lopinavir/ritonavir, ribavirin, chloroquine phosphate, and arbidol have been proven to possess effectiveness in the general treatment of COVID-19, and tocilizumab (biological drugs) is used in severe/critical immunotherapy in accordance with the guidelines of the Chinese Diagnosis and Treatment Protocol on the government website (Trial Version 7) (https://china.huanqiu.com/article/9CaKrnKpIEe. Available on October 19, 2020). Most AvDs currently on the market are oral tablets. Poor pharmacokinetic profiles and high resistance are already known to the main disadvantages of these AvDs. Therefore, loading antiviral drugs into advanced delivery systems, such as lipid-based, macromolecule-based, and nanoparticle-based systems, may conducive to overcoming the abovementioned disadvantages. In this review, 16 typical AvDs currently on the market are investigated by retrieving their data from available databases (Table 1).
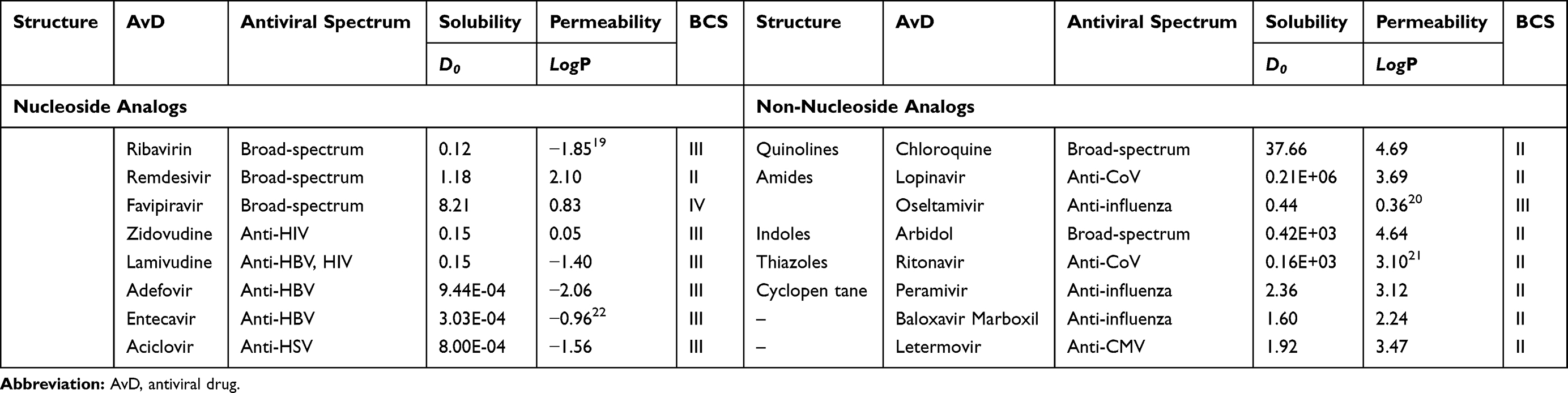 |
Table 1 Solubility and Permeability of AvDs |
Solubility, Permeability and Structural Properties of AvDs
Solubility and Permeability Limits of AvDs
The dose number (D0) and oil-water partition coefficient (log P) were employed to estimate the biopharmaceutics classification system (BCS) for AvDs. The D0 value > 1 is the defining criterion of low solubility and log P ≤ 1.632 is the defining criterion of low permeability.23 According to the solubility and permeability, the AvDs are classified into 3 groups (Figure 2). Among these drugs, 50% belong to BCS II with low solubility, 44% belong to BCS III with low permeability, 6% belong to BCS IV with low solubility and low permeability. These parameters illustrate the low absorption of AvDs in vivo.
 |
Figure 2 The classification ratio of antiviral drugs (AvDs) according to the biopharmaceutics classification system (BCS) criteria. |
Structural Characteristics of AvDs
According to molecular structure, AvDs are divided into 2 types (Figure 3). (1) NA-AvD, which are modified nucleosides with a structure that mimics the structure of natural nucleosides. NA-AvDs are recognized by cellular or viral enzymes and lead to disruption/termination of replication or other biological processes due to incorrect structural modifications.24 NA-AvDs are further classified into 3 subtypes: (i) purine NA-AvDs (eg, adefovir, entecavir and acyclovir), (ii) pyrimidine NA-AvDs (eg, zidovudine and lamivudine), and (iii) other NA-AvDs (eg, ribavirin, remdesivir and favipiravir). (2) NN-AvDs, include 4 subtypes: (i) quinolines, such as chloroquine phosphate, (ii) amides, such as lopinavir, oseltamivir (neuraminidase inhibitor: cyclohexene derivative) and palamivir (neuraminidase inhibitor: cyclopentane derivative), (iii) indoles, such as abidol, (iv) thiazoles (ritonavir), and (v) others (baloxavir marboxil and letermovir).
 |
Figure 3 Structures of AvDs. Basic structures of (A) adenine nucleotide analog, (B) guanine nucleotide analog, (C) thymine nucleotide analog, (D) cytosine, (E) quinolones, (F) indoles, (G) thiazoles nucleotide analog; (H) baloxavir marboxil; (I) letermovir. |
Relationship Between Structure and Solubility/Permeability of AvDs
Seventy-five percent of the abovementioned NA-AvDs (such as ribavirin, adefovir, entecavir, acyclovir, zidovudine, lamivudine) and approximately 12% of the NN-AvDs (such as oseltamivir) had low permeability, probably due to the hydroxyl groups in their molecular structures, which increase their polarity. Also, 25% of NA-AvDs (such as remdesivir, favipiravir) and 88% of NN-AvDs (such as chloroquine, lopinavir, arbidol, ritonavir, peramivir, baloxavir marboxil and letermovir) had low solubility due to their hydrophobic macromolecular structures.
Pharmaceutical Technology to Improve Solubility and Permeability
Oral drugs require sufficient solubility and intestinal absorption to enable drug molecules to reach action sites and exert therapeutic effects.25 Solubility is a prerequisite to confirm drug absorption and clinical response for drugs given orally.23 Permeability represents the speed and extent of oral drug diffusion through the mucus layer and then through the submucosa and epithelial cell barriers into the blood or lymphatic circulation.26 Currently, numerous advanced pharmaceutical technologies have been applied to increase the solubility and permeability of AvDs, including adding auxiliary ingredients (such as latent solvents and penetration enhancers) and applying cutting-edge preparation methods (such as inclusion technology, solid dispersion technology and micronization technology and nanotechnology).
Pharmaceutical Technology to Increase Both Solubility and Permeability
Adding Auxiliary Ingredients
Lopinavir solid dispersions were developed by using Soluplus as a polymeric solubilizer.27 An in vitro characterization study showed that Soluplus solubilized lopinavir in water almost linearly as a function of concentration by creating H-bonds of water with the drug carbonyl group and forming micelles in water. A Caco-2 cell transport study demonstrated that Soluplus significantly enhanced the permeability of lopinavir through the rat intestine via H-bond or micelle formation and P-glycoprotein (P-gp) inhibition. The bioavailability of lopinavir in Soluplus matrixed extrudate was 3.70-fold that of lopinavir crystal. Soluplus (polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol grafted copolymer) is a new amphiphilic nonionic medicinal polymer material that not only changes the interface state of the solution system but also increases the solubility of poorly soluble drugs.28
Preparing Polymeric Micelles
The polymeric micelles enhanced the solubility and permeability of acyclovir.29 The apparent solubility value (1.39 mg/mL) of acyclovir polymeric micelles was 1.36-fold that of acyclovir. The amount of acyclovir in the polymeric micelles that passed through the cornea in 6 h was approximately 10 times greater and the lag time was apparently shorter than that of aqueous solution. Polymeric micelles are colloidal structures of block copolymers: hydrophobic fragments form the spherical inner core, encapsulating poorly water-soluble drugs, while hydrophilic fragments form the outer shell.30
Preparing Solid Dispersion
A lyophilized milk-based solid dispersion was developed to enhance the solubility and permeability of ritonavir.31 Ritonavir was dispersed in an amorphous polymer matrix and existed primarily in a molecularly dispersed state.32 This formulation (drug:carrier mass ratio of 1:4) exhibited higher dissolution efficiency (~55.26 ± 1.29%, representing a 10-fold increase compared to pure ritonavir). Ex vivo permeation research indicated that the permeation extent of ritonavir formulation (33~75% w/w) was ~1.5~3.7-fold greater than that of pure ritonavir (~20%).
Pharmaceutical Technology to Increase Solubility Alone
Preparing Cyclodextrin Inclusion
The use of γ-cyclodextrin (a cyclodextrin derivatization) resulted in an 87-fold increase in lopinavir solubilization.33 Cyclodextrin is a class of cyclic oligosaccharides of α (1→4) glucopyranosides, having a hydrophilic outer surface and a considerably less hydrophilic central cavity that enable it to form complexes with drug molecules, thereby enhancing the aqueous solubility and bioavailability of the drug.34
Preparing Nanosuspension
The use of ritonavir nanosuspension increased up the dissolution rate of ritonavir. The area under the plasma concentration-time curve (AUC) values of ritonavir nanosuspension were 1.40-, 6.16- and 12.40-fold higher, and the maximum plasma concentration (Cmax) values were 1.90-, 3.23- and 8.91-fold higher, than those of the commercial product, physical mixture and coarse powder of ritonavir, respectively.35 Nanosuspensions containing surfactants or polymers as stabilizers may be employed to address drug delivery issues.36
Preparing a self-microemulsifying drug delivery system (SMEDDS). The solid SMEDDS tablets (completely dissolved in 60 min) markedly enhanced the drug dissolution rate (30%), Cmax (160.63%) and oral bioavailability (196.46%) compared to free ritonavir.37 SMEDDS is a promising approach to deliver lipophilic drugs due to their self-dispersion characteristics. The small droplet sizes observed upon dispersion have shown that drug absorption benefited from the large interfacial area.38
Pharmaceutical Technology to Increase Permeability Alone
Adding Penetration Enhancer
Caco-2 cell permeation studies showed that the permeability of acyclovir increases by 30 to 40 times in the presence of chitosan.39 Chitosan is an unbranched binary heteropolysaccharide consisting of the two units N-acetyl-d-glucosamine and d-glucosamine, which can increase or accelerate drug penetration.17,40
Adding an Absorption Enhancer
Gelucire 44/14 (at a concentration of 0.05% or 0.1% w/v) is able to increase the apparent permeability coefficient (Papp) by 6.47-fold and promote ocular bioavailability by 5.40-fold compared to free ribavirin.41 Gelucires are a series of amphiphilic pharmaceutical excipients that are widely used as powerful solubilization agents and bioavailability enhancers via oral and topical routes.
Preparing Prodrugs
Oseltamivir carboxylate formed a valyl amino acid prodrug via an isopropyl-methylenedioxy linker. This oseltamivir prodrug had a 9-fold enhanced Papp value in Caco-2 cells compared to that of the parent drug.42 The ethanol and butanol prodrugs of lamivudine increased permeability 2- and 10-fold, respectively.43
Metabolic Enzyme, Reaction and Metabolite of AvDs
AvDs are converted into active ingredients or metabolites with high polarity and high-water solubility, mainly in the liver and intestine. Phase I biotransformation involves oxidation, reduction or hydrolysis reaction of the functional group of drug molecules. Phase II biotransformation (combination reaction) combines the polar groups produced by phase I with endogenous components in the body through covalent bonds.23,44 The produced combination, possessing high polarity, is easy to dissolve in water and discharge from the body. Renal clearance is mainly controlled by membrane transport proteins and is an important elimination pathway for antiviral agents.
Enzymatic System Related to Metabolism of AvDs
The main metabolic enzymes of AvDs are microsomal enzymes existing in the liver, lung, kidney, small intestine, placenta and skin, mainly in intestinal epithelial cells and hepatocytes. Cytochrome P450 enzyme system (CYP450) mainly exists on the smooth endoplasmic reticulum and mitochondria of hepatocytes and in the small intestinal epithelia or the proximal tubules of the kidneys to a lesser extent. CYP450 is mainly involved in phase I reactions of AvDs.45 Approximately 40–70% of all clinical drugs are subjected to glucuronidation reactions metabolized by uridine diphosphate-glucuronosyl transferases (UGTs) in humans.46 UGTs are a superfamily of membrane-bound enzymes that catalyze the formation of a chemical bond between a nucleophilic O-, N-, S-, or C atom and uridine-5’-diphosphate-α-D-glucuronic acid (UDPGA).47
Metabolic Reaction and Metabolite in the Liver and Intestine
Metabolic Reaction of NA-AvDs
NA-AvDs enter cells through specific plasma membrane nucleoside transporters, such as the SLC22, SLC15, SLC28 and SLC29 gene family.48 After entering the cells, NA-AvDs are phosphorylated by cellular nucleoside kinase to form nucleoside monophosphate, diphosphate and triphosphate. Nucleoside triphosphates are the active form of NA-AvDs, which work by inhibiting cellular or viral enzymes (such as DNA/RNA polymerase).49 Most NA-AvDs are activated in vivo through 5’-phosphorylation. NA-AvDs are subjected to phosphorylation (7/8, ie, 7 of a total of 8), glucuronidation (1/8), oxidation (1/8), and reduction (1/8) (Table 2).
 |
Table 2 Main Metabolic Pathway of AvDs in Liver and Intestine |
Purine NA-AvDs: (1) Phosphorylation. Adefovir with hydroxyl groups was phosphorylated to the diphosphate compound, which competes with deoxyadenosine triphosphate kinase for incorporation by HBV reverse transcriptase to exert its antiviral effect.55 Entecavir undergoes intracellular phosphorylation to form di- and triphosphate metabolites by the natural substrate deoxyguanosine triphosphate and is then incorporated into HBV DNA polymerase to inhibit replication.56 (2) Oxidation. Acyclovir with the primary hydroxyl group in the side chain underwent oxidation, resulting in the formation of active carboxy-acyclovir.62
Pyrimidine NA-AvDs: (1) Phosphorylation. Zidovudine undergoes intracellular phosphorylation to form monophosphate, diphosphate and active triphosphate compounds by thymidine, thymidylate kinase and nucleoside diphosphate kinase, respectively.53 Lamivudine formed monophosphate, diphosphate and active 5’-triphosphate by deoxycytidine kinase, deoxycytidylate kinase and nucleoside diphosphate kinase, in turn.54 (2) Glucuronidation. Zidovudine underwent glucuronidation by UGT2B7 to form inactive metabolites excreted in urine.53 (3) Reduction. CYP450s and CYP450 reductase have been applied in the reduction of the azido moiety of zidovudine. The metabolite 30-amino-3’-deoxythymidine of zidovudine has been shown to be approximately 5- to 7-fold more toxic to human hematopoietic progenitor cells.53
Other NA-AvDs: Phosphorylation. Favipiravir first forms ribonucleoside 5’-monophosphate and then forms ribonucleosides 5’-diphosphate and 5’-triphosphate under the action of guanine phosphoribosyltransferase. In addition, another active metabolite, nicotinamide adenosine favipiravir, was formed by nicotinamide mononucleotide adenylyltransferase.52 Remdesivir is metabolized through the following 3 pathways: forming nucleoside triphosphate by phosphorylation, forming alanine metabolite by dephenylization, and forming nucleoside monophosphate by deamination.51 Ribavirin forms 5’-phosphorylation compounds by adenosine kinase catalysis.63 Phosphorylated ribavirin metabolites have exhibited broad antiviral activity.50
Metabolic Reaction of NN-AvDs
The whole 8 NN-AvDs investigated in this article are subjected to oxidation (3/8), reduction (2/8), hydrolysis (1/8) and conjugation (1/8).
Oxidation: Lopinavir is mainly catalyzed by CYP3A to undergo oxidative metabolism. The predominant metabolic site was carbon-4 of the cyclic urea moiety, with subsequent secondary metabolism occurring on the diphenyl core moiety.58 Arbidol contains sulfide, which forms sulfinylarbidol by sulfoxidation. The oxidation was catalyzed by flavin-containing monooxygenases.60 Ritonavir undergoes several oxidations, including hydroxylation at the isopropyl group by CYP2D6 to form the major metabolite and the oxidation of the thiazole rings on the eastern and western side of the molecule catalyzed by CYP2J2.61 CYP3A4 was the major isoform involved in arbidol metabolism in the liver and intestines.60
Reduction: Chloroquine was rapidly dealkylated into the pharmacologically active desethylchloroquine, bisdesethylchloroquine and 7-chloro-4-aminoquinoline. The main metabolic enzymes were two major isoforms, CYP3As and CYP2D6.57 Ritonavir underwent dealkylation by CYP3A4 to form demethylation metabolites.61
Hydrolysis: Oseltamivir was metabolized to GS4071, (3R,4R,5S)-3-(1-ethylpropyloxy)-4-acetamido-5-amino-cyclohexene-1-carboxylate (an active neuraminidase inhibitor) through ester hydrolysis.59
Conjugation: Arbidol was conjugated with glucuronide and sulfate via free hydroxyl groups on the indole ring by UGT1A9.64 The obtained conjugates were major metabolites in human urine.
Pharmacological Activity of AvDs and Their Delivery Systems
Pharmacological Activity of AvDs
AvDs usually reduce viral synthesis by interfering with the synthesis cycle of viral RNA (Figure 4). Among all 14 AvDs, 5 AvDs are broad-spectrum AvDs, 3 AvDs are mainly for HBV, 2 are mainly for HIV, 2 are mainly for CoV, 2 are mainly for influenza and 1 is for HSV (Table 3). In particular, half of the abovementioned AvDs, including ribavirin,65 remdesivir,66 favipiravir,67 chloroquine,68 lopinavir, ritonavir69 and arbidol,12 exhibited potential efficacy for COVID-19 treatment. Ribavirin interfered with the replication of DNA and RNA viruses and boosted the antiviral Th1 arm of the immune system to regulate T cells. In addition, ribavirin was proven to be effective for COVID-19 in several clinical trials.70 Recently, remdesivir in the triphosphate form was found to compete with the natural counterpart ATP and cause SARS-CoV RNA synthesis arrest at a specific position; thus, remdesivir might resist COVID-19.71 Favipiravir was effective in reducing the SARS-CoV-2 infection in vitro.12 Favipiravir stopped the disease progression of COVID-19 by inhibiting and clearing SARS-CoV-2 virus to achieve good treatment outcomes in COVID-19 patients.72 Chloroquine was a potential drug for COVID-19, since it altered the crosstalk of SARS-CoV-2 molecules with target cells because the inhibition capability of p38 mitogen-activated protein kinase (MAPK) interfered with proteolytic processing of the M protein and altered virion assembly and budding.73 Both lopinavir and ritonavir bound well to the SARS-CoV 3C-like protease.74 The combination of lopinavir and ritonavir represent a potential treatment for COVID-19. Arbidol effectively acted against SARS-CoV-2 in vivo and in vitro study.75
 |
 |
 |
 |
Table 3 Pharmacological Activities of AvDs |



(1) Five AvDs exhibited broad-spectrum antiviral effects. Three NA-AvDs (ribavirin,99 remdesivir100 and favipiravir101) are inhibitors of RNA-dependent RNA polymerase. NN-AvD chloroquine69 inhibited phosphorylation of MAPK in THP-1 (a human monocytic leukemia) cells, as well as caspase-1, and then blocked the virus replication cycle.102 The NN-AvD arbidol103 had broad-spectrum activity, since it interacted with both membranes and with viral/cellular proteins.104
(2) Three AvDs exhibited anti-HBV effects. Three NA-AvDs (lamivudine,85 adefovir,86 entecavir87) effectively inhibited the replication of HBV virus. Lamivudine also reduced viral load and reversed fibrosis, and entecavir was also a highly selective inhibitor of HBV DNA polymerase.105
(3) Two AvDs exhibited anti-HIV effects. Two NA-AvDs (zidovudine106 and lamivudine107 are inhibitors of reverse transcriptase enzymes. Zidovudine also inhibited viral replication by interfering with chain elongation of the viral DNA.108 Lamivudine also exerted antiviral effects by acting as a DNA chain terminator.107
(4) Two AvDs exhibited anti-CoV effects. Two NN-AvDs (lopinavir109 and ritonavir82) were combined to treat coronavirus infections. Lopinavir was a protease inhibitor which mainly inhibited the 3C-like protease of CoV-virus and modulated apoptosis in human cells.110 While ritonavir inhibited the CYP3A mediated metabolism of lopinavir and thereby potentiated the serum level of lopinavir.111
(5) Two AvDs exhibited anti-influenza effects. Two NN-AvDs contained oseltamivir112 and peramivir96). The former mainly targeted a glycoprotein neuraminidase on the surface of influenza virus.113 Peramivir is an efficacious nucleoside analog inhibitor to treat influenza.96
(6) 1 AvDs exhibited anti-HSV effects. NA-AvD acyclovir88 was activated by viral thymidine kinase and then di- and tri-phosphorylated by cellular kinases. The active tri-phosphorylated forms of acyclovir specifically interfered with viral DNA polymerase and caused chain termination.88
Effect of Metabolism on Pharmacological Activity
The in vivo metabolization of AvDs had a large influence on viral replication processes and pharmacological function.
(1) Generation of active metabolites. Eight inactive or low-activity AvDs were transformed into active or high-activity forms through metabolism in the body.
Typically, NA-AvDs are prodrugs that must be converted to nucleotide triphosphate metabolites to exert antiviral activity. Ribavirin was phosphorylated to form monophosphate by adenosine kinase and triphosphate by nucleoside mono- and diphosphate kinases.114 The triphosphate metabolite of remdesivir markedly increased the plasma half-life time (t1/2) value (14 h versus 0.39 h for remdesivir) and antiviral activity (> 40 times) compared to the free drug.115 Favipiravir showed no inhibitory effect on influenza, whereas its active metabolite (favipiravir triphosphate) strongly inhibited influenza virus RNA polymerase.116 Zidovudine was phosphorylated at the intracellular site, and zidovudine triphosphate was the inhibitor of reverse transcriptase activity required for viral replication.117 Lamivudine was phosphorylated to lamivudine triphosphate, which was able to inhibit viral reverse transcriptase and terminate proviral DNA chain extension due to the lack of the 3’-hydroxyl group required for nucleic acid replication.118 Entecavir-formed triphosphate inside cells inhibited replication of the hepatitis B virus.119
In the case of NN-AvDs, desethylchloroquine was an active metabolite of chloroquine. Both chloroquine and its metabolite acted against Zika virus at low concentrations (ie, at the micromolar level).120 Oseltamivir was converted into oseltamivir carboxylate (a potent neuraminidase inhibitor).121 The active metabolites of arbidol, sulfinylarbidol and sulfonylarbidol, increased the t1/2 values (25 h, 26 h versus 15.7 h for arbidol) and peak time (Tmax) values (13 h, 19 h versus 1.38 h), respectively.122
(2) Generation of inactive/low-active metabolites. Zidovudine was metabolized quickly to the inactive form glucuronide with a t1/2 value of 1 h.123 Lopinavir was primarily mediated by CYP3A enzymes and yielded metabolites that were less potent as protease inhibitors.124
(3) Generation of toxic metabolites. Zidovudine’s metabolite (30-amino-3’-deoxythymidine) was approximately 5- to 7-fold more toxic to human hematopoietic progenitor cells.53
Delivery System to Improve Pharmacological Activity of AvDs
Most AvDs had poor solubility/permeability and low oral bioavailability. Appropriate drug delivery systems might be able to overcome the shortcomings of AvDs and improve pharmacological activity.
(1) Preparing nanocarriers to lessen the side effects of AvDs. Ribavirin caused hemolytic anemia due to the accumulation inside red blood cells. Poly(glycerol-adipate) nanoparticles (NPs) delivered ribavirin to the targeted liver and subsequently decreased the red blood cell uptake rate, which overcame the side effects of ribavirin (hemolytic anemia caused by ribavirin due to its accumulation inside red blood cells).125 Lamivudine has low bioavailability in the brain (0.05–1.14%) and cannot kill viruses completely. Mannosylated polymeric NPs improved brain bioavailability and targeted mannose receptors on the macrophage surface to improve the therapeutic outcome and reduce toxicity.126 Oseltamivir loaded into the surface of selenium NPs improved antiviral activity and increased the viability of cells infected with a virus to 83.2%.127
(2) Preparing nanocarriers to extend the dose interval of AvDs. Zidovudine had low bioavailability and biological t1/2 value that led to dose-dependent anemia and first-pass metabolism. Amide-functionalized alginate NPs were used to encapsulate zidovudine to realize slow and sustained drug release.123
(3) Other delivery systems to improve pharmacological activity of AvDs. Acyclovir was loaded onto activated carbon particles. The highly porous carbon structures trapped virions, blocked infection and thus improved efficacy with acyclovir.128 Ribavirin was coupled to macromolecular carriers and delivered the drug to the targeted site liver, which may reduce systemic complications. Hemoglobin-ribavirin conjugates had greater antiviral activity on both isolated hepatocytes and macrophages, and they significantly reduced viral replication at 1 μM, while free ribavirin was ineffective at the same concentration.129
Pharmacokinetic Characteristics of AvDs and Their Delivery System
Pharmacokinetic Behavior of AvDs
Most AvDs belong to BCS II, III or IV types with low solubility/permeability. Therefore, their AUC, Cmax, tmax, t1/2 or mean retention time (MRT) values are low (Table 4). These adverse pharmacokinetic behaviors are often unfavorable to the pharmacological action of AvDs. The bioavailabilities of orally administered adefovir were 1% in monkeys and 8–11% in rats. The low bioavailability was mainly attributed to the low passive permeability across the intestinal membrane.130 It is crucial to apply appropriate drug delivery systems to improve the bioavailability and enhance the pharmacological effects of AvDs.
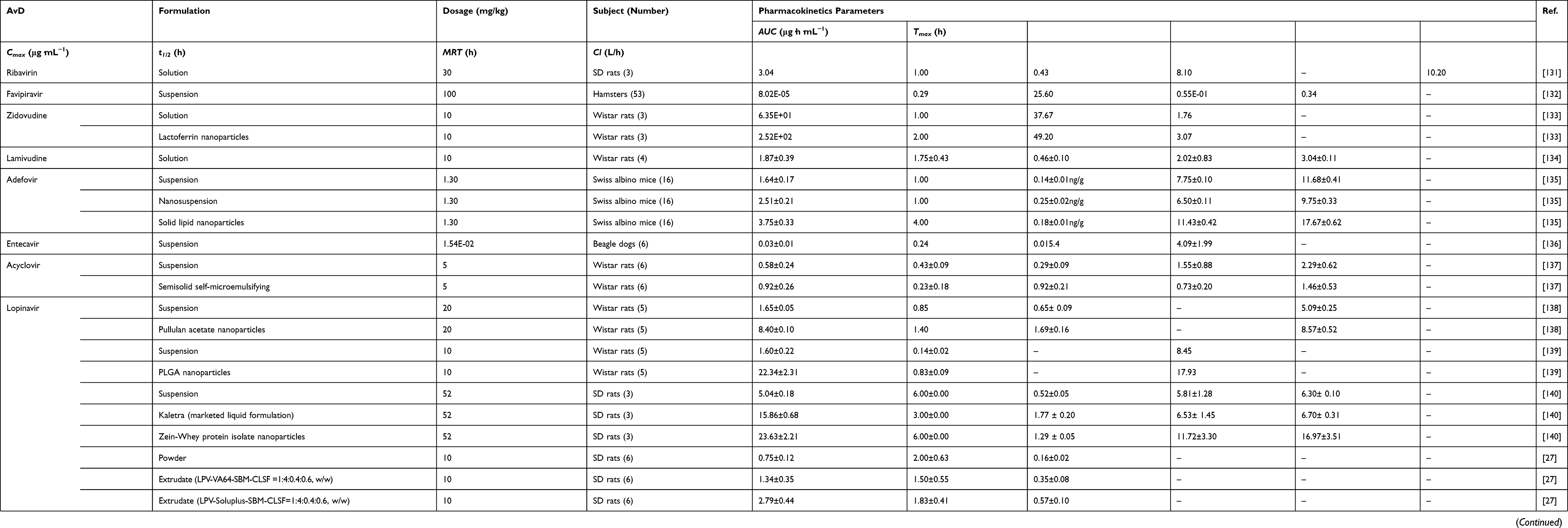 |
Table 4 Main Pharmacokinetic Properties of AvDs via Oral Administration |
Nanotechnology to Improve Pharmacokinetics of AvDs
Delivery System to Improve Pharmacokinetics
Iron-catechol-based nanoscale coordination polymers with antiretroviral ligand functionalization served as a promising strategy for the bioavailability enhancement of zidovudine. These polymers offered long-lasting drug release and improved colloidal stabilities, as well as enhanced cellular uptake (ie, an increase of up to 50-fold).144
The proliposomes increased the MRT of adefovir dipivoxil in the liver by approximately 3-fold compared with the adefovir suspension.145
The poly (lactic acid)-poly (ethylene glycol)-ligand NPs targeted the intestinal transporter PepT1 and increased permeability by 2.7-fold compared with free acyclovir.146
The triglyceride-mimetic superficially modified mesoporous silica NPs increased the AUC, Cmax and MRT values of lopinavir by 9.65-, 3.87- and 2.70-fold, respectively. The NPs improved poor solubility and avoided the first-pass metabolism of oral lopinavir to achieve high oral bioavailability with no side effects.147 The lopinavir-loaded bioadhesive protein NPs increased the oral bioavailability by 4- and 1.5-fold compared to the free lopinavir suspension and lopinavir/ritonavir formulations, respectively.140 The poly (lactic-co-glycolic acid) NPs increased the permeability and oral bioavailability of lopinavir by 3.04- and 13.9-fold in rats.139 The lopinavir in hydrophobically modified pullulan NPs was metabolized to a lesser extent in the gut. The NPs increased the bioavailability twofold.148
Effect of Prodrugs on Pharmacokinetics of AvDs
Prodrugs usually offer a versatile strategy to overcome the flaws of antiviral drugs. The prodrugs strategy improved the pharmacokinetic properties, efficacy and safety profile of many viable drugs.149 Effective prodrug strategies include ester prodrugs, targeted delivery prodrugs, macromolecular prodrugs and nucleoside conjugates.16 Zidovudine prodrug (the ester conjugation of zidovudine with ursodeoxycholic acid) permeated considerably more efficiently, and remained in murine macrophages longer, than the parent drug. The MRTs for zidovudine and its prodrug were 6.5 min and 19.6 min, respectively.150 Entecavir ester prodrugs prolonged the therapeutic period. After subcutaneous injection of the entecavir prodrug in beagle dogs, the plasma drug concentration was markedly protracted (T1/2 is 129.3 h) with a lower maximum plasma concentration (Cmax is 4.7 ng/mL) compared to entecavir (oral administration, T1/2 is 4.09 h and Cmax is 15.4 ng/mL).136 The chloroquine prodrug hydroxychloroquine had higher accumulation in cells and a longer elimination half-life, resulting in a more effect against SARS-CoV-2 infection.151
Effect of Administration Route on Pharmacokinetics of AvDs
The current primary administration route for AvDs is oral administration, which has the advantages of convenience, safety and cost-effectiveness. However, the oral bioavailability of most AvDs was not satisfactory due to poor solubility and permeability. Therefore, it was very important to choose the appropriate administration route152 according to the treatment needs and safety assessment to maximize the efficacy of AvDs (Table 5).
 |
Table 5 Main Pharmacokinetic Properties of AvDs via Intravenous Administration |
Ribavirin nasal spray consisting of spray-dried excipient particles was suitable for nasal deposition. This method provided effective mucosal adhesion and penetration enhancement. In vivo results confirmed that the agglutination rate was nearly 6 times higher than conventional intravenous administration, suggesting that the preparation has the potential to deliver a highly brain-targeted antiviral from the nose to the brain.160 Adefovir suspension after intravenous administration increased the AUC 5.45-fold compared with oral administration.135 After subcutaneous injection of entecavir in beagle dogs, the Cmax (4.70 ng/mL) was protracted, and the t1/2 value was lengthened (129.30 h) compared to oral administration (15.40ng/mL, 4.1 h).136
After intravenous administration of acyclovir solution, there was 90-fold higher Cmax (~26.23 μg/mL) and considerably shorter Tmax (~8.00 min) compared to that of acyclovir suspension by oral administration (Cmax was ~0.29 μg/mL and Tmax was ~26.00 min).137 After intravenous administration of oseltamivir solution92 or peramivir solution,141 there were 1.50- and 2.26-fold AUC increases compared to oral administration.
Conclusion and Prospects
As the main resource in the effort to treat viruses, AvDs have advantages such as convenient use, accurate dosage, clear targets and strong antiviral efficacy. However, insufficient solubility/permeability of AvDs (most belong to BCS II and III), has a great impact on their oral absorption and results in lower oral bioavailability and requires multiple medications. A better understanding of the in vivo metabolism and pharmacokinetic process of AvDs may help researchers to develop new formulations to overcome these problems. In this review, we investigated the structure-based metabolic reactions of AvDs mainly in the liver and intestine. Phosphorylation (one conjugation reaction) is the most common reaction of NA-AvDs; other reactions, such as oxidation and reduction, also occur, but no hydrolysis occurs. For NN-AvDs, in addition to oxidation, reduction and conjugation reactions, hydrolysis also occurs. Enzymatic systems (CYPs and phosphokinase) are mainly produced by epithelial cells. Most AvDs are activated, and a small number are inactivated or maintain activity through metabolic reactions. The pharmacological activity and pharmacokinetics of AvDs are improved by loading into suitable delivery systems with nanotechnology, prodrug strategy and administration route consideration. The mechanisms of action of AvDs are generally to inhibit the key enzymes (such as RNA-dependent RNA polymerase, reverse transcriptase and nucleoside reverse transcriptase) of virus synthesis and consequently block viral synthesis.
Notably, some AvDs (such as ribavirin, remdesivir, favipiravir, chloroquine, lopinavir and ritonavir) exhibit potential as treatments for COVID-19. Several new antiviral drugs have been approved for marketing, such as bictegravir sodium, emtricitabine and tenofovir alafenamide fumarate tablets (Biktarvy®, a nucleotide reverse transcriptase inhibitor); baloxavir marboxil tablets (Xofluza®, a polymerase acidic endonuclease inhibitor) and letermovir injections (Prevymis®, viral terminase inhibitor) have been developed rapidly, and several antiviral drugs that are undergoing clinical trials may also contribute to the global management of antiviral infections (such as albuvirtide and elsulfavirine). In addition, Regeneron’s REGN-COV2 antibody cocktail and Eli Lilly’s LY-CoV555 are being used in the treatment of the president of the US, and their clinical trials have proven that the drugs have anti-COVID-19 efficacy. In the future, new antiviral drugs in suitable delivery systems are expected to perform better than old ones.
Abbreviations
AvD, antiviral drug; BCS, biopharmaceutical classification system; COVID-19, coronavirus disease 2019; CYP450, cytochrome P450 enzyme system; dATP, deoxyadenosine triphosphate; D0, dose number; log P, oil-water partition coefficient; MAPK, p38 mitogen-activated protein kinase; MERS, Middle East respiratory syndrome; NA-AvD, nucleoside analog of antiviral drug; NN-AvD, non-nucleoside analog of antiviral drug; NP, nanoparticle; SARS, severe acute respiratory syndrome; SMEDDS, semisolid self-microemulsifying drug delivery system; Papp, apparent permeability coefficient; P-gp, P-glycoprotein; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2; Tmax, peak time; t1/2, plasma half-life time; UDPGA, uridine-5’-diphosphate-α-D-glucuronic acid; UGT, uridine diphosphate-glucuronosyl transferase; WHO, world health organization.
Acknowledgments
This research is partially supported by grants from Chongqing Science and Technology Association (2020-11), Chongqing Science and Technology Committee (cstc2015jcyjBX0027, cstc2017shmsA130028) and Chongqing Education Committee (CYS20212, CY160406).
Disclosure
Daojun Pu is an employee of Southwest Pharmaceutical Limited Company. The authors declare no other potential conflicts of interest for this work and no financial support.
References
1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in china. Nature. 2020;579:265–269. doi:10.1021/acs.biochem.0c00160
2. The Lancet Infectious Diseases. Covid-19, a pandemic or not? Lancet Infect Dis. 2020;20(4):383.
3. Conti P, Ronconi G, Caraffa A, et al. Induction of pro-inflammatory cytokines (il-1 and il-6) and lung inflammation by coronavirus-19 (covi-19 or sars-cov-2): anti-inflammatory strategies. J Biol Regul Homeost Agents. 2020;34:327–331.
4. Conti P, Caraffa A, Gallenga CE, et al. Il-1 induces throboxane-a2 (txa2) in covid-19 causing inflammation and micro-thrombi: inhibitory effect of the il-1 receptor antagonist (il-1ra). J Biol Regul Homeost Agents. 2020;34:1623–1627.
5. Kritas SK, Ronconi G, Caraffa A, Gallenga CE, Ross R, Conti P. Mast cells contribute to coronavirus-induced inflammation: new anti-inflammatory strategy. J Biol Regul Homeost Agents. 2020;34:9–14.
6. Sun J, He WT, Wang L, et al. Covid-19: epidemiology, evolution, and cross-disciplinary perspectives. Trends Mol Med. 2020;26:483–495. doi:10.1016/j.molmed.2020.02.008
7. Vellingiri B, Jayaramayya K, Iyer M, et al. Covid-19: a promising cure for the global panic. Sci Total Environ. 2020;725:138277. doi:10.1016/j.scitotenv.2020.138277
8. Pastorino G, Cornara L, Soares S, Rodrigues F, Oliveira M. Liquorice (glycyrrhiza glabra): a phytochemical and pharmacological review. Phytother Res. 2018;32:2323–2339. doi:10.1002/ptr.6178
9. Oo A, Teoh BT, Sam SS, Bakar SA, Zandi K. Baicalein and baicalin as zika virus inhibitors. Arch Virol. 2019;164:585–593. doi:10.1007/s00705-018-4083-4
10. Huh J, Song JH, Kim SR, et al. Lignan dimers from forsythia viridissima roots and their antiviral effects. J Nat Prod. 2019;82:232–238. doi:10.1021/acs.jnatprod.8b00590
11. Zhao J, Li Y, He D, et al. Natural oral anticancer medication in small ethanol nanosomes coated with a natural alkaline polysaccharide. ACS Appl Mater Interfaces. 2020;12:16159–16167. doi:10.1021/acsami.0c02788
12. Wang M, Cao R, Zhang L, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-ncov) in vitro. Cell Res. 2020;30:269–271. doi:10.1038/s41422-020-0282-0
13. Zhang ZJ, Morris-Natschke SL, Cheng YY, Lee KH, Li RT. Development of anti-influenza agents from natural products. Med Res Rev. 2020;40:2290–2338. doi:10.1002/med.21707
14. Visser LJ, Aloise C, Swatek KN, et al. Dissecting distinct proteolytic activities of fmdv lpro implicates cleavage and degradation of rlr signaling proteins, not its deisgylase/dub activity, in type i interferon suppression. PLoS Pathog. 2020;16:e1008702. doi:10.1371/journal.ppat.1008702
15. Tiwari V, Beer JC, Sankaranarayanan NV, Swanson-Mungerson M, Desai UR. Discovering small-molecule therapeutics against sars-cov-2. Drug Discov Today. 2020;25:1535–1544. doi:10.1016/j.drudis.2020.06.017
16. Sinokrot H, Smerat T, Najjar A, Karaman R. Advanced prodrug strategies in nucleoside and non-nucleoside antiviral agents: a review of the recent five years. Molecules. 2017;22:1736.
17. Gu J, Huang Y, Yan Z, et al. Biomimetic membrane-structured nanovesicles carrying a supramolecular enzyme to cure lung cancer. ACS Appl Mater Interfaces. 2020;12:31112–31123. doi:10.1021/acsami.0c06207
18. Yang L, Zhang Y, Xie J, et al. Biomimetic polysaccharide-cloaked lipidic nanovesicles/microassemblies for improving the enzymatic activity and prolonging the action time for hyperuricemia treatment. Nanoscale. 2020;12:15222–15235. doi:10.1039/D0NR02651D
19. Goodarzi N, Barazesh Morgani A, Abrahamsson B, et al. Biowaiver monographs for immediate release solid oral dosage forms: ribavirin. J Pharm Sci. 2016;105:1362–1369. doi:10.1016/j.xphs.2016.01.017
20. Widmer N, Meylan P, Ivanyuk A, Aouri M, Decosterd LA, Buclin T. Oseltamivir in seasonal, avian h5n1 and pandemic 2009 a/h1n1 influenza: pharmacokinetic and pharmacodynamic characteristics. Clin Pharmacokinet. 2010;49:741–765. doi:10.2165/11534730-000000000-00000
21. Aungst BJ, Nguyen NH, Bulgarelli JP, Oates-Lenz K. The influence of donor and reservoir additives on caco-2 permeability and secretory transport of hiv protease inhibitors and other lipophilic compounds. Pharm Res. 2000;17:1175–1180. doi:10.1023/A:1026402410783
22. Li X, Wang X, Jiang Q, Chi F, Liu Q, Zhang T. The delivery of arbidol by salt engineering: synthesis, physicochemical properties and pharmacokinetics. Drug Dev Ind Pharm. 2016;43:151–159. doi:10.1080/03639045.2016.1225755
23. Yang J, Li K, He D, et al. Toward a better understanding of metabolic and pharmacokinetic characteristics of low-solubility, low-permeability natural medicines. Drug Metab Rev. 2020;52:19–43. doi:10.1080/03602532.2020.1714646
24. Seley-Radtke KL, Yates MK. The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antiviral Res. 2018;154:66–86.
25. Volpe DA. Advances in cell-based permeability assays to screen drugs for intestinal absorption. Expert Opin Drug Discov. 2020;15:539–549. doi:10.1080/17460441.2020.1735347
26. Babadi D, Dadashzadeh S, Osouli M, Daryabari MS, Haeri A. Nanoformulation strategies for improving intestinal permeability of drugs: a more precise look at permeability assessment methods and pharmacokinetic properties changes. J Control Release. 2020;321:669–709. doi:10.1016/j.jconrel.2020.02.041
27. Zi P, Zhang C, Ju C, et al. Solubility and bioavailability enhancement study of lopinavir solid dispersion matrixed with a polymeric surfactant - soluplus. Eur J Pharm Sci. 2019;134:233–245. doi:10.1016/j.ejps.2019.04.022
28. Ahire E, Thakkar S, Darshanwad M, Misra M. Parenteral nanosuspensions: a brief review from solubility enhancement to more novel and specific applications. Acta Pharm Sin B. 2018;8:733–755. doi:10.1016/j.apsb.2018.07.011
29. Varela-Garcia A, Concheiro A, Alvarez-Lorenzo C. Soluplus micelles for acyclovir ocular delivery: formulation and cornea and sclera permeability. Int J Pharm. 2018;552:39–47. doi:10.1016/j.ijpharm.2018.09.053
30. Lembo D, Donalisio M, Civra A, Argenziano M, Cavalli R. Nanomedicine formulations for the delivery of antiviral drugs: a promising solution for the treatment of viral infections. Expert Opin Drug Deliv. 2018;15:93–114. doi:10.1080/17425247.2017.1360863
31. Dhore PW, Dave VS, Saoji SD, Bobde YS, Mack C, Raut NA. Enhancement of the aqueous solubility and permeability of a poorly water soluble drug ritonavir via lyophilized milk-based solid dispersions. Pharm Dev Technol. 2016;22:90–102. doi:10.1080/10837450.2016.1193193
32. Huang Y, Dai WG. Fundamental aspects of solid dispersion technology for poorly soluble drugs. Acta Pharm Sin B. 2014;4:18–25. doi:10.1016/j.apsb.2013.11.001
33. Adeoye O, Conceicao J, Serra PA, et al. Cyclodextrin solubilization and complexation of antiretroviral drug lopinavir: in silico prediction; effects of derivatization, molar ratio and preparation method. Carbohydr Polym. 2020;227:115287. doi:10.1016/j.carbpol.2019.115287
34. Conceicao J, Adeoye O, Cabral-Marques HM, Lobo JMS. Cyclodextrins as excipients in tablet formulations. Drug Discov Today. 2018;23:1274–1284. doi:10.1016/j.drudis.2018.04.009
35. Karakucuk A, Teksin ZS, Eroglu H, Celebi N. Evaluation of improved oral bioavailability of ritonavir nanosuspension. Eur J Pharm Sci. 2019;131:153–158. doi:10.1016/j.ejps.2019.02.028
36. Jacob S, Nair AB, Shah J. Emerging role of nanosuspensions in drug delivery systems. Biomater Res. 2020;24:3. doi:10.1186/s40824-020-0184-8
37. Deshmukh A, Kulkarni S. Solid self-microemulsifying drug delivery system of ritonavir. Drug Dev Ind Pharm. 2014;40:477–487. doi:10.3109/03639045.2013.768632
38. Vithani K, Jannin V, Pouton CW, Boyd BJ. Colloidal aspects of dispersion and digestion of self-dispersing lipid-based formulations for poorly water-soluble drugs. Adv Drug Deliv Rev. 2019;142:16–34. doi:10.1016/j.addr.2019.01.008
39. Kubbinga M, Augustijns P, García MA, et al. The effect of chitosan on the bioaccessibility and intestinal permeability of acyclovir. Eur J Pharm Biopharm. 2019;136:147–155. doi:10.1016/j.ejpb.2019.01.021
40. Wan S, He D, Yuan Y, Yan Z, Zhang X, Zhang J. Chitosan-modified lipid nanovesicles for efficient systemic delivery of l-asparaginase. Colloids Surf B Biointerfaces. 2016;143:278–284. doi:10.1016/j.colsurfb.2016.03.046
41. Liu R, Liu Z, Zhang C, Zhang B. Gelucire44/14 as a novel absorption enhancer for drugs with different hydrophilicities: in vitro and in vivo improvement on transcorneal permeation. J Pharm Sci. 2011;100:3186–3195. doi:10.1002/jps.22540
42. Incecayir T, Sun J, Tsume Y, et al. Carrier-mediated prodrug uptake to improve the oral bioavailability of polar drugs: an application to an oseltamivir analogue. J Pharm Sci. 2016;105:925–934. doi:10.1016/j.xphs.2015.11.036
43. Gualdesi MS, Brinon MC, Quevedo MA. Intestinal permeability of lamivudine (3tc) and two novel 3tc prodrugs. Experimental and theoretical analyses. Eur J Pharm Sci. 2012;47:965–978. doi:10.1016/j.ejps.2012.10.002
44. Zhou Y, Zhang M, He D, et al. Uricase alkaline enzymosomes with enhanced stabilities and anti-hyperuricemia effects induced by favorable microenvironmental changes. Sci Rep. 2016;6:20136. doi:10.1038/srep20136
45. Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis. 2017;21:1–20. doi:10.1016/j.cld.2016.08.001
46. Zhao J, Liu S, Hu X, et al. Improved delivery of natural alkaloids into lung cancer through woody oil-based emulsive nanosystems. Drug Deliv. 2018;25:1426–1437. doi:10.1080/10717544.2018.1474970
47. Afsar NA, Bruckmueller H, Werk AN, Nisar MK, Ahmad HR, Cascorbi I. Implications of genetic variation of common drug metabolizing enzymes and abc transporters among the pakistani population. Sci Rep. 2019;9:7323. doi:10.1038/s41598-019-43736-z
48. van Nuland M, Rosing H, Huitema ADR, Beijnen JH. Predictive value of microdose pharmacokinetics. Clin Pharmacokinet. 2019;58:1221–1236. doi:10.1007/s40262-019-00769-x
49. Eyer L, Nencka R, de Clercq E, Seley-Radtke K, Ruzek D. Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antivir Chem Chemother. 2018;26:2040206618761299. doi:10.1177/2040206618761299
50. Urtishak KA, Wang LS, Culjkovic-Kraljacic B, et al. Targeting eif4e signaling with ribavirin in infant acute lymphoblastic leukemia. Oncogene. 2019;38:2241–2262. doi:10.1038/s41388-018-0567-7
51. Warren TK, Jordan R, Lo MK, et al. Therapeutic efficacy of the small molecule gs-5734 against ebola virus in rhesus monkeys. Nature. 2016;531:381–385. doi:10.1038/nature17180
52. Huchting J, Vanderlinden E, Van Berwaer R, Meier C, Naesens L. Cell line-dependent activation and antiviral activity of t-1105, the non-fluorinated analogue of t-705 (favipiravir). Antiviral Res. 2019;167:1–5. doi:10.1016/j.antiviral.2019.04.002
53. Ghodke Y, Anderson PL, Sangkuhl K, Lamba J, Altman RB, Klein TE. Pharmgkb summary. Pharmacogenet Genomics. 2012;22:891–894. doi:10.1097/FPC.0b013e32835879a8
54. Halling Folkmar Andersen A, Tolstrup M. The potential of long-acting, tissue-targeted synthetic nanotherapy for delivery of antiviral therapy against hiv infection. Viruses. 2020;12:412. doi:10.3390/v12040412
55. Ray AS, Vela JE, Olson L, Fridland A. Effective metabolism and long intracellular half life of the anti-hepatitis b agent adefovir in hepatic cells. Biochem Pharmacol. 2004;68:1825–1831. doi:10.1016/j.bcp.2004.07.010
56. Murata K, Tsukuda S, Suizu F, et al. Immunomodulatory mechanism of acyclic nucleoside phosphates in treatment of hepatitis b virus infection. Hepatology. 2020;71:1533–1545. doi:10.1002/hep.30956
57. Smit C, Peeters MYM, van den Anker JN, Knibbe CAJ. Chloroquine for sars-cov-2: implications of its unique pharmacokinetic and safety properties. Clin Pharmacokinet. 2020;59:659–669. doi:10.1007/s40262-020-00891-1
58. Kraft JC, McConnachie LA, Koehn J, et al. Mechanism-based pharmacokinetic (mbpk) models describe the complex plasma kinetics of three antiretrovirals delivered by a long-acting anti-hiv drug combination nanoparticle formulation. J Control Release. 2018;275:229–241. doi:10.1016/j.jconrel.2018.02.003
59. Hu Y, Chen B, Lei Z, et al. Synthesis and biological evaluation of nh2-sulfonyl oseltamivir analogues as influenza neuraminidase inhibitors. Molecules. 2019;24:2176. doi:10.3390/molecules24112176
60. Hulseberg CE, Fénéant L, Szymańska-de Wijs KM, et al. Arbidol and other low-molecular-weight drugs that inhibit lassa and ebola viruses. J Virol. 2019;93. doi:10.1128/JVI.02185-18
61. Kaspera R, Kirby BJ, Sahele T, et al. Investigating the contribution of cyp2j2 to ritonavir metabolism in vitro and in vivo. Biochem Pharmacol. 2014;91:109–118. doi:10.1016/j.bcp.2014.06.020
62. Prasse C, Wagner M, Schulz R, Ternes TA. Oxidation of the antiviral drug acyclovir and its biodegradation product carboxy-acyclovir with ozone: kinetics and identification of oxidation products. Environ Sci Technol. 2012;46:2169–2178. doi:10.1021/es203712z
63. Nayar U, Sadek J, Reichel J, et al. Identification of a nucleoside analog active against adenosine kinase–expressing plasma cell malignancies. J Clin Invest. 2017;127:2066–2080. doi:10.1172/JCI83936
64. Fang -Z-Z, Tosh DK, Tanaka N, et al. Metabolic mapping of a3 adenosine receptor agonist mrs5980. Biochem Pharmacol. 2015;97:215–223. doi:10.1016/j.bcp.2015.07.007
65. Hung IF-N, Lung K-C, Tso EY-K, et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with covid-19: an open-label, randomised, Phase 2 trial. Lancet. 2020;395:1695–1704. doi:10.1016/S0140-6736(20)31042-4
66. Grein J, Ohmagari N, Shin D, et al. Compassionate use of remdesivir for patients with severe covid-19. N Engl J Med. 2020;382:2327–2336. doi:10.1056/NEJMoa2007016
67. Yamamura H, Matsuura H, Nakagawa J, Fukuoka H, Domi H, Chujoh S. Effect of favipiravir and an anti-inflammatory strategy for covid-19. Crit Care. 2020;24:413. doi:10.1186/s13054-020-03137-5
68. Li X, Wang Y, Agostinis P, et al. Is hydroxychloroquine beneficial for covid-19 patients? Cell Death Dis. 2020;11:512. doi:10.1038/s41419-020-2721-8
69. Hazafa A, Ur-Rahman K, Haq IU, et al. The broad-spectrum antiviral recommendations for drug discovery against covid-19. Drug Metab Rev. 2020;52:408–424. doi:10.1080/03602532.2020.1770782
70. Khalili JS, Zhu H, Mak NSA, Yan Y, Zhu Y. Novel coronavirus treatment with ribavirin: groundwork for an evaluation concerning covid-19. J Med Virol. 2020;92:740–746. doi:10.1002/jmv.25798
71. Gordon CJ, Tchesnokov EP, Feng JY, Porter DP, Gotte M. The antiviral compound remdesivir potently inhibits rna-dependent rna polymerase from middle east respiratory syndrome coronavirus. J Biol Chem. 2020;295:4773–4779. doi:10.1074/jbc.AC120.013056
72. Cai Q, Yang M, Liu D, et al. Experimental treatment with favipiravir for covid-19: an open-label control study. Engineering (Beijing). 20206(10):1192–1198. doi:10.1016/j.eng.2020.03.007
73. Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for covid-19? Int J Antimicrob Agents. 2020;55:105938. doi:10.1016/j.ijantimicag.2020.105938
74. Nutho B, Mahalapbutr P, Hengphasatporn K, et al. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry. 2020;59:1769–1779.
75. Wang X, Cao R, Zhang H, et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of sars-cov-2 in vitro. Cell Discov. 2020;6:28. doi:10.1038/s41421-020-0169-8
76. Bi Y, Wong G, Liu Y, Liu L, Gao GF, Shi Y. Ribavirin is effective against drug-resistant h7n9 influenza virus infections. Protein Cell. 2016;7:611–614. doi:10.1007/s13238-016-0287-0
77. Coelmont L, Paeshuyse J, Windisch MP, De Clercq E, Bartenschlager R, Neyts J. Ribavirin antagonizes the in vitro anti-hepatitis c virus activity of 2’-c-methylcytidine, the active component of valopicitabine. Antimicrob Agents Chemother. 2006;50:3444–3446.
78. Mirabelli C, Jaspers M, Boon M, et al. Differential antiviral activities of respiratory syncytial virus (rsv) inhibitors in human airway epithelium. J Antimicrob Chemother. 2018;73:1823–1829. doi:10.1093/jac/dky089
79. Oestereich L, Rieger T, Ludtke A, et al. Efficacy of favipiravir alone and in combination with ribavirin in a lethal, immunocompetent mouse model of lassa fever. J Infect Dis. 2016;213:934–938.
80. Oestereich L, Rieger T, Neumann M, et al. Evaluation of antiviral efficacy of ribavirin, arbidol, and t-705 (favipiravir) in a mouse model for Crimean-Congo hemorrhagic fever. PLoS Negl Trop Dis. 2014;8:e2804. doi:10.1371/journal.pntd.0002804
81. Franco EJ, Rodriquez JL, Pomeroy JJ, Hanrahan KC, Brown AN. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir Chem Chemother. 2018;26:2040206618807580. doi:10.1177/2040206618807580
82. Sheahan TP, Sims AC, Leist SR, et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against mers-cov. Nat Commun. 2020;11:222. doi:10.1038/s41467-019-13940-6
83. Agostini ML, Andres EL, Sims AC, et al. Coronavirus susceptibility to the antiviral remdesivir (gs-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9:e00221–e00218. doi:10.1128/mBio.00221-18
84. Frezza C, Grelli S, Federico M, Marino-Merlo F, Mastino A, Macchi B. Testing anti-hiv activity of antiretroviral agents in vitro using flow cytometry analysis of cem-gfp cells infected with transfection-derived hiv-1 nl4-3. J Med Virol. 2016;88:979–986. doi:10.1002/jmv.24418
85. Liu X, Xu Z, Hou C, et al. Inhibition of hepatitis b virus replication by targeting ribonucleotide reductase m2 protein. Biochem Pharmacol. 2016;103:118–128. doi:10.1016/j.bcp.2016.01.003
86. Wiemer AJ, Wiemer DF. Prodrugs of phosphonates and phosphates: crossing the membrane barrier. Top Curr Chem. 2015;360:115–160.
87. Charlton MR, Alam A, Shukla A, et al. An expert review on the use of tenofovir alafenamide for the treatment of chronic hepatitis b virus infection in asia. J Gastroenterol. 2020;55:811–823. doi:10.1007/s00535-020-01698-4
88. Michaelis M, Kleinschmidt MC, Bojkova D, Rabenau HF, Wass MN, Cinatl J. Omeprazole increases the efficacy of acyclovir against herpes simplex virus type 1 and 2. Front Microbiol. 2019;10:2790. doi:10.3389/fmicb.2019.02790
89. Vincent MJ, Bergeron E, Benjannet S, et al. Chloroquine is a potent inhibitor of sars coronavirus infection and spread. Virol J. 2005;2:69. doi:10.1186/1743-422X-2-69
90. de Wilde AH, Jochmans D, Posthuma CC, et al. Screening of an fda-approved compound library identifies four small-molecule inhibitors of middle east respiratory syndrome coronavirus replication in cell culture. Antimicrob Agents Chemother. 2014;58:4875–4884. doi:10.1128/AAC.03011-14
91. Delvecchio R, Higa LM, Pezzuto P, et al. Chloroquine, an endocytosis blocking agent, inhibits zika virus infection in different cell models. Viruses. 2016;8:322.
92. Shin JS, Ku KB, Jang Y, et al. Comparison of anti-influenza virus activity and pharmacokinetics of oseltamivir free base and oseltamivir phosphate. J Microbiol. 2017;55:979–983.
93. Du Q, Gu Z, Leneva I, et al. The antiviral activity of arbidol hydrochloride against herpes simplex virus type ii (hsv-2) in a mouse model of vaginitis. Int Immunopharmacol. 2019;68:58–67.
94. Wang Y, Ding Y, Yang C, et al. Inhibition of the infectivity and inflammatory response of influenza virus by arbidol hydrochloride in vitro and in vivo (mice and ferret). Biomed Pharmacother. 2017;91:393–401. doi:10.1016/j.biopha.2017.04.091
95. Pecheur EI, Borisevich V, Halfmann P, et al. The synthetic antiviral drug arbidol inhibits globally prevalent pathogenic viruses. J Virol. 2016;90:3086–3092. doi:10.1128/JVI.02077-15
96. Wang PC, Chiu DC, Jan JT, et al. Peramivir conjugates as orally available agents against influenza h275y mutant. Eur J Med Chem. 2018;145:224–234. doi:10.1016/j.ejmech.2017.12.072
97. Noshi T, Kitano M, Taniguchi K, et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase pa subunit. Antiviral Res. 2018;160:109–117. doi:10.1016/j.antiviral.2018.10.008
98. Bowman LJ, Melaragno JI, Brennan DC. Letermovir for the management of cytomegalovirus infection. Expert Opin Investig Drugs. 2017;26:235–241. doi:10.1080/13543784.2017.1274733
99. Totura AL, Bavari S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin Drug Discov. 2019;14:397–412. doi:10.1080/17460441.2019.1581171
100. Sheahan TP, Sims AC, Graham RL, et al. Broad-spectrum antiviral gs-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med. 2017;9:eaal3653. doi:10.1126/scitranslmed.aal3653
101. Furuta Y, Komeno T, Nakamura T. Favipiravir (t-705), a broad spectrum inhibitor of viral rna polymerase. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93:449–463. doi:10.2183/pjab.93.027
102. Quiros Roldan E, Biasiotto G, Magro P, Zanella I. The possible mechanisms of action of 4-aminoquinolines (chloroquine/hydroxychloroquine) against sars-cov-2 infection (covid-19): a role for iron homeostasis? Pharmacol Res. 2020;158:104904. doi:10.1016/j.phrs.2020.104904
103. Deng L, Li C, Zeng Q, et al. Arbidol combined with lpv/r versus lpv/r alone against corona virus disease 2019: a retrospective cohort study. J Infect. 2020;81:e1–e5. doi:10.1016/j.jinf.2020.03.002
104. McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C. Candidate drugs against sars-cov-2 and covid-19. Pharmacol Res. 2020;157:104859. doi:10.1016/j.phrs.2020.104859
105. Zannella A, Marignani M, Begini P. Hematological malignancies and hbv reactivation risk: suggestions for clinical management. Viruses. 2019;11:858. doi:10.3390/v11090858
106. Morandi E, Tanasescu R, Tarlinton RE, Constantin-Teodosiu D, Gran B. Do antiretroviral drugs protect from multiple sclerosis by inhibiting expression of ms-associated retrovirus? Front Immunol. 2018;9:3092. doi:10.3389/fimmu.2018.03092
107. Quercia R, Perno CF, Koteff J, et al. Twenty-five years of lamivudine: current and future use for the treatment of hiv-1 infection. J Acquir Immune Defic Syndr. 2018;78:125–135. doi:10.1097/QAI.0000000000001660
108. Fischl MA, Richman DD, Hansen N, et al. The safety and efficacy of zidovudine (azt) in the treatment of subjects with mildly symptomatic human immunodeficiency virus type 1 (hiv) infection. A double-blind, placebo-controlled trial. The aids clinical trials group. Ann Intern Med. 1990;112:727–737. doi:10.7326/0003-4819-112-10-727
109. Yan D, Liu XY, Zhu YN, et al. Factors associated with prolonged viral shedding and impact of lopinavir/ritonavir treatment in hospitalised non-critically ill patients with sars-cov-2 infection. Eur Respir J. 2020;56:2000799. doi:10.1183/13993003.00799-2020
110. Chan JF, Yao Y, Yeung ML, et al. Treatment with lopinavir/ritonavir or interferon-beta1b improves outcome of mers-cov infection in a nonhuman primate model of common marmoset. J Infect Dis. 2015;212:1904–1913. doi:10.1093/infdis/jiv392
111. Lecronier M, Beurton A, Burrel S, et al. Comparison of hydroxychloroquine, lopinavir/ ritonavir,and standard of care in critically ill patients with sars-cov-2 pneumonia: an opportunistic retrospective analysis. Crit Care. 2020;24:418. doi:10.1186/s13054-020-03117-9
112. Jang Y, Shin JS, Yoon YS, et al. Salinomycin inhibits influenza virus infection by disrupting endosomal acidification and viral matrix protein 2 function. J Virol. 2018;92:e01441. doi:10.1128/JVI.01441-18
113. Neri-Bazan RM, Garcia-Machorro J, Mendez-Luna D, et al. Design, in silico studies, synthesis and in vitro evaluation of oseltamivir derivatives as inhibitors of neuraminidase from influenza a virus h1n1. Eur J Med Chem. 2017;128:154–167. doi:10.1016/j.ejmech.2017.01.039
114. Todt D, Walter S, Brown RJ, Steinmann E. Mutagenic effects of ribavirin on hepatitis e virus-viral extinction versus selection of fitness-enhancing mutations. Viruses. 2016;8:283. doi:10.3390/v8100283
115. Avataneo V, de Nicolo A, Cusato J, et al. Development and validation of a uhplc-ms/ms method for quantification of the prodrug remdesivir and its metabolite gs-441524: a tool for clinical pharmacokinetics of sars-cov-2/covid-19 and ebola virus disease. J Antimicrob Chemother. 2020;75:1772–1777. doi:10.1093/jac/dkaa152
116. Shiraki K, Daikoku T. Favipiravir, an anti-influenza drug against life-threatening rna virus infections. Pharmacol Ther. 2020;209:107512. doi:10.1016/j.pharmthera.2020.107512
117. Pagadala NS. Azt acts as an anti-influenza nucleotide triphosphate targeting the catalytic site of a/pr/8/34/h1n1 rna dependent rna polymerase. J Comput Aided Mol Des. 2019;33:387–404. doi:10.1007/s10822-019-00189-w
118. Bertoletti N, Chan AH, Schinazi RF, Yin YW, Anderson KS. Structural insights into the recognition of nucleoside reverse transcriptase inhibitors by hiv-1 reverse transcriptase: first crystal structures with reverse transcriptase and the active triphosphate forms of lamivudine and emtricitabine. Protein Sci. 2019;28:1664–1675. doi:10.1002/pro.3681
119. Lee HW, Park JY, Ahn SH. An evaluation of entecavir for the treatment of chronic hepatitis b infection in adults. Expert Rev Gastroenterol Hepatol. 2016;10:177–186. doi:10.1586/17474124.2016.1125781
120. Han Y, Pham HT, Xu H, Quan Y, Mesplede T. Antimalarial drugs and their metabolites are potent zika virus inhibitors. J Med Virol. 2019;91:1182–1190. doi:10.1002/jmv.25440
121. Chen Y, Ke M, Xu J, Lin C. Simulation of the pharmacokinetics of oseltamivir and its active metabolite in normal populations and patients with hepatic cirrhosis using physiologically based pharmacokinetic modeling. AAPS PharmSciTech. 2020;21:98. doi:10.1208/s12249-020-1638-y
122. Deng P, Zhong D, Yu K, Zhang Y, Wang T, Chen X. Pharmacokinetics, metabolism, and excretion of the antiviral drug arbidol in humans. Antimicrob Agents Chemother. 2013;57:1743–1755. doi:10.1128/AAC.02282-12
123. Joshy KS, Susan MA, Snigdha S, Nandakumar K, Laly AP, Sabu T. Encapsulation of zidovudine in pf-68 coated alginate conjugate nanoparticles for anti-hiv drug delivery. Int J Biol Macromol. 2018;107:929–937. doi:10.1016/j.ijbiomac.2017.09.078
124. van Waterschoot RAB, Ter Heine R, Wagenaar E, et al. Effects of cytochrome p450 3a (cyp3a) and the drug transporters p-glycoprotein (mdr1/abcb1) and mrp2 (abcc2) on the pharmacokinetics of lopinavir. Br J Pharmacol. 2010;160:1224–1233. doi:10.1111/j.1476-5381.2010.00759.x
125. Abd-Rabou AA, Bharali DJ, Mousa SA. Viramidine-loaded galactosylated nanoparticles induce hepatic cancer cell apoptosis and inhibit angiogenesis. Appl Biochem Biotechnol. 2020;190:305–324. doi:10.1007/s12010-019-03090-2
126. Patel BK, Parikh RH, Patel N. Targeted delivery of mannosylated-plga nanoparticles of antiretroviral drug to brain. Int J Nanomedicine. 2018;13:97–100. doi:10.2147/IJN.S124692
127. Zhong J, Xia Y, Hua L, et al. Functionalized selenium nanoparticles enhance the anti-ev71 activity of oseltamivir in human astrocytoma cell model. Artif Cells Nanomed Biotechnol. 2019;47:3485–3491. doi:10.1080/21691401.2019.1640716
128. Yadavalli T, Ames J, Agelidis A, et al. Drug-encapsulated carbon (decon): a novel platform for enhanced drug delivery. Sci Adv. 2019;5:eaax0780. doi:10.1126/sciadv.aax0780
129. Levy GA, Adamson G, Phillips MJ, et al. Targeted delivery of ribavirin improves outcome of murine viral fulminant hepatitis via enhanced anti-viral activity. Hepatology. 2006;43:581–591. doi:10.1002/hep.21072
130. Giesler KE, Marengo J, Liotta DC. Reduction sensitive lipid conjugates of tenofovir: synthesis, stability, and antiviral activity. J Med Chem. 2016;59:7097–7110. doi:10.1021/acs.jmedchem.6b00428
131. Lin CC, Yeh LT, Luu T, Lourenco D, Lau JY. Pharmacokinetics and metabolism of [(14)c]ribavirin in rats and cynomolgus monkeys. Antimicrob Agents Chemother. 2003;47:1395–1398. doi:10.1128/AAC.47.4.1395-1398.2003
132. Gowen BB, Sefing EJ, Westover JB, et al. Alterations in favipiravir (t-705) pharmacokinetics and biodistribution in a hamster model of viral hemorrhagic fever. Antiviral Res. 2015;121:132–137. doi:10.1016/j.antiviral.2015.07.003
133. Kumar P, Lakshmi YS, C. B, Golla K, Kondapi AK. Improved safety, bioavailability and pharmacokinetics of zidovudine through lactoferrin nanoparticles during oral administration in rats. PLoS One. 2015;10:e0140399. doi:10.1371/journal.pone.0140399
134. Nirogi R, Kandikere V, Komarneni P, et al. Exploring dried blood spot sampling technique for simultaneous quantification of antiretrovirals: lamivudine, stavudine and nevirapine in a rodent pharmacokinetic study. Biomed Chromatogr. 2012;26:1472–1481. doi:10.1002/bmc.2718
135. Dodiya S, Chavhan S, Korde A, Sawant KK. Solid lipid nanoparticles and nanosuspension of adefovir dipivoxil for bioavailability improvement: formulation, characterization, pharmacokinetic and biodistribution studies. Drug Dev Ind Pharm. 2012;39:733–743. doi:10.3109/03639045.2012.694889
136. Ho MJ, Lee DR, Im SH, et al. Microsuspension of fatty acid esters of entecavir for parenteral sustained delivery. Int J Pharm. 2018;543:52–59. doi:10.1016/j.ijpharm.2018.03.042
137. Djekic L, Janković J, Rašković A, Primorac M. Semisolid self-microemulsifying drug delivery systems (smeddss): effects on pharmacokinetics of acyclovir in rats. Eur J Pharm Sci. 2018;121:287–292. doi:10.1016/j.ejps.2018.06.005
138. Ravi PR, Vats R, Dalal V, Murthy AN. A hybrid design to optimize preparation of lopinavir loaded solid lipid nanoparticles and comparative pharmacokinetic evaluation with marketed lopinavir/ritonavir coformulation. J Pharm Pharmacol. 2014;66:912–926. doi:10.1111/jphp.12217
139. Joshi G, Kumar A, Sawant K. Bioavailability enhancement, caco-2 cells uptake and intestinal transport of orally administered lopinavir-loaded plga nanoparticles. Drug Deliv. 2016;23:3492–3504. doi:10.1080/10717544.2016.1199605
140. Islam MS, Reineke J, Kaushik R, et al. Bioadhesive food protein nanoparticles as pediatric oral drug delivery system. ACS Appl Mater Interfaces. 2019;11:18062–18073. doi:10.1021/acsami.9b00152
141. Lei M, Gan W, Sun Y. Hplc-ms/ms analysis of peramivir in rat plasma: elimination of matrix effect using the phospholipid-removal solid-phase extraction method. Biomed Chromatogr. 2018;32:e4103. doi:10.1002/bmc.4103
142. Koshimichi H, Ishibashi T, Wajima T. Population pharmacokinetics of baloxavir marboxil in japanese pediatric influenza patients. J Pharm Sci. 2019;108:3112–3117. doi:10.1016/j.xphs.2019.04.010
143. Kropeit D, Scheuenpflug J, Erb-Zohar K, et al. Pharmacokinetics and safety of letermovir, a novel anti-human cytomegalovirus drug, in patients with renal impairment. Br J Clin Pharmacol. 2017;83:1944–1953. doi:10.1111/bcp.13292
144. Solórzano R, Tort O, García-Pardo J, et al. Versatile iron–catechol-based nanoscale coordination polymers with antiretroviral ligand functionalization and their use as efficient carriers in hiv/aids therapy. Biomater Sci. 2019;7:178–186. doi:10.1039/C8BM01221K
145. Abdelbary GA, Amin MM, Zakaria MY, El Awdan SA. Adefovir dipivoxil loaded proliposomal powders with improved hepatoprotective activity: formulation, optimization, pharmacokinetic, and biodistribution studies. J Liposome Res. 2017;28:259–274. doi:10.1080/08982104.2017.1363228
146. Gourdon B, Chemin C, Moreau A, et al. Functionalized pla-peg nanoparticles targeting intestinal transporter pept1 for oral delivery of acyclovir. Int J Pharm. 2017;529:357–370. doi:10.1016/j.ijpharm.2017.07.024
147. Mao Y, Feng S, Li S, et al. Chylomicron-pretended nano-bio self-assembling vehicle to promote lymphatic transport and galts target of oral drugs. Biomaterials. 2019;188:173–186. doi:10.1016/j.biomaterials.2018.10.012
148. Ravi PR, Vats R, Balija J, Adapa SP, Aditya N. Modified pullulan nanoparticles for oral delivery of lopinavir: formulation and pharmacokinetic evaluation. Carbohydr Polym. 2014;110:320–328. doi:10.1016/j.carbpol.2014.03.099
149. Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17:559–587.
150. Dalpiaz A, Fogagnolo M, Ferraro L, et al. Bile salt-coating modulates the macrophage uptake of nanocores constituted by a zidovudine prodrug and enhances its nose-to-brain delivery. Eur J Pharm Biopharm. 2019;144:91–100. doi:10.1016/j.ejpb.2019.09.008
151. Yao X, Ye F, Zhang M, et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (sars-cov-2). Clin Infect Dis. 2020;71:732–739. doi:10.1093/cid/ciaa237
152. Zhong M, Feng Y, Liao H, et al. Azithromycin cationic non-lecithoid nano/microparticles improve bioavailability and targeting efficiency. Pharm Res. 2014;31:2857–2867. doi:10.1007/s11095-014-1382-7
153. Wannachaiyasit S, Chanvorachote P, Nimmannit U. A novel anti-hiv dextrin-zidovudine conjugate improving the pharmacokinetics of zidovudine in rats. AAPS PharmSciTech. 2008;9:840–850. doi:10.1208/s12249-008-9122-0
154. Senanayake TH, Gorantla S, Makarov E, Lu Y, Warren G, Vinogradov SV. Nanogel-conjugated reverse transcriptase inhibitors and their combinations as novel antiviral agents with increased efficacy against hiv-1 infection. Mol Pharm. 2015;12:4226–4236. doi:10.1021/acs.molpharmaceut.5b00424
155. Dutta L, Mukherjee B, Chakraborty T, et al. Lipid-based nanocarrier efficiently delivers highly water soluble drug across the blood-brain barrier into brain. Drug Deliv. 2018;25:504–516. doi:10.1080/10717544.2018.1435749
156. Li CL, Hsieh CH, Tsai TH. Preclinical pharmacokinetics of lamivudine and its interaction with schisandra chinensis extract in rats. ACS Omega. 2020;5:1997–2004. doi:10.1021/acsomega.9b03922
157. Ayoub MM, Elantouny NG, El-Nahas HM, Ghazy FES. Injectable plga adefovir microspheres; the way for long term therapy of chronic hepatitis-b. Eur J Pharm Sci. 2018;118:24–31. doi:10.1016/j.ejps.2018.03.016
158. Zhang C, Wang A, Wang H, et al. Entecavir-loaded poly (lactic-co-glycolic acid) microspheres for long-term therapy of chronic hepatitis-b: preparation and in vitro and in vivo evaluation. Int J Pharm. 2019;560:27–34. doi:10.1016/j.ijpharm.2019.01.052
159. Perazzolo S, Shireman LM, McConnachie LA, et al. Integration of computational and experimental approaches to elucidate mechanisms of first-pass lymphatic drug sequestration and long-acting pharmacokinetics of the injectable triple-hiv drug combination tlc-art 101. J Pharm Sci. 2020;109:1789–1801. doi:10.1016/j.xphs.2020.01.016
160. Giuliani A, Balducci AG, Zironi E, et al. In vivo nose-to-brain delivery of the hydrophilic antiviral ribavirin by microparticle agglomerates. Drug Deliv. 2018;25:376–387. doi:10.1080/10717544.2018.1428242
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.