")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 9
Antitumor Effects and Mechanisms of Metabolic Syndrome Medications on Hepatocellular Carcinoma
Authors Oura K , Morishita A, Tani J, Masaki T
Received 3 October 2022
Accepted for publication 4 December 2022
Published 14 December 2022 Volume 2022:9 Pages 1279—1298
DOI https://doi.org/10.2147/JHC.S392051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Kyoko Oura, Asahiro Morishita, Joji Tani, Tsutomu Masaki
Department of Gastroenterology and Neurology, Faculty of Medicine, Kagawa University, Kagawa, Japan
Correspondence: Kyoko Oura, Department of Gastroenterology and Neurology, Kagawa University, 1750-1 Ikenobe, Miki, Kida, Kagawa, Japan, Tel +81-87-891-2156, Fax +81-87-891-2158, Email [email protected]
Abstract: Liver cancer has a high incidence and mortality rate worldwide, with hepatocellular carcinoma (HCC) being the most common histological type. With the decrease in the number of newly infected patients and the spread of antiviral therapy, hepatitis virus-negative chronic liver diseases including steatohepatitis are increasingly accounting for a large proportion of HCC, and an important clinical characteristic is the high prevalence of metabolic syndrome including hypertension, type 2 diabetes (T2D), dyslipidemia, and obesity. Since patients with steatohepatitis are less likely to undergo surveillance for early detection of HCC, they may be diagnosed at an advanced stage and have worse prognosis. Therefore, treatment strategies for patients with HCC caused by steatohepatitis, especially in advanced stages, become increasingly important. Further, hypertension, T2D, and dyslipidemia may occur as side effects during systemic treatment, and there will be increasing opportunities to prescribe metabolic syndrome medications, not only for originally comorbid diseases, but also for adverse events during HCC treatment. Interestingly, epidemiological studies have shown that patients taking some metabolic syndrome medications are less likely to develop various types of cancers, including HCC. Basic studies have also shown that these drugs have direct antitumor effects on HCC. In particular, angiotensin II receptor blockers (a drug group for treating hypertension), biguanides (a drug group for treating T2D), and statins (a drug group for treating dyslipidemia) have shown to elucidate antitumor effects against HCC. In this review, we focus on the antitumor effects of metabolic syndrome medications on HCC and their mechanisms based on recent literature. New therapeutic agents are also increasingly being reported. Analysis of the antitumor effects of metabolic syndrome medications on HCC and their mechanisms will be doubly beneficial for HCC patients with metabolic syndrome, and the use of these medications may be a potential strategy against HCC.
Keywords: hypertension, angiotensin II receptor blockers, diabetes, biguanide, dyslipidemia, statin
Introduction
In 2021, liver cancer had the sixth highest incidence and the third highest mortality rate of all cancer types worldwide.1,2 Hepatocellular carcinoma (HCC) is the most common major histologic type of primary liver cancer, accounting for over 90% of cases.3 Despite the development of therapeutic modalities, HCC holds one of the poorest cancer prognoses due to the difficulty of early detection, resistance to anticancer drugs, and high recurrence rate, with a 5-year survival rate of 15–38%.4–6 The occurrence of HCC is strongly related to high hepatitis virus infection rates, including hepatitis B virus (HBV) and hepatitis C virus (HCV) infection. For instance, HBV-induced chronic hepatitis (CH) is the main cause of HCC in China, Southeast Asia, and Central and South Africa, while HCV-induced CH is the main cause of HCC in Japan and Southern Europe.3 Although the details of association between hepatitis viruses and carcinogenesis are still unclear, clinical data exist to support these findings. HBV carriers are at a higher risk of developing HCC at higher HBV load,7 while reports show that HCV elimination with interferon or direct-acting antivirals was effective in reducing HCC occurrence.8,9 Furthermore, HCC is associated with high rates of CH and cirrhosis due to the persistence of neuroinflammatory responses from hepatocytes, a major cause of hepatocarcinogenesis. Multiple factors are intricately involved, including the persistence of immune-mediated inflammation,10 their associated genetic mutations, and altered intracellular signaling.11 However, the occurrence of HCC without cirrhosis is common in the elderly, which may be related to age-related changes in the immune response.
Although most cases of HCC are caused by hepatitis viruses, 5–20% of HCC patients in Japan are negative for both HBV and HCV.12,13 The major causative factors of HCC are alcoholic liver injury, nonalcoholic fatty liver disease (NAFLD), autoimmune hepatitis, and aflatoxin exposure.3,14 With declining numbers of new HBV and HCV infections and the widespread use of antiviral therapies, the proportion of HCC caused by hepatitis virus infection has recently been on the decline, whereas the number of hepatocarcinogenesis cases caused by alcoholic or nonalcoholic steatohepatitis (NASH) has been increasing.15 Our epidemiological study of 802 HCC patients treated in our Department (Kagawa University Hospital, Japan) over a 15-year period from 2003 to 2017 also showed an increase in hepatitis virus-negative HCC including steatohepatitis with the proportion gradually increasing to 11.8% in the early period, 32.9% in the middle period, and 41.1% in the late period.16 Their important clinical characteristics include a high prevalence of metabolic syndrome, with 47.5% having hypertension, 42.0% having type 2 diabetes (T2D), and 47% having obesity. Furthermore, patients who are not infected with hepatitis virus are less likely to undergo surveillance for early detection of HCC, and therefore may be diagnosed at an advanced stage and have a poorer prognosis. Consequently, treatment strategies for patients with HCC caused by steatohepatitis will become more important, especially for advanced stage cases.
Systemic therapy of advanced HCC that is unresectable due to major vascular invasion and/or metastasis generally involves immune checkpoint inhibitors and molecular targeted agents with several currently available drugs including atezolizumab/bevacizumab combination therapy for first-line therapy and sorafenib, lenvatinib, and other drugs for second-line therapy.17,18 However, hypertension, T2D, and dyslipidemia may occur as side effects during these systemic therapies; in the future, there will be more opportunities to prescribe metabolic syndrome medications not only for originally comorbid conditions, but also for adverse events during HCC treatment.
Interestingly, epidemiological studies have shown that patients taking several metabolic syndrome medications are less likely to develop various types of cancers.19–22 There are also basic studies that have showed the direct antitumor effects of metabolic syndrome medications on various cancer cells.23–28 Analysis of these antitumor effects on HCC and their mechanisms will be doubly beneficial for HCC patients who have metabolic syndrome. Further, preclinical studies and clinical trials suggest that regimens that include therapeutic immunotherapies targeting programmed death-1 (PD1), such as the atorolimumab/bevacizumab combination, may be less effective against NASH-induced HCC,29,30 and metabolic syndrome drugs may provide adjuvant antitumor effects through an entirely different mechanism. In this review, based on recent literature, we summarize the association between HCC development and metabolic syndrome, including obesity, hypertension, T2D, and dyslipidemia. We also focus on the antitumor effects of various metabolic syndromes on HCC and their mechanisms and discuss their therapeutic applications.
Obesity and Liver Disease
Obesity is characterized by chronic accumulation of excess body fat caused by genetics, environmental factors, comorbidities, and certain medical treatment such as hormone therapy.31,32 It was shown that more than 700 million adults, or approximately 15% of all adults worldwide, were obese in 2020, and the number is expected to increase rapidly.32 Obesity is an independent risk factor for progression of many diseases, including T2D, cardiovascular disease, hypertension, dyslipidemia, and NAFLD.31 Recent reports have also linked it to an increased risk of various cancers, including HCC.33 The pathophysiology of NAFLD can lead to HCC development not caused by the hepatitis virus, on the continuum to metabolic syndrome, including hypertension, diabetes, dyslipidemia, and obesity is shown in Figure 1.34 NAFLD progresses to NASH characterized by hepatocyte ballooning, apoptosis, accumulation of Mallory–Denk bodies, and inflammation in the liver parenchyma and portal vein and ultimately leads to irreversible cirrhosis and hepatocellular carcinogenesis.
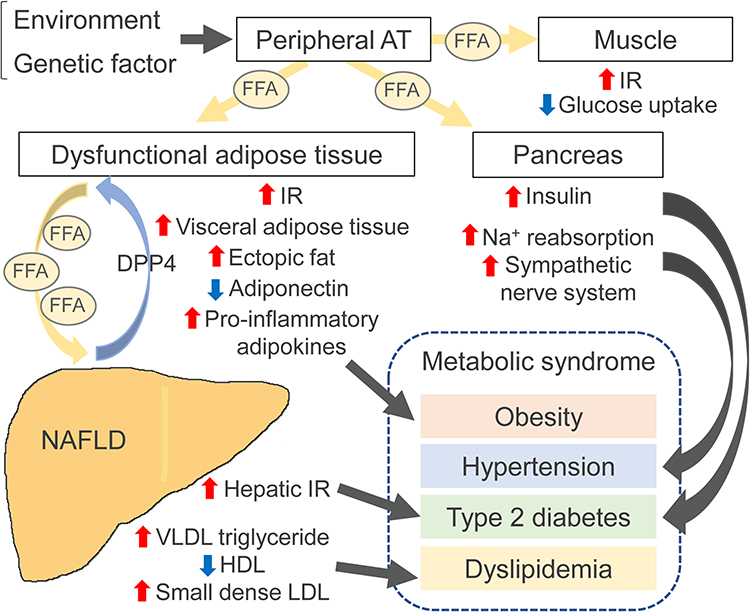 |
Figure 1 Pathophysiology of nonalcoholic fatty liver disease (NAFLD) on the continuum to metabolic syndrome. When environmental and genetic factors induce weight gain, increased mobilization of free fatty acids (FFAs) from subcutaneous adipose tissue (AT) results in accumulation of visceral and ectopic fat. In the muscle, increased accumulation of FFAs promotes insulin resistance (IR) and inhibits insulin-mediated glucose uptake. FFAs leaking into the pancreas cause β-cell dysfunction and hyperglycemia. Insulin resistance (IR) promotes lipolysis in dysfunctional AT and increases the flux of FFAs to the liver, promoting hepatic glucose production, lipogenesis, release of very low-density lipoproteins (VLDLs), and dyslipidemia. These global IR conditions can lead to hyperinsulinemia; they promote sodium reabsorption and lead to hypertension. Inflamed dysfunctional AT increases IR and releases levels of inflammatory adipokines while decreasing anti-inflammatory adiponectin levels. In the liver, triglycerides and toxic metabolites induce lipotoxicity, mitochondrial dysfunction, and endoplasmic reticulum stress, leading to hepatocyte damage, apoptosis, and fibrosis. These dysfunctional hepatocytes synthesize and secrete dipeptidyl peptidase 4 (DPP4), which promotes AT macrophage inflammation and further causes IR. |
While treatment for obesity and related chronic liver disease primarily consists of lifestyle modifications focused on weight management, patients with moderate to severe obesity or mild obesity refractory to lifestyle therapy should be considered for pharmacotherapy. Orlistat is a gastrointestinal lipase inhibitor that modestly reduces body weight by limiting the absorption of fat from the intestinal tract, but has been shown to reduce intrahepatic inflammation and fibrosis in steatohepatitis.35 Combination weight-reduction therapies, including phentermine/topiramate and naltrexone-bupropion do not show a preventive effect on HCC, but may be of clinical value because weight reduction is associated with a decrease in intrahepatic lipid accumulation. Liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist used in the treatment of T2D, helps obese patients lose weight by reducing food intake. Liraglutide prevents progression from NAFLD to HCC occurrence in mice with obesity and streptozotocin-induced diabetes.36 Promising therapeutic approaches target adiposity, hepatitis, and fibrosis through multiple mechanisms of action, such as GLP-1, glucagon receptor, and glucose-dependent insulinotropic polypeptide. There is limited evidence to conclude whether pharmacological treatment of obesity prevents HCC; further preclinical studies and clinical trials on humans are warranted to validate its role in the prevention of hepatocarcinogenesis.
Hypertension and Liver Disease
Hypertension is one of the major diseases in the metabolic syndrome, along with T2D, dyslipidemia, and obesity, and it affects approximately 30% of the general population. It results from a combination of multiple factors, including genetic predisposition and environmental risk factors such as excessive salt intake, obesity, smoking, lack of exercise, and stress.37 Hypertension can not only cause ischemic heart disease and cerebrovascular disease, but it is also associated with NAFLD, which encompasses a continuous spectrum leading to NASH with advanced cirrhosis and HCC. Approximately 49.5% of hypertensive patients have NAFLD, indicating a significantly higher prevalence of hypertension in NAFLD patients compared to general population.38,39 Several prospective studies have also shown that NAFLD is an independent risk factor for the development of hypertension after adjustment for T2D, dyslipidemia, obesity, and other systemic metabolic disorders.38,40,41 Interestingly, another report has shown that persistence of NAFLD over a 5-year observation period increased the risk of developing hypertension. Meanwhile, the occurrence of hypertension is not increased in cases with improved imaging findings of fatty liver.42 It is unclear from the clinical evidence whether NAFLD is a consequence or a cause of hypertension.
Furthermore, it has been shown that NAFLD causes several effects such as hepatitis, insulin resistance, and renin-angiotensin system (RAS)-sympathetic nervous system (SNS) activation, which have been shown to be important physiological mechanisms that lead to hypertension.43,44 In patients with NAFLD, cardiac and autonomic functions are significantly impaired, independent of SNS, and blood levels of tumor necrosis factor (TNF)-α and cytokeratin 18, which are markers of liver damage, are elevated; therefore, activation of the RAS is shown to be a major mechanism for the progression of hypertension.45 Via the production of angiotensinogen in the liver and kidney, cytokines such as TNF-α also promote systemic and local angiotensin (Ang) II production and Ang II-dependent hypertension.46 In addition, several cytokines, such as retinol binding protein 4 and fetuin A, are upregulated in patients with NAFLD, and have been optimized to cause hepatitis by activating toll-like receptor (TLR)-4 dependent inflammatory pathways.47 However, TLR4 activation can also promote cardiovascular and renal pro-inflammatory cytokines and reactive oxygen species, which may adversely affect hypertension.48 Furthermore, another report suggests that NAFLD is independently related to the development of chronic liver disease; local kidney inflammation appears to cause hypertension.49
In general, blood pressure is often low in the terminal stages of cirrhosis via hemodynamic and blood bioactive substances, but in other cases of chronic liver diseases complicated by hypertension, the usual antihypertensive drugs are used. In patients with severe hepatic dysfunction, blood levels of antihypertensive drugs in hepatic metabolism are increased, necessitating dose reduction. Non-selective β-blockers, such as propranolol, decrease portal blood pressure and reduce the incidence of gastrointestinal bleeding and the risk of death in patients with cirrhosis.50 RAS inhibitors, such as angiotensin converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs), have the potential to reduce liver fibrosis during the transition from CH to cirrhosis.51 Other studies showed that RAS inhibitors are effective in improving pathophysiological responses, including liver fibrosis in patients with NAFLD;52,53 therefore, RAS inhibitors may be best suited as antihypertensive agents for patients with chronic liver disease, especially NAFLD.
Prevention of HCC Occurrence with Antihypertensive Drugs
Since obesity and NAFLD promote hypertension and affect carcinogenesis, hypertension itself is suggested to have no independent role in the development and progression of HCC; however, there is evidence for preventive and antitumor effects of hypertensive drugs against HCC, independent of their effect on blood pressure. In recent years, there has been a growing number of clinical studies that have examined the association between the risk of HCC development and antihypertensive drugs, such as RAS inhibitors and β-blockers (Table 1). Recent systematic reviews suggest that RAS inhibitors alone or in combination significantly reduce HCC recurrence, although they do not prolong patient survival.54 Although a case-control study examining the association between RAS inhibitor use and the development of HCC found no significant findings overall, a woman receiving 30 or more cumulative defined daily doses (cDDDs) of RAS inhibitors had a significantly lower incidence of HCC in a subgroup analysis.55 Furthermore, patients without T2D and with RAS inhibitor cDDD of 1800 or higher had significantly reduced the development of HCC compared to those with no RAS inhibitor exposure; this suggests that the risk of HCC occurrence may be lower with higher cumulative doses. Other reports found positive results in patients receiving therapeutic interventions for HCC: it was showed that overall survival (OS) in HCC patients treated with sorafenib and RAS inhibitors was prolonged.56 HCC patients treated with radiofrequency ablation (RFA) also reported significantly longer OS and disease-free survival in cases that had received ARBs in the previous two years at least, while those treated with ACE inhibitors did not.57 On the other hand, there are several studies showing negative results regarding the effect of RAS inhibitors in preventing the development of HCC.58–61 Interestingly, the use of RAS inhibitors rather increased the HCC occurrence in HCV-infected patients without cirrhosis, T2D, or dyslipidemia.58 In a study of post-tumor resection of HCV-related HCC patients, the ARB-treated group did not have an OS advantage over the control groups, but cirrhosis patients prescribed other antihypertensive drugs had a significantly shorter OS than those prescribed ARB.61 Several interventional studies examined the effects of ACE inhibitors alone or in combination with other drugs in patients after RFA; these showed that ACE inhibitors reduced the risk of HCC recurrence in combination with branched-chain amino acids (BCAAs) or vitamin K, but no significant OS benefit was observed.62–64 Thus, although the results for patient survival with RAS inhibitors appear to be contradictory, this accumulating evidence suggests that RAS inhibitors may work to reduce the occurrence of HCC.
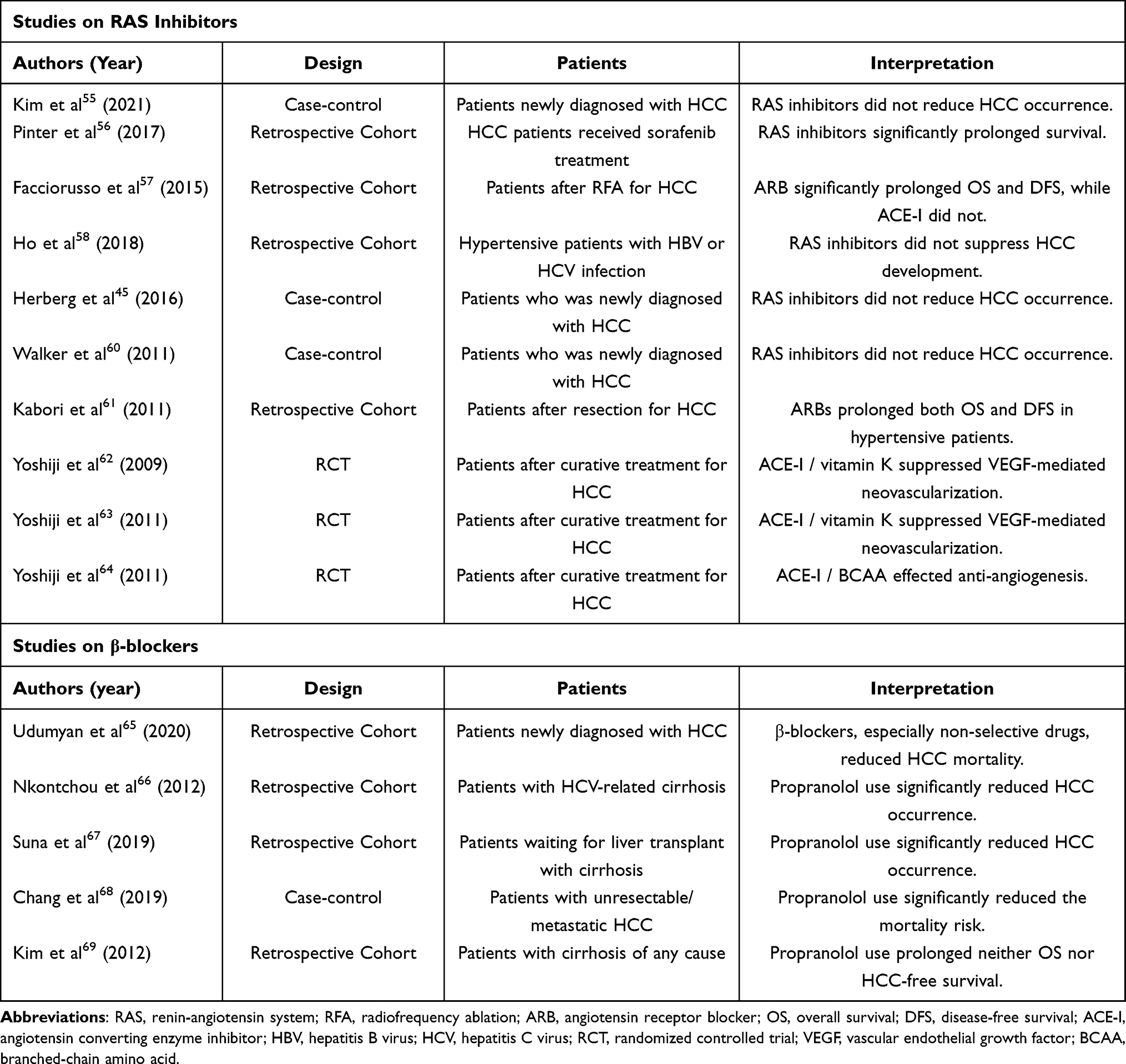 |
Table 1 Clinical Studies on the Prevention of Hepatocellular Carcinoma (HCC) by Antihypertensive Drugs |
There are some remarkable studies on whether the use of β-blockers benefits patients after HCC treatment or puts them at a high risk of carcinogenesis. In a large cohort study, β-blocker use reduced mortality from HCC, and a greater inverse correlation was observed, especially with respect to non-selective β-blocker use.65 In a retrospective long-term observation study, propranolol treatment was the only independent prognostic factor associated with the HCC development in patients with HCV-related cirrhosis and esophageal varices.66 Another cohort study of patients with uncompensated cirrhosis awaiting liver transplantation found that the cause and stages of cirrhosis were similar in the propranolol-treated and control groups, but the HCC occurrence was significantly reduced in the propranolol-treated patients. This result supported the fact that propranolol treatment prevented the development of HCC in patients awaiting liver transplantation.67 In a study investigating the long-term prognosis of patients with unresectable HCC, propranolol was found to significantly reduce mortality risk by 22% and improve OS after performing a multivariate Cox regression analysis on HCC mortality.68 Conversely, another study showed that a low dose of propranolol in patients with cirrhosis did not make a significant difference in HCC development and OS.69 This evidence regarding the prevention of HCC by β-blockers may not only reflect its direct antitumor effect, but could result from an improvement in portal hypertension; caution should be exercised in interpreting these results.
Antitumor Effects and Mechanisms of RAS Inhibitors on HCC
In recent years, several experimental data have been presented examining the antitumor effects of RAS inhibitors, including ARBs and ACE inhibitors, on HCC (Table 2). In our previous study, we evaluated the antitumor effects of several ARBs, including telmisartan, valsartan, irbesartan, and losartan, on HCC using cell lines.70 Only telmisartan showed antitumor effects on poorly differentiated HCC cell lines, such as HLE, HLF, and HepG2, but not on HuH-7 and PLC/PRF/5. The main mechanism of the antitumor effect was activation of AMP-activated protein kinase (AMPK) and inhibition of mammalian target of rapamycin (mTOR) pathway, resulting in decreased expression of cyclin D1 and G1 arrest.70 Earlier studies have shown that staphylococcal nuclease domain containing-1 (SND1), known to promote tumorigenesis of HCC cells, increases Ang II type 1 receptor (AT1R) levels. Furthermore, losartan suppressed the migration and invasion of Hep3B and QGY-7703, suggesting that SND1 inhibitors and ARBs may be an effective therapeutic strategy against advanced HCC.71 In a study using the rat hepatoma cell line, which were transfected with a plasmid producing non-secreted angiotensinogen, losartan inhibited cell growth.72 Candesartan was as effective as losartan in competing with Ang II-AT1R interactions, but did not inhibit cell growth. These in vitro data can be conflicting, but studies using animal models can help clarify the antitumor effects and mechanisms of ARBs. For example, a study examining the antitumor effects both in vitro and in vivo showed that candesartan did not affect the growth of HCC cell lines including LO2, SMMG7721, and HepG2, while in a xenograft mouse model with SMMG7721, candesartan showed tumor suppression by decreasing the expression of vascular endothelial growth factor (VEGF)-A.73 Using an animal model of Wistar male rats fed with a 24-week choline-deficient, L-amino acid-defined (CDAA) diet to induce liver cirrhosis and liver carcinogenesis, another author found that telmisartan treatment suppressed liver carcinogenesis by reducing HIF-α and VEGF expression.74
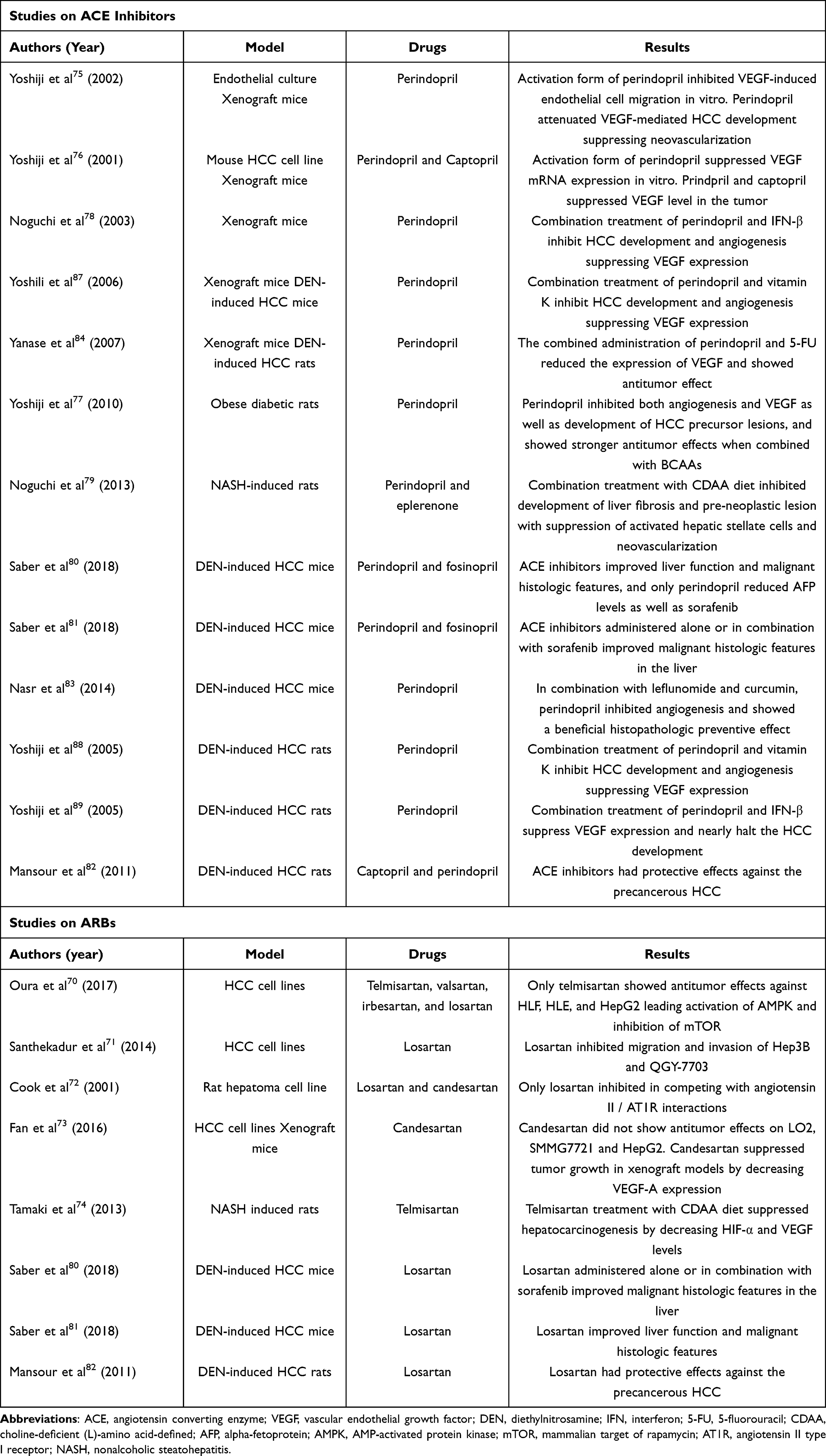 |
Table 2 Experimental Studies on the Antitumor Effects of Renin-Angiotensin System (RAS) Inhibitors Against Hepatocellular Carcinoma (HCC) |
The antitumor effects of ACE inhibitors have been validated by several animal experiments. Using xenograft mice models with HCC cell lines, perindopril was found to significantly attenuate VEGF-mediated tumor development suppressing neovascularization at a clinically comparable low dose.75,76 The same authors also used obese, diabetic rats treated with dimethylnitrosamine and found that perindopril inhibited both angiogenesis and VEGF expression, as well as the development of HCC precursor lesions, and showed stronger antitumor effects when combined with BCAAs.77
The combination of ACE inhibitors with other angiogenesis-related drugs has often been used to enhance antitumor effects against HCC. Combined administration of perindopril and interferon (IFN)-β at clinically equivalent low doses in xenograft mice with HCC cell lines has been shown to inhibit HCC development and angiogenesis by suppressing VEGF expression.78 Using male Fisher-344 rats receiving a modified choline-deficient, low-methionine diet, the same authors also showed that combination treatment with perindopril and eplerenone inhibited development of liver fibrosis and pre-neoplastic lesion with suppression of activated hepatic stellate cells and neovascularization.79
Furthermore, previous basic studies using animal models of diethylnitrosamine (DEN)-induced hepatocarcinogenesis also support the evidence associated with premalignant changes of RAS inhibitors on HCC. A study comparing the effects of RAS inhibitors, including perindopril, fosinopril, and losartan, on DEN-induced HCC in mice with standard therapy using sorafenib showed that RAS inhibitors improved liver function and malignant histologic features, while perindopril or sorafenib reduced alpha-fetoprotein (AFP) levels.80 The main mechanisms of these were through inactivation of the NFκB pathway, which induced TNF-α and reduced transforming growth factor (TGF)-β1 levels, leading to lower VEGF and matrix metalloproteinase (MMP)-2 levels. However, in another study, the same authors reported that perindopril, fosinopril, and losartan, administered alone or in combination with sorafenib, markedly improved liver tissue in DEN-induced HCC mice, but were not associated with prolonged OS due to the adverse effects of DEN on other organs. They concluded that HCC mortality assessment in such animal models may be unsuitable.81 Animal studies in rats with DEN-induced HCC suggest that RAS inhibitors, including captopril, perindopril, and losartan, have similar protective effects against the precancerous stages of HCC.82 Treatment of captopril or losartan caused a remarkable decrease in AFP levels and nearly halved VEGF, TGF-β, and fibroblast growth factor levels, only in rats with accelerated hepatocarcinogenesis. Another group focused on the antitumor effects by combinations of angiogenesis inhibitors on HCC and reported that, when combining perindopril, leflunomide, and curcumin, the active principle of turmeric more potently inhibited angiogenesis and showed a beneficial histopathologic preventive effect against DEN-induced HCC in mice.83 As an effective therapeutic strategy, the combination of angiogenesis inhibitors with conventional chemotherapeutic agents provides synergistic anticancer effects. Although perindopril and 5-fluorouracil (5-FU) did not have a significant inhibitory effect on HCC growth when used at low doses, their combined administration reduced the expression of VEGF and suppressed tumor growth in xenograft mice with BNL-HCC cells.84 Furthermore, even in DEN-treated rats, this combination treatment markedly suppressed the development of precancerous HCC lesions.
Furthermore, Vitamin K is a reprehensive drug that has been shown to have antitumor effects against HCC,85,86 and, in combination with perindopril, has inhibited tumor growth in xenograft mice with HCC cells and inhibited hepatocarcinogenesis in DEN-induced HCC mice and rats.87,88 The same authors also reported that perindopril, when used in combination with IFN-β, could suppress VEGF expression and nearly halt HCC development in DEN-induced rats.89 These reports suggest that ACE inhibitors may exert stronger antitumor effects in combination with other angiogenesis inhibitors or standard treatments for HCC, which may provide clues for therapeutic applications.
Type 2 Diabetes (T2D) and Liver Disease
T2D is characterized by a disruption of glucose homeostasis and defective insulin action in many target tissues, including the liver, muscles, and pancreas.90 T2D affects 1 in 11 adults, or 463 million people, globally.91 Patients with T2D are at more than twice the risk of progressive fibrosis, cirrhosis-related complications, and liver disease mortality compared to individuals without T2D. Furthermore, these patients show higher risk of severe liver diseases than patients with any other diseases, including obesity, hypertension, and dyslipidemia.92 A longer history of metabolic dysfunction has been shown to be related to more progressive liver fibrosis in NAFLD patients.93 In turn, NAFLD patients are more likely to have T2D, which is caused by insulin resistance and damaged islet cell function.91 Individuals diagnosed with NAFLD have a two-fold higher risk of T2D94 and a higher risk of developing cardiovascular disease95,96 and hepatocarcinogenesis,97 especially when associated with T2D.
In clinical studies investigating the risk factor of cancers in patients with T2D, elevated levels of the potent mitogen insulin-like growth factor (IGF)-1 have been reported, which may contribute to cancer development.98 In addition, an association between T2D and carcinogenesis has been suggested in several organs such as the endometrium, breast, pancreas, liver, stomach, and liver.99 For instance, the risk of biliary tract cancer is increased in patients with T2D,100 while the prevalence of prostate cancer is decreased in patients with T2D.101 T2D is often accompanied by dyslipidemia and obesity, which further increases the risk of cancer development, especially of most site-specific cancers.102 A strong positive correlation with endometrial and renal cancers was reported, while a weak one with bladder, prostate, and stomach cancers was reported.103,104 Interestingly, the incidence of lung cancer was inversely correlated with T2D and obesity.103 T2D is also closely associated with the prevalence of HCC. Studies in diverse populations with T2D have reported that T2D increases the HCC occurrence by two to three times; the risk of HCC was significantly higher in males than in females.105 Furthermore, the risk of HCC may increase with a longer duration of T2D,106 but the association between T2D severity and the HCC occurrence remains unknown.
In T2D patients, insulin resistance and hyperinsulinemia are important mechanisms of liver disease progression. As the T2D progresses, chronic hyperglycemia and failure of peripheral tissues to respond to circulating insulin leads to insulin resistance. Hyperinsulinemia caused by impaired glucose metabolism of insulin in the skeletal muscle and the liver increases the production of IGF-1 and promotes hepatocyte proliferation and inhibition of apoptosis.107 In addition, insulin resistance and hyperinsulinemia have been reported to be closely associated with the development of HCC resulting from NAFLD.108 Among other factors in the pathogenesis of T2D, inflammatory cytokines,109 oxidative stress,110 gut microbiota abnormalities,111,112 angiogenesis,113 and autophagy114 influence development and progression of HCC.
Suppression of HCC Occurrence by T2D Medication
Several T2D drugs associated with cancer have been reported. As noted above, insulin has tumor growth effects, and the use of insulin secretagogues and insulin preparation may increase the risk of cancer. The use of sulfonylureas (SU), insulin secretagogues, increased the risk of cancer,115 with a reported cancer risk being 1.78 times higher in SU users than in metformin users.116 Research results on insulin preparations and cancer risk have been inconsistent, with past studies reporting an increase in cancer risk, specifically in breast cancer among insulin glargine users,117,118 while others have found no association.119 Addressing the limitations and biases of previous studies, a recent study found that there appear to be differences in cancer risk by cancer type and duration of treatment.120 Specifically in liver cancer, the study had shown a lower risk of carcinogenesis in men who had been treated with insulin for three to four years.120
Of the oral glucose-lowering drugs, metformin most commonly affects the incidence of HCC (Table 3). In a pioneering study, metformin use was associated with decreased cancer risk, reporting an odds ratio of 0.86 (95% confidence interval (CI): 0.73–1.02) for cancer occurrence.121 Regarding HCC occurrence, a retrospective case-control study including 610 HCC patients, 618 cirrhosis patients, and 1696 controls reported that metformin use was related to the lower risk of HCC occurrence compared with SU or insulin use.122 Another hospital-based study including 420 HCC patients and 1104 controls reported that SU or insulin use was associated with the highest risk for HCC occurrence, while metformin or glitazone use reduced HCC risk by 70% in patients with T2D.123 In addition, in a large cohort study including 19,349 diabetes patients and 77,396 controls, patients with T2D had a two-fold higher incidence of HCC than controls, and those treated with either metformin or glitazone had a significantly lower incidence of HCC than those treated with other drugs.124 Several recent meta-analyses support these results. In one consisting of five case-control studies, three cohort studies, and two randomized controlled trials (RCTs), it was shown that patients treated with metformin had approximately 50% less HCC occurrence than those treated with SU, gulitazone, or insulin.125 In another meta-analysis including one RCT, four cohort studies, and eight case-control studies enrolling approximately 480,000 T2D patients, metformin use decreased the risk of HCC incidence, and interestingly, insulin use was conversely associated with an increased risk of HCC occurrence.126 It should be noted, however, that there have been conflicting results from an observational study showing no association between the use of hypoglycemic drugs, including metformin, and incidence of all cancers, including HCC.127 How metformin decreases the risk of HCC development remains unclear, and larger RCTs are needed.
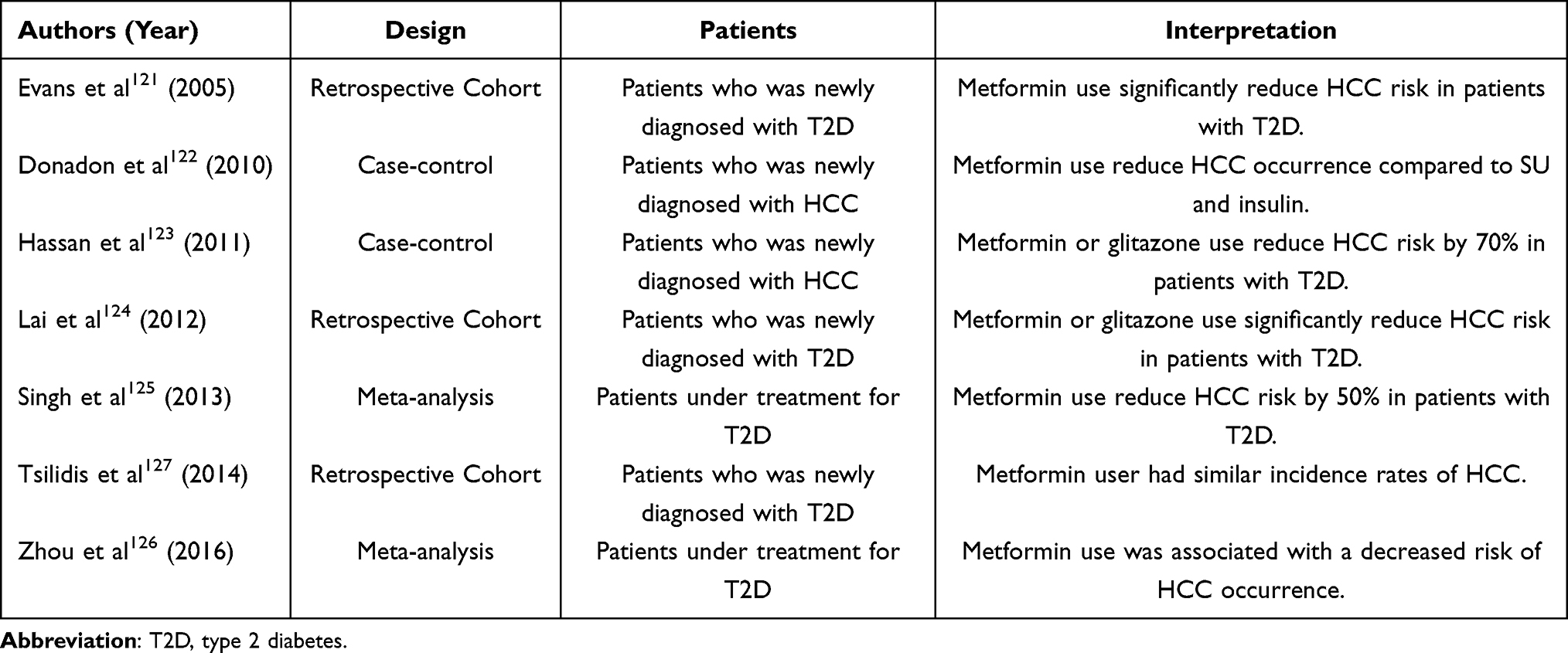 |
Table 3 Clinical Studies on the Prevention of Hepatocellular Carcinoma (HCC) by Metformin |
Dipeptidyl peptidase-4 (DPP-4) has seen a rapid expansion in clinical use over the past decade, which acts by lowering blood glucose by inhibiting the degradation of incretin;128,129 cases presenting inhibition of DPP-4 show elevated levels of both endogenous glucose-dependent insulinotropic polypeptide-1 (GLP-1) and GLP-2, which play crucial roles in cancer progression and metastasis.130 Though the appropriateness of long-term DPP-4 inhibitor use is debatable, some studies suggest that diabetic patients treated with DPP-4 inhibitors do not have a higher risk of cancer development than those treated with placebo or other drugs.131,132 Although there is not much epidemiologic evidence on the risk of developing HCC, one study, comparing the risk of HCC in adults with T2D and HCV-related CH who received DPP-4 inhibitor therapy versus those who did not, showed that DPP-4 inhibitor use suppressed the HCC occurrence.133 In a cohort study of propensity score-matched DPP-4 inhibitor users and non-users in patients with compensated liver cirrhosis, DPP-4 inhibitor use caused the development of decompensated cirrhosis and hepatic failure.134
Sodium/glucose cotransporter-2 (SGLT-2) is a protein involved in glucose reabsorption in the renal tubules. SGLT-2 inhibitors are effective against T2D135 which selectively inhibit renal glucose reabsorption, thereby increasing urinary glucose excretion and lowering plasma glucose levels.136 A meta-analysis based on evidence from short-term RCTs showed that SGLT2 inhibitors did not significantly increase overall cancer risk compared to placebo or other drugs.137 However, empagliflozin may increase the risk of bladder cancer and canagliflozin may decrease the risk of gastrointestinal cancers.137 In another meta-analysis incorporating 27 clinical trials, use of SGLT-2 inhibitors did not increase the risk of developing any common malignancies, including prostate, skin, breast, gastrointestinal tract, bladder, respiratory airways, kidney, pancreas, female genital tract, and liver cancer.138 Although there are no ongoing clinical trials on the use of SGLT-2 inhibitors in HCC patients, there are several clinical trials of SGLT-2 inhibitors in NASH that are expected to shed further light on its potential clinical benefit in patients with NASH-associated HCC.139–141
Antitumor Effects and Mechanism of T2D Drugs on HCC
Metformin is not only suggested to have cancer-inhibitory effects in many cohort and case-control studies, but it is also the T2D drug whose antitumor mechanisms have been most investigated in basic and animal studies in recent years. In general, the antitumor effects of metformin are assumed to be mediated by mechanisms such as activating AMPK, suppressing mammalian target of rapamycin (mTOR), inhibiting human epithelial growth factor receptor 2 (HER2) expression, suppressing angiogenesis, arresting the cell cycle, and inducing apoptosis.142 Several basic studies have demonstrated a variety of antitumor effects, including direct inhibition of tumor growth and induction of apoptosis in HCC (Table 4). Among the effects of metformin on cancer cell proliferation, activation of AMPK in the liver, muscle, and adipocytes has been shown to inhibit HCC proliferation by suppressing the upregulation of IGF-2 molecules and IGF-1 receptors.143 In one in vitro and in vivo study with HCC cell lines, metformin was shown to reduce HCC growth and invasion through PI3K/AKT/mTOR pathway and to promote antitumor effects by inducing apoptosis and autophagy.144 A genetic HCC mouse model experiment of effects on apoptotic pathways showed that metformin reduced tumor size and induced apoptosis by decreasing myeloid cell leukemia 1 (MCL-1) and phosphorylated eukaryotic initiation factor 4E and (elF4E)-binding protein 1 (4E-BP1) levels.145 In another in vitro study using HCC cell lines, metformin induced apoptosis by upregulating AMPK phosphorylation and p53 expression and activating miR-23a, a functional target of forkhead box protein A1 (FOXA1). The inhibition of p53 suppressed miR-23a upregulation by metformin, indicating that the AMPK/p53 signaling is involved in the induction of miR-23a.146 We have also shown in previous in vitro and in vivo studies that metformin inhibits HCC growth and induces G1 cell cycle arrest via microRNA changes.147,148 In addition, recent studies using multiple mouse models of NASH have shown that NASH causes changes in the inflammatory phenotype of hepatic CD8+ T cells, blunting the efficacy of PD-1 therapy; however, metformin treatment restores the efficacy of anti-PD-1 therapy against NASH-induced liver cancer.149 Thus, investigating the interaction between the immune checkpoint inhibitor and metformin will contribute to improvement in the prognosis of patients with advanced HCC-related T2D and NASH, which is expected to increase in the future.
 |
Table 4 Experimental Studies on the Antitumor Effects of Metformin Against Hepatocellular Carcinoma (HCC) |
A basic study on the antitumor effects of DPP-4 inhibitors on HCC showed that anagliptin and vildagliptin did not affect the proliferation of Huh-7 and Li-7 cell lines in vitro and had no effect on cell cycle-related proteins such as p21, p27kip1, cyclin-dependent kinase 2 (CDK2), and retinoblastoma protein (Rb).150 However, both anagliptin and vildagliptin inhibited xenograft HCC growth by natural killer and T-cell tumor accumulation in vivo. Furthermore, sitagliptin has improved the efficiency and duration of tumor-specific T-cell responses when used in combination with anti-programmed cell death 1 (PD1) blockade immunotherapy and other therapies. In an in vivo study using a tumor transplant mouse model, sitagliptin or anti-PD1 antibody monotherapy was shown to delay HCC growth. Interestingly, complete tumor regression was observed with sitagliptin plus anti-PD1 administration.151 Tumor from sitagliptin-treated mice showed a remarkable change in the number of CD8+ T cells, promoting the transport of CD8+ T lymphocytes into the tumor. The study also indicated higher CD8+ T-cell infiltration in HCC tissue from patients treated with sitagliptin compared to that in patients not treated with it, suggesting that sitagliptin may improve the efficacy of PD1 blockade immunotherapy.
Among SGLT2 inhibitors, there has been some evidence regarding the antitumor effect of canagliflozin on HCC. In a report regarding the cytotoxic and antitumor effects of canagliflozin in combination with doxorubicin, canagliflozin significantly increased the cytotoxicity of doxorubicin in HepG2 cell line and enhanced the cellular uptake of doxorubicin by lowering the P-glycoprotein level.152 In vivo analysis using the xenograft mouse model also showed that canagliflozin significantly increased the antitumor effects of doxorubicin. The same authors also elucidated the effects of canagliflozin on HCC development under hypoxia and showed that canagliflozin significantly inhibited hypoxia-induced metastasis, angiogenesis, and metabolic reprogramming in HCC cell lines by targeting the Akt/mammalian target of rapamycin (mTOR) pathway and inhibiting the accumulation of hypoxia-inducible factor 1-α (HIF-1α) protein.153 Another basic study showed that canagliflozin inhibited cell proliferation in HepG2 cell lines and that incubation with canagliflozin followed by exposure to γ-radiation more potently inhibited cell growth and clonogenic survival by disabling signaling pathways that contribute to metabolic reprogramming and tumor progression, leading to radio resistance and treatment failure.154
Dyslipidemia and Liver Disease
Excess fat in the body is stored in hepatocytes in the form of lipid droplets covered with several structural proteins, which progress to chronic liver disease.155,156 NAFLD develops from abnormalities in lipid metabolism, including systemic lipolysis, increased liver free fatty acid (FFA) uptake and very low-density lipoprotein synthesis, and decreased FFA oxidation and triglyceride (TG) export.157,158 These alterations in lipid metabolism are associated with oxidative stress and liver inflammation in NAFLD patients, as well as the abnormal production of adipokines including resistin, visfatin, adiponectin, leptin, and retinol binding protein 4 (RBP4).159,160
Different lipid profiles, including TG and total cholesterol (TC) including low-density lipoprotein cholesterol (LDL-C) and high-density lipoprotein cholesterol (HDL-C) appear to have different risks of HCC development in patients with dyslipidemia. In the general population, low TC levels are strongly associated with a high risk of HCC development;161–165 for every 39 mg/Dl increase in TC, about 50% reduction in HCC occurrence was observed.163 Only a few studies have examined the association between other lipids and HCC, but low levels of TG and LDL-C are generally associated with a high risk of HCC occurrence, while the association with HDL-C levels was unknown.161,162 Furthermore, in patients with chronic liver disease, as in the general population, TC levels have been shown to be inversely associated with the risk of HCC occurrence, although relatively few reports have shown the association between other lipid profiles and HCC occurrence. In patients with viral hepatitis (including HBV and HCV), NAFLD, and cirrhosis, higher TC levels were associated with a decreased risk of HCC occurrence.162,166–169 The presence of chronic liver disease is associated with altered lipid metabolism, and serum TC levels in HCC patients were lower than healthy controls,170–173 while lower TC levels were associated with severity of liver disease.163
Suppression of HCC Occurrence of Dyslipidemia Drugs
Statins are one of the most important lipid-lowering agents, acting by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-CoA), the rate-limiting enzyme in cholesterol biosynthesis. Statins not only significantly reduce the risk of cardiovascular morbidity and mortality, but have recently been shown to be effective against NASH and have even been associated with reduced mortality from cancer.174 Several studies have reported that statin use decreased the risk of HCC development in patients with viral hepatitis and NAFLD.166–168,175–181 A recent meta-analysis reported that statin use in patients with chronic liver disease reduced the risk of HCC occurrence with a hazard ratio of 0.57.181 However, observational studies in the general population found no benefit of statin in preventing HCC occurrence,163,182 nor did an RCT of the statin use for the presentation of cardiovascular disease.183,184 Hypocholesterolemia in the natural course without statin use may be a potential risk factor for HCC development.161–165 Since lower cholesterol would result in less frequent statin use, caution should be exercised in assessing the beneficial effects of statins against HCC.
Antitumor Effects and Mechanism of Dyslipidemia Drugs on HCC
Although the mechanisms by which statins exert their antitumor effects on HCC are not yet fully elucidated, several reports have provided evidence for interrelated molecular pathways (Table 5). Statins inhibit cholesterol biogenesis by suppressing the conversion of HMG-CoA to mevalonic acid (MVA), as well as the production of derivatives of the MVA pathway, which has important effects on cell growth differentiation, membrane integrity, motility, signal transduction and other growth signals. Thus, statin administration produces antiproliferative, apoptosis-promoting, and anti-angiogenic effects.185,186
 |
Table 5 Experimental Studies on the Antitumor Effects of Statins Against Hepatocellular Carcinoma (HCC) |
Certain statins generally inhibit cancer cell growth through inhibition of HMG-CoA reductase, followed by reduction of isoprenoid. Cerivastatin had been shown to inhibit Ras- and Rho-mediated cell proliferation,187 while lovastatin-inhibited activation of the proteasome pathway and stabilizes p21 and p27.188 In the liver, simvastatin and lovastatin have also been shown to inhibit hepatic astrocyte proliferation and their collagen steady-state levels.189 An in vivo study showed that pravastatin inhibited p21ras isoprenylation in a rat model of N-nitrosomorpholine-induced hepatocarcinogenesis and the development of neoplastic liver nodule formation by inhibiting cell proliferation and inducing apoptosis.190 Conversely, lovastatin induced cell cycle arrest by inhibiting G1/S and G2/M transitions. Furthermore, induction of apoptosis is an important mechanism of tumor suppression of statins; simvastatin has been shown to induce Bax expression and inhibit Bcl-2 expression in several cancer cell lines including HCC,191 thereby promoting DNA fragmentation. Interestingly, statin-mediated apoptosis was observed only in cancer cells, while non-cancerous fibroblasts showed no signs of apoptosis. Another report showed that the antitumor effect of statins was associated with the overexpression of p53. For instance, the HuH-7 cell line, which overexpresses p53, was sensitized to statin-induced apoptosis by stable knockdown of endogenous p53.192 In addition to inhibiting cell proliferation and inducing apoptosis, angiogenesis was an important mechanism of antitumor effects. Several studies in various cancer types have shown that statins inhibit cell migration and proliferation.193,194 In HCC, simvastatin decreases tumor cell proliferation in a dose-dependent manner, impairs tumor cell adhesion to the endothelial cell monolayer, and decreases tumor cell invasion.195 However, there are few reports of statins inhibiting angiogenesis in HCC.
A recent study involving two in vivo rat models of HCC induced with DEN and hexachlorobenzene (HCB) reported that atorvastatin and simvastatin inhibit HCC growth by regulating TGF-β1 and thyroid hormones.196 There are also increasing number of reports that statins improve sorafenib resistance in HCC, and simvastatin inhibited the HIF-1α/peroxisome proliferator-activated receptor-γ (PPAR-γ)/pyruvate kinase 2 axis, resulting in decreased proliferation and increased apoptosis in HCC cells, which can re-sensitize HCC cells to sorafenib.197 According to another report, inactivation of hypoxia-induced Yes associate-protein by statins improved hypoxic resistance to sorafenib in HCC cells.198
Regarding dyslipidemia drugs other than statins, such as bezafibrate, these can potentiate the antitumor effects of PD-1 antibodies against other cancer types, including colorectal cancer, and regulate PPAR-γ coactivator 1α, a molecule that exhibits mitochondrial activity.199 However, there is virtually no evidence of antitumor effects against HCC, and further basic studies are needed.
Conclusion
Metabolic syndrome, including hypertension, T2D, dyslipidemia, and obesity, is associated with the development of HCC. In addition, these diseases can develop as adverse events during systemic therapy for advanced HCC. Interestingly, some metabolic syndrome medications show antitumor effects against HCC, while others do not. Our current review provides valuable evidence on the metabolic syndrome medications that may have an inhibitory effect on the development and progression of HCC in patients with chronic liver disease, including steatohepatitis, that may develop metabolic syndrome as a comorbidity. Various mechanisms have been reported for the antitumor effects of metabolic syndrome medications, not all of which have been elucidated in basic studies. Analysis of these mechanisms is beneficial for HCC patients with metabolic syndrome, and metabolic syndrome medications may contribute to potential therapeutic strategies.
Acknowledgments
We would like to thank Editage for English language editing.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN Estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
4. Zhang G, Li R, Deng Y, Zhao L. Conditional survival of patients with hepatocellular carcinoma: results from the surveillance, epidemiology, and end results registry. Expert Rev Gastroenterol Hepatol. 2018;12(5):515–523. doi:10.1080/17474124.2018.1453806
5. Altekruse SF, Henley SJ, Cucinelli JE, McGlynn KA. Changing hepatocellular carcinoma incidence and liver cancer mortality rates in the United States. Am J Gastroenterol. 2014;109(4):542–553. doi:10.1038/ajg.2014.11
6. Xu L, Kim Y, Spolverato G, Gani F, Pawlik TM. Racial disparities in treatment and survival of patients with hepatocellular carcinoma in the United States. Hepatobiliary Surg Nutr. 2016;5(1):43–52. doi:10.3978/j.issn.2304-3881.2015.08.05
7. Tseng TC, Liu CJ, Yang HC, et al. High levels of hepatitis B surface antigen increase risk of hepatocellular carcinoma in patients with low HBV load. Gastroenterology. 2012;142(5):1140–1149e3; quiz e13–4. doi:10.1053/j.gastro.2012.02.007
8. Ikeda K, Saitoh S, Arase Y, et al. Effect of interferon therapy on hepatocellular carcinogenesis in patients with chronic hepatitis type C: a long-term observation study of 1643 patients using statistical bias correction with proportional hazard analysis. Hepatology. 1999;29(4):1124–1130. doi:10.1002/hep.510290439
9. Carrat F, Fontaine H, Dorival C, et al. Clinical outcomes in patients with chronic hepatitis C after direct-acting antiviral treatment: a prospective cohort study. Lancet. 2019;393(10179):1453–1464. doi:10.1016/S0140-6736(18)32111-1
10. Cho HJ, Cheong JY. Role of immune cells in patients with hepatitis B Virus-related hepatocellular carcinoma. Int J Mol Sci. 2021;22(15):1. doi:10.3390/ijms22158011
11. Niu ZS, Niu XJ, Wang WH. Genetic alterations in hepatocellular carcinoma: an update. World J Gastroenterol. 2016;22(41):9069–9095. doi:10.3748/wjg.v22.i41.9069
12. Nagaoki Y, Hyogo H, Aikata H, et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol Res. 2012;42(4):368–375. doi:10.1111/j.1872-034X.2011.00929.x
13. Nishikawa H, Osaki Y. Non-B, non-C hepatocellular carcinoma (review). Int J Oncol. 2013;43(5):1333–1342. doi:10.3892/ijo.2013.2061
14. Zhang W, He H, Zang M, et al. Genetic features of aflatoxin-associated hepatocellular carcinoma. Gastroenterology. 2017;153(1):249–262 e2. doi:10.1053/j.gastro.2017.03.024
15. Huang DQ, Singal AG, Kono Y, Tan DJH, El-Serag HB, Loomba R. Changing global epidemiology of liver cancer from 2010 to 2019: NASH is the fastest growing cause of liver cancer. Cell Metab. 2022;34(7):969–977e2. doi:10.1016/j.cmet.2022.05.003
16. Oura K, Takuma K, Nakahara M, et al. Clinical characteristics of hepatocellular carcinoma for the past 15 years. J Kagawa Phys Assoc. 2021;57:10–19.
17. Kudo M, Kawamura Y, Hasegawa K, et al. Management of hepatocellular carcinoma in japan: JSH consensus statements and recommendations 2021 update. Liver Cancer. 2021;10(3):181–223. doi:10.1159/000514174
18. European Association for the Study of the Liver. Electronic address eee, European Association for the study of the L. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236. doi:10.1016/j.jhep.2018.03.019
19. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012;7(3):e33411. doi:10.1371/journal.pone.0033411
20. Kasuga M, Ueki K, Tajima N, et al. Report of the Japan Diabetes Society/Japanese Cancer Association Joint Committee on diabetes and cancer. Cancer Sci. 2013;104(7):965–976. doi:10.1111/cas.12203
21. Tadic M, Cuspidi C, Belyavskiy E, Grassi G. Intriguing relationship between antihypertensive therapy and cancer. Pharmacol Res. 2019;141:501–511. doi:10.1016/j.phrs.2019.01.037
22. Lee MMY, Docherty KF, Sattar N, et al. Renin-angiotensin system blockers, risk of SARS-CoV-2 infection and outcomes from CoViD-19: systematic review and meta-analysis. Eur Heart J Cardiovasc Pharmacother. 2022;8(2):165–178. doi:10.1093/ehjcvp/pvaa138
23. Morale MG, Tamura RE, Rubio IGS. Metformin and Cancer hallmarks: molecular mechanisms in thyroid, prostate and head and neck cancer models. Biomolecules. 2022;12(3):1. doi:10.3390/biom12030357
24. Ye J, Qi L, Chen K, et al. Metformin induces TPC-1 cell apoptosis through endoplasmic reticulum stress-associated pathways in vitro and in vivo. Int J Oncol. 2019;55(1):331–339. doi:10.3892/ijo.2019.4820
25. Amable G, Martinez-Leon E, Picco ME, et al. Metformin inhibits beta-catenin phosphorylation on Ser-552 through an AMPK/PI3K/Akt pathway in colorectal cancer cells. Int J Biochem Cell Biol. 2019;112:88–94. doi:10.1016/j.biocel.2019.05.004
26. Almaimani RA, Aslam A, Ahmad J, et al. In vivo and in vitro enhanced tumoricidal effects of metformin, active vitamin D3, and 5-fluorouracil triple therapy against colon cancer by modulating the PI3K/Akt/PTEN/mTOR network. Cancers. 2022;14(6):1. doi:10.3390/cancers14061538
27. Ren D, Qin G, Zhao J, et al. Metformin activates the STING/IRF3/IFN-beta pathway by inhibiting AKT phosphorylation in pancreatic cancer. Am J Cancer Res. 2020;10(9):2851–2864.
28. Tang G, Guo J, Zhu Y, et al. Metformin inhibits ovarian cancer via decreasing H3K27 trimethylation. Int J Oncol. 2018;52(6):1899–1911. doi:10.3892/ijo.2018.4343
29. Pfister D, Nunez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592(7854):450–456. doi:10.1038/s41586-021-03362-0
30. Pinter M, Pinato DJ, Ramadori P, Heikenwalder M. NASH and hepatocellular carcinoma: immunology and immunotherapy. Clin Cancer Res. 2022;2022. doi:10.1158/1078-0432.CCR-21-1258
31. Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH. The disease burden associated with overweight and obesity. JAMA. 1999;282(16):1523–1529. doi:10.1001/jama.282.16.1523
32. Zunica ERM, Heintz EC, Axelrod CL, Kirwan JP. Obesity Management in the primary prevention of hepatocellular carcinoma. Cancers. 2022;14(16). doi:10.3390/cancers14164051
33. Lauby-Secretan B, Scoccianti C, Loomis D, et al. Body fatness and cancer--viewpoint of the IARC working group. N Engl J Med. 2016;375(8):794–798. doi:10.1056/NEJMsr1606602
34. Godoy-Matos AF, Silva Junior WS, Valerio CM. NAFLD as a continuum: from obesity to metabolic syndrome and diabetes. Diabetol Metab Syndr. 2020;12:60. doi:10.1186/s13098-020-00570-y
35. Ye J, Wu Y, Li F, et al. Effect of orlistat on liver fat content in patients with nonalcoholic fatty liver disease with obesity: assessment using magnetic resonance imaging-derived proton density fat fraction. Therap Adv Gastroenterol. 2019;12:1756284819879047. doi:10.1177/1756284819879047
36. Kojima M, Takahashi H, Kuwashiro T, et al. Glucagon-like peptide-1 receptor agonist prevented the progression of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. Int J Mol Sci. 2020;21(16). doi:10.3390/ijms21165722
37. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. Hypertension. 2018;71(6):e13–e115. doi:10.1161/HYP.0000000000000065
38. Zhao YC, Zhao GJ, Chen Z, She ZG, Cai J, Li H. Nonalcoholic fatty liver disease: an emerging driver of hypertension. Hypertension. 2020;75(2):275–284. doi:10.1161/HYPERTENSIONAHA.119.13419
39. Lorbeer R, Bayerl C, Auweter S, et al. Association between MRI-derived hepatic fat fraction and blood pressure in participants without history of cardiovascular disease. J Hypertens. 2017;35(4):737–744. doi:10.1097/HJH.0000000000001245
40. Bonnet F, Gastaldelli A, Pihan-le Bars F, et al. Gamma-glutamyltransferase, fatty liver index and hepatic insulin resistance are associated with incident hypertension in two longitudinal studies. J Hypertens. 2017;35(3):493–500. doi:10.1097/HJH.0000000000001204
41. Zhou K, Cen J. The fatty liver index (FLI) and incident hypertension: a longitudinal study among Chinese population. Lipids Health Dis. 2018;17(1):214. doi:10.1186/s12944-018-0858-6
42. Sung KC, Wild SH, Byrne CD. Development of new fatty liver, or resolution of existing fatty liver, over five years of follow-up, and risk of incident hypertension. J Hepatol. 2014;60(5):1040–1045. doi:10.1016/j.jhep.2014.01.009
43. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol. 2018;68(2):335–352. doi:10.1016/j.jhep.2017.09.021
44. Oikonomou D, Georgiopoulos G, Katsi V, et al. Non-alcoholic fatty liver disease and hypertension: coprevalent or correlated? Eur J Gastroenterol Hepatol. 2018;30(9):979–985. doi:10.1097/MEG.0000000000001191
45. Houghton D, Zalewski P, Hallsworth K, et al. The degree of hepatic steatosis associates with impaired cardiac and autonomic function. J Hepatol. 2019;70(6):1203–1213. doi:10.1016/j.jhep.2019.01.035
46. Satou R, Penrose H, Navar LG. Inflammation as a regulator of the renin-angiotensin system and blood pressure. Curr Hypertens Rep. 2018;20(12):100. doi:10.1007/s11906-018-0900-0
47. Meex RCR, Watt MJ. Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol. 2017;13(9):509–520. doi:10.1038/nrendo.2017.56
48. Nunes KP, de Oliveira AA, Mowry FE, Biancardi VC. Targeting toll-like receptor 4 signalling pathways: can therapeutics pay the toll for hypertension? Br J Pharmacol. 2019;176(12):1864–1879. doi:10.1111/bph.14438
49. Sinn DH, Kang D, Jang HR, et al. Development of chronic kidney disease in patients with non-alcoholic fatty liver disease: a cohort study. J Hepatol. 2017;67(6):1274–1280. doi:10.1016/j.jhep.2017.08.024
50. Cheng JW, Zhu L, Gu MJ, Song ZM. Meta analysis of propranolol effects on gastrointestinal hemorrhage in cirrhotic patients. World J Gastroenterol. 2003;9(8):1836–1839. doi:10.3748/wjg.v9.i8.1836
51. Kim G, Kim J, Lim YL, Kim MY, Baik SK. Renin-angiotensin system inhibitors and fibrosis in chronic liver disease: a systematic review. Hepatol Int. 2016;10(5):819–828. doi:10.1007/s12072-016-9705-x
52. Yokohama S, Yoneda M, Haneda M, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40(5):1222–1225. doi:10.1002/hep.20420
53. Goh GB, Pagadala MR, Dasarathy J, et al. Renin-angiotensin system and fibrosis in non-alcoholic fatty liver disease. Liver Int. 2015;35(3):979–985. doi:10.1111/liv.12611
54. Barone M, Viggiani MT, Losurdo G, Principi M, Leo AD. Systematic review: renin-angiotensin system inhibitors in chemoprevention of hepatocellular carcinoma. World J Gastroenterol. 2019;25(20):2524–2538. doi:10.3748/wjg.v25.i20.2524
55. Kim KM, Roh JH, Lee S, Yoon JH. Do renin-angiotensin system inhibitors reduce risk for hepatocellular carcinoma?: a nationwide nested case-control study. Clin Res Hepatol Gastroenterol. 2021;45(4):101510. doi:10.1016/j.clinre.2020.07.015
56. Pinter M, Weinmann A, Worns MA, et al. Use of inhibitors of the renin-angiotensin system is associated with longer survival in patients with hepatocellular carcinoma. United European Gastroenterol J. 2017;5(7):987–996. doi:10.1177/2050640617695698
57. Facciorusso A, Del Prete V, Crucinio N, et al. Angiotensin receptor blockers improve survival outcomes after radiofrequency ablation in hepatocarcinoma patients. J Gastroenterol Hepatol. 2015;30(11):1643–1650. doi:10.1111/jgh.12988
58. Ho CM, Lee CH, Lee MC, et al. Comparative effectiveness of angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers in chemoprevention of hepatocellular carcinoma: a nationwide high-risk cohort study. BMC Cancer. 2018;18(1):401. doi:10.1186/s12885-018-4292-y
59. Hagberg KW, Sahasrabuddhe VV, McGlynn KA, Jick SS. Does Angiotensin-converting enzyme inhibitor and beta-blocker use reduce the risk of primary liver cancer? A case-control study using the U.K. clinical practice research datalink. Pharmacotherapy. 2016;36(2):187–195. doi:10.1002/phar.1704
60. Walker AJ, West J, Grainge MJ, Card TR. Angiotensin converting enzyme inhibitors and hepatocellular carcinoma incidence in the general practice research database. Cancer Causes Control. 2011;22(12):1743–1747. doi:10.1007/s10552-011-9837-1
61. Kaibori M, Ishizaki M, Matsui K, Kitade H, Matsui Y, Kwon AH. Evaluation of metabolic factors on the prognosis of patients undergoing resection of hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26(3):536–543. doi:10.1111/j.1440-1746.2010.06439.x
62. Yoshiji H, Noguchi R, Toyohara M, et al. Combination of vitamin K2 and angiotensin-converting enzyme inhibitor ameliorates cumulative recurrence of hepatocellular carcinoma. J Hepatol. 2009;51(2):315–321. doi:10.1016/j.jhep.2009.04.011
63. Yoshiji H, Noguchi R, Ikenaka Y, et al. Soluble VEGF receptor-2 may be a predictive marker of anti-angiogenic therapy with clinically available safe agents. Oncol Lett. 2011;2(1):69–73. doi:10.3892/ol.2010.196
64. Yoshiji H, Noguchi R, Ikenaka Y, et al. Combination of branched-chain amino acids and angiotensin-converting enzyme inhibitor suppresses the cumulative recurrence of hepatocellular carcinoma: a randomized control trial. Oncol Rep. 2011;26(6):1547–1553. doi:10.3892/or.2011.1433
65. Udumyan R, Montgomery S, Duberg AS, et al. Beta-adrenergic receptor blockers and liver cancer mortality in a national cohort of hepatocellular carcinoma patients. Scand J Gastroenterol. 2020;55(5):597–605. doi:10.1080/00365521.2020.1762919
66. Nkontchou G, Aout M, Mahmoudi A, et al. Effect of long-term propranolol treatment on hepatocellular carcinoma incidence in patients with HCV-associated cirrhosis. Cancer Prev Res. 2012;5(8):1007–1014. doi:10.1158/1940-6207.CAPR-11-0450
67. Suna N, Ozer Etik D, Ocal S, Selcuk H. Effect of propranolol treatment on the incidence of hepatocellular carcinoma in patients waiting for liver transplant with cirrhosis: a retrospective, surveillance study in a tertiary center. Exp Clin Transplant. 2019;17(5):632–637. doi:10.6002/ect.2018.0321
68. Chang PY, Chung CH, Chang WC, et al. The effect of propranolol on the prognosis of hepatocellular carcinoma: a nationwide population-based study. PLoS One. 2019;14(5):e0216828. doi:10.1371/journal.pone.0216828
69. Kim TW, Kim HJ, Chon CU, et al. Is there any vindication for low dose nonselective beta-blocker medication in patients with liver cirrhosis? Clin Mol Hepatol. 2012;18(2):203–212. doi:10.3350/cmh.2012.18.2.203
70. Oura K, Tadokoro T, Fujihara S, et al. Telmisartan inhibits hepatocellular carcinoma cell proliferation in vitro by inducing cell cycle arrest. Oncol Rep. 2017;38(5):2825–2835. doi:10.3892/or.2017.5977
71. Santhekadur PK, Akiel M, Emdad L, et al. Staphylococcal nuclease domain containing-1 (SND1) promotes migration and invasion via angiotensin II type 1 receptor (AT1R) and TGFbeta signaling. FEBS Open Bio. 2014;4:353–361. doi:10.1016/j.fob.2014.03.012
72. Cook JL, Zhang Z, Re RN. In vitro evidence for an intracellular site of angiotensin action. Circ Res. 2001;89(12):1138–1146. doi:10.1161/hh2401.101270
73. Fan F, Tian C, Tao L, et al. Candesartan attenuates angiogenesis in hepatocellular carcinoma via downregulating AT1R/VEGF pathway. Biomed Pharmacother. 2016;83:704–711. doi:10.1016/j.biopha.2016.07.039
74. Tamaki Y, Nakade Y, Yamauchi T, et al. Angiotensin II type 1 receptor antagonist prevents hepatic carcinoma in rats with nonalcoholic steatohepatitis. J Gastroenterol. 2013;48(4):491–503. doi:10.1007/s00535-012-0651-7
75. Yoshiji H, Yoshii J, Ikenaka Y, et al. Suppression of the renin-angiotensin system attenuates vascular endothelial growth factor-mediated tumor development and angiogenesis in murine hepatocellular carcinoma cells. Int J Oncol. 2002;20(6):1227–1231.
76. Yoshiji H, Kuriyama S, Kawata M, et al. The angiotensin-I-converting enzyme inhibitor perindopril suppresses tumor growth and angiogenesis: possible role of the vascular endothelial growth factor. Clin Cancer Res. 2001;7(4):1073–1078.
77. Yoshiji H, Noguchi R, Kaji K, et al. Attenuation of insulin-resistance-based hepatocarcinogenesis and angiogenesis by combined treatment with branched-chain amino acids and angiotensin-converting enzyme inhibitor in obese diabetic rats. J Gastroenterol. 2010;45(4):443–450. doi:10.1007/s00535-009-0158-z
78. Noguchi R, Yoshiji H, Kuriyama S, et al. Combination of interferon-beta and the angiotensin-converting enzyme inhibitor, perindopril, attenuates murine hepatocellular carcinoma development and angiogenesis. Clin Cancer Res. 2003;9(16 Pt 1):6038–6045.
79. Noguchi R, Yoshiji H, Ikenaka Y, et al. Dual blockade of angiotensin-II and aldosterone suppresses the progression of a non-diabetic rat model of steatohepatitis. Hepatol Res. 2013;43(7):765–774. doi:10.1111/hepr.12008
80. Saber S, Mahmoud AAA, Goda R, Helal NS, El-Ahwany E, Abdelghany RH. Perindopril, fosinopril and losartan inhibited the progression of diethylnitrosamine-induced hepatocellular carcinoma in mice via the inactivation of nuclear transcription factor kappa-B. Toxicol Lett. 2018;295:32–40. doi:10.1016/j.toxlet.2018.05.036
81. Saber S, Mahmoud A, Helal N, El-Ahwany E, Abdelghany R. Liver protective effects of renin-angiotensin system inhibition have no survival benefits in hepatocellular carcinoma induced by repetitive administration of diethylnitrosamine in mice. Open Access Maced J Med Sci. 2018;6(6):955–960. doi:10.3889/oamjms.2018.167
82. Mansour MA, Al-Ismaeel H, Al-Rikabi AC, Al-Shabanah OA. Comparison of angiotensin converting enzyme inhibitors and angiotensin II type 1 receptor blockade for the prevention of premalignant changes in the liver. Life Sci. 2011;89(5–6):188–194. doi:10.1016/j.lfs.2011.06.002
83. Nasr M, Selima E, Hamed O, Kazem A. Targeting different angiogenic pathways with combination of curcumin, leflunomide and perindopril inhibits diethylnitrosamine-induced hepatocellular carcinoma in mice. Eur J Pharmacol. 2014;723:267–275. doi:10.1016/j.ejphar.2013.11.022
84. Yanase K, Yoshiji H, Ikenaka Y, et al. Synergistic inhibition of hepatocellular carcinoma growth and hepatocarcinogenesis by combination of 5-fluorouracil and angiotensin-converting enzyme inhibitor via anti-angiogenic activities. Oncol Rep. 2007;17(2):441–446.
85. Otsuka M, Kato N, Shao RX, et al. Vitamin K2 inhibits the growth and invasiveness of hepatocellular carcinoma cells via protein kinase A activation. Hepatology. 2004;40(1):243–251. doi:10.1002/hep.20260
86. Kakizaki S, Sohara N, Sato K, et al. Preventive effects of vitamin K on recurrent disease in patients with hepatocellular carcinoma arising from hepatitis C viral infection. J Gastroenterol Hepatol. 2007;22(4):518–522. doi:10.1111/j.1440-1746.2007.04844.x
87. Yoshiji H, Kuriyama S, Noguchi R, et al. Amelioration of carcinogenesis and tumor growth in the rat liver by combination of vitamin K2 and angiotensin-converting enzyme inhibitor via anti-angiogenic activities. Oncol Rep. 2006;15(1):155–159.
88. Yoshiji H, Kuriyama S, Noguchi R, et al. Combination of vitamin K2 and the angiotensin-converting enzyme inhibitor, perindopril, attenuates the liver enzyme-altered preneoplastic lesions in rats via angiogenesis suppression. J Hepatol. 2005;42(5):687–693. doi:10.1016/j.jhep.2004.12.025
89. Yoshiji H, Noguchi R, Kuriyama S, Yoshii J, Ikenaka Y. Combination of interferon and angiotensin-converting enzyme inhibitor, perindopril, suppresses liver carcinogenesis and angiogenesis in mice. Oncol Rep. 2005;13(3):491–495.
90. Srivastava SP, Goodwin JE. Cancer biology and prevention in diabetes. Cells. 2020;9(6). doi:10.3390/cells9061380
91. Tanase DM, Gosav EM, Costea CF, et al. The Intricate relationship between Type 2 Diabetes Mellitus (T2DM), Insulin Resistance (IR), and Nonalcoholic Fatty Liver Disease (NAFLD). J Diabetes Res. 2020;2020:3920196. doi:10.1155/2020/3920196
92. Jarvis H, Craig D, Barker R, et al. Metabolic risk factors and incident advanced liver disease in non-alcoholic fatty liver disease (NAFLD): a systematic review and meta-analysis of population-based observational studies. PLoS Med. 2020;17(4):e1003100. doi:10.1371/journal.pmed.1003100
93. Pitisuttithum P, Chan WK, Piyachaturawat P, et al. Predictors of advanced fibrosis in elderly patients with biopsy-confirmed nonalcoholic fatty liver disease: the GOASIA study. BMC Gastroenterol. 2020;20(1):88. doi:10.1186/s12876-020-01240-z
94. Ballestri S, Zona S, Targher G, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31(5):936–944. doi:10.1111/jgh.13264
95. Brouwers M, Simons N, Stehouwer CDA, Isaacs A. Non-alcoholic fatty liver disease and cardiovascular disease: assessing the evidence for causality. Diabetologia. 2020;63(2):253–260. doi:10.1007/s00125-019-05024-3
96. Koo BK, Allison MA, Criqui MH, Denenberg JO, Wright CM. The association between liver fat and systemic calcified atherosclerosis. J Vasc Surg. 2020;71(1):204–211 e4. doi:10.1016/j.jvs.2019.03.044
97. Fujiwara N, Qian T, Koneru B, Hoshida Y. Omics-derived hepatocellular carcinoma risk biomarkers for precision care of chronic liver diseases. Hepatol Res. 2020;50(7):817–830. doi:10.1111/hepr.13506
98. Vigneri P, Frasca F, Sciacca L, Pandini G, Vigneri R. Diabetes and cancer. Endocr Relat Cancer. 2009;16(4):1103–1123. doi:10.1677/ERC-09-0087
99. Abudawood M. Diabetes and cancer: a comprehensive review. J Res Med Sci. 2019;24:94. doi:10.4103/jrms.JRMS_242_19
100. Shebl FM, Andreotti G, Rashid A, et al. Diabetes in relation to biliary tract cancer and stones: a population-based study in Shanghai, China. Br J Cancer. 2010;103(1):115–119. doi:10.1038/sj.bjc.6605706
101. Waters KM, Henderson BE, Stram DO, Wan P, Kolonel LN, Haiman CA. Association of diabetes with prostate cancer risk in the multiethnic cohort. Am J Epidemiol. 2009;169(8):937–945. doi:10.1093/aje/kwp003
102. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–1638. doi:10.1056/NEJMoa021423
103. Garcia-Jimenez C, Gutierrez-Salmeron M, Chocarro-Calvo A, Garcia-Martinez JM, Castano A, De la Vieja A. From obesity to diabetes and cancer: epidemiological links and role of therapies. Br J Cancer. 2016;114(7):716–722. doi:10.1038/bjc.2016.37
104. Gutierrez-Salmeron M, Chocarro-Calvo A, Garcia-Martinez JM, de la Vieja A, Garcia-Jimenez C. Epidemiological bases and molecular mechanisms linking obesity, diabetes, and cancer. Endocrinol Diabetes Nutr. 2017;64(2):109–117. doi:10.1016/j.endinu.2016.10.005
105. Ohkuma T, Peters SAE, Woodward M. Sex differences in the association between diabetes and cancer: a systematic review and meta-analysis of 121 cohorts including 20 million individuals and one million events. Diabetologia. 2018;61(10):2140–2154. doi:10.1007/s00125-018-4664-5
106. El-Serag HB, Tran T, Everhart JE. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology. 2004;126(2):460–468. doi:10.1053/j.gastro.2003.10.065
107. Shi T, Kobara H, Oura K, Masaki T. Mechanisms underlying hepatocellular carcinoma progression in patients with type 2 diabetes. J Hepatocell Carcinoma. 2021;8:45–55. doi:10.2147/JHC.S274933
108. Streba LA, Vere CC, Rogoveanu I, Streba CT. Nonalcoholic fatty liver disease, metabolic risk factors, and hepatocellular carcinoma: an open question. World J Gastroenterol. 2015;21(14):4103–4110. doi:10.3748/wjg.v21.i14.4103
109. Hamouda HA, Mansour SM, Elyamany MF. Vitamin D combined with pioglitazone mitigates type-2 diabetes-induced hepatic injury through targeting inflammation, apoptosis, and oxidative stress. Inflammation. 2022;45(1):156–171. doi:10.1007/s10753-021-01535-7
110. Zhang Y, Wang H, Zhang L, Yuan Y, Yu D. Codonopsis lanceolata polysaccharide CLPS alleviates high fat/high sucrose diet-induced insulin resistance via anti-oxidative stress. Int J Biol Macromol. 2020;145:944–949. doi:10.1016/j.ijbiomac.2019.09.185
111. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69(1):107–120. doi:10.1002/hep.30036
112. Gurung M, Li Z, You H, et al. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine. 2020;51:102590. doi:10.1016/j.ebiom.2019.11.051
113. Kawaguchi T, Nakano D, Okamura S, et al. Spontaneous regression of hepatocellular carcinoma with reduction in angiogenesis-related cytokines after treatment with sodium-glucose cotransporter 2 inhibitor in a cirrhotic patient with diabetes mellitus. Hepatol Res. 2019;49(4):479–486. doi:10.1111/hepr.13247
114. Yoo J, Jeong IK, Ahn KJ, Chung HY, Hwang YC. Fenofibrate, a PPARalpha agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism. 2021;120:154798. doi:10.1016/j.metabol.2021.154798
115. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009;52(9):1766–1777. doi:10.1007/s00125-009-1440-6
116. Hsieh MC, Lee TC, Cheng SM, Tu ST, Yen MH, Tseng CH. The influence of type 2 diabetes and glucose-lowering therapies on cancer risk in the Taiwanese. Exp Diabetes Res. 2012;2012:413782. doi:10.1155/2012/413782
117. Hemkens LG, Grouven U, Bender R, et al. Risk of malignancies in patients with diabetes treated with human insulin or insulin analogues: a cohort study. Diabetologia. 2009;52(9):1732–1744. doi:10.1007/s00125-009-1418-4
118. Jonasson JM, Ljung R, Talback M, Haglund B, Gudbjornsdottir S, Steineck G. Insulin glargine use and short-term incidence of malignancies-a population-based follow-up study in Sweden. Diabetologia. 2009;52(9):1745–1754. doi:10.1007/s00125-009-1444-2
119. Fagot JP, Blotiere PO, Ricordeau P, Weill A, Alla F, Allemand H. Does insulin glargine increase the risk of cancer compared with other basal insulins?: a French nationwide cohort study based on national administrative databases. Diabetes Care. 2013;36(2):294–301. doi:10.2337/dc12-0506
120. But A, De Bruin ML, Bazelier MT, et al. Cancer risk among insulin users: comparing analogues with human insulin in the CARING five-country cohort study. Diabetologia. 2017;60(9):1691–1703. doi:10.1007/s00125-017-4312-5
121. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi:10.1136/bmj.38415.708634.F7
122. Donadon V, Balbi M, Mas MD, Casarin P, Zanette G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. 2010;30(5):750–758. doi:10.1111/j.1478-3231.2010.02223.x
123. Hassan MM, Curley SA, Li D, et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer. 2010;116(8):1938–1946. doi:10.1002/cncr.24982
124. Lai SW, Chen PC, Liao KF, Muo CH, Lin CC, Sung FC. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: a population-based cohort study. Am J Gastroenterol. 2012;107(1):46–52. doi:10.1038/ajg.2011.384
125. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108(6):881–91; quiz 892. doi:10.1038/ajg.2013.5
126. Zhou YY, Zhu GQ, Wang Y, et al. Systematic review with network meta-analysis: statins and risk of hepatocellular carcinoma. Oncotarget. 2016;7(16):21753–21762. doi:10.18632/oncotarget.7832
127. Tsilidis KK, Capothanassi D, Allen NE, et al. Metformin does not affect cancer risk: a cohort study in the U.K. Clinical practice research datalink analyzed like an intention-to-treat trial. Diabetes Care. 2014;37(9):2522–2532. doi:10.2337/dc14-0584
128. Florentin M, Kostapanos MS, Papazafiropoulou AK. Role of dipeptidyl peptidase 4 inhibitors in the new era of antidiabetic treatment. World J Diabetes. 2022;13(2):85–96. doi:10.4239/wjd.v13.i2.85
129. Yin R, Xu Y, Wang X, Yang L, Zhao D. Role of dipeptidyl peptidase 4 inhibitors in antidiabetic treatment. Molecules. 2022;27(10). doi:10.3390/molecules27103055
130. Nagel AK, Ahmed-Sarwar N, Werner PM, Cipriano GC, Van Manen RP, Brown JE. Dipeptidyl peptidase-4 inhibitor-associated pancreatic carcinoma: a review of the FAERS database. Ann Pharmacother. 2016;50(1):27–31. doi:10.1177/1060028015610123
131. Zhao M, Chen J, Yuan Y, et al. Dipeptidyl peptidase-4 inhibitors and cancer risk in patients with type 2 diabetes: a meta-analysis of randomized clinical trials. Sci Rep. 2017;7(1):8273. doi:10.1038/s41598-017-07921-2
132. Overbeek JA, Bakker M, van der Heijden A, van Herk-sukel MPP, Herings RMC, Nijpels G. Risk of dipeptidyl peptidase-4 (DPP-4) inhibitors on site-specific cancer: a systematic review and meta-analysis. Diabetes Metab Res Rev. 2018;34(5):e3004. doi:10.1002/dmrr.3004
133. Hsu WH, Sue SP, Liang HL, et al. Dipeptidyl peptidase 4 inhibitors decrease the risk of hepatocellular carcinoma in patients with chronic hepatitis c infection and type 2 diabetes mellitus: a nationwide study in Taiwan. Front Public Health. 2021;9:711723. doi:10.3389/fpubh.2021.711723
134. Yen FS, Wei JC, Yip HT, Hwu CM, Hou MC, Hsu CC. Dipeptidyl peptidase-4 inhibitors may accelerate cirrhosis decompensation in patients with diabetes and liver cirrhosis: a nationwide population-based cohort study in Taiwan. Hepatol Int. 2021;15(1):179–190. doi:10.1007/s12072-020-10122-1
135. Zou H, Zhou B, Xu G. SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovasc Diabetol. 2017;16(1):65. doi:10.1186/s12933-017-0547-1
136. Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8(8):495–502. doi:10.1038/nrendo.2011.243
137. Tang H, Dai Q, Shi W, Zhai S, Song Y, Han J. SGLT2 inhibitors and risk of cancer in type 2 diabetes: a systematic review and meta-analysis of randomised controlled trials. Diabetologia. 2017;60(10):1862–1872. doi:10.1007/s00125-017-4370-8
138. Dicembrini I, Nreu B, Mannucci E, Monami M. Sodium-glucose co-transporter-2 (SGLT-2) inhibitors and cancer: a meta-analysis of randomized controlled trials. Diabetes Obes Metab. 2019;21(8):1871–1877. doi:10.1111/dom.13745
139. Kuchay MS, Krishan S, Mishra SK, et al. Effect of empagliflozin on liver fat in patients with type 2 diabetes and nonalcoholic fatty liver disease: a randomized controlled trial (E-LIFT trial). Diabetes Care. 2018;41(8):1801–1808. doi:10.2337/dc18-0165
140. Ito D, Shimizu S, Inoue K, et al. Comparison of ipragliflozin and pioglitazone effects on nonalcoholic fatty liver disease in patients with type 2 diabetes: a randomized, 24-week, open-label, active-controlled Trial. Diabetes Care. 2017;40(10):1364–1372. doi:10.2337/dc17-0518
141. Shibuya T, Fushimi N, Kawai M, et al. Luseogliflozin improves liver fat deposition compared to metformin in type 2 diabetes patients with non-alcoholic fatty liver disease: a prospective randomized controlled pilot study. Diabetes Obes Metab. 2018;20(2):438–442. doi:10.1111/dom.13061
142. Abd El-Fattah EE, Zakaria AY. Metformin modulate immune fitness in hepatocellular carcinoma: molecular and cellular approach. Int Immunopharmacol. 2022;109:108889. doi:10.1016/j.intimp.2022.108889
143. Vacante F, Senesi P, Montesano A, Paini S, Luzi L, Terruzzi I. Metformin counteracts HCC progression and metastasis enhancing KLF6/p21 expression and downregulating the IGF axis. Int J Endocrinol. 2019;2019:7570146. doi:10.1155/2019/7570146
144. Sun R, Zhai R, Ma C, Miao W. Combination of aloin and metformin enhances the antitumor effect by inhibiting the growth and invasion and inducing apoptosis and autophagy in hepatocellular carcinoma through PI3K/AKT/mTOR pathway. Cancer Med. 2020;9(3):1141–1151. doi:10.1002/cam4.2723
145. Bhat M, Yanagiya A, Graber T, et al. Metformin requires 4E-BPs to induce apoptosis and repress translation of Mcl-1 in hepatocellular carcinoma cells. Oncotarget. 2017;8(31):50542–50556. doi:10.18632/oncotarget.10671
146. Sun Y, Tao C, Huang X, et al. Metformin induces apoptosis of human hepatocellular carcinoma HepG2 cells by activating an AMPK/p53/miR-23a/FOXA1 pathway. Onco Targets Ther. 2016;9:2845–2853. doi:10.2147/OTT.S99770
147. Miyoshi H, Kato K, Iwama H, et al. Effect of the anti-diabetic drug metformin in hepatocellular carcinoma in vitro and in vivo. Int J Oncol. 2014;45(1):322–332. doi:10.3892/ijo.2014.2419
148. Fujita K, Iwama H, Miyoshi H, et al. Diabetes mellitus and metformin in hepatocellular carcinoma. World J Gastroenterol. 2016;22(27):6100–6113. doi:10.3748/wjg.v22.i27.6100
149. Wabitsch S, McCallen JD, Kamenyeva O, et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. 2022;77(3):748–760. doi:10.1016/j.jhep.2022.03.010
150. Nishina S, Yamauchi A, Kawaguchi T, et al. Dipeptidyl peptidase 4 inhibitors reduce hepatocellular carcinoma by activating lymphocyte chemotaxis in mice. Cell Mol Gastroenterol Hepatol. 2019;7(1):115–134. doi:10.1016/j.jcmgh.2018.08.008
151. Huang XY, Zhang PF, Wei CY, et al. Circular RNA circMET drives immunosuppression and anti-PD1 therapy resistance in hepatocellular carcinoma via the miR-30-5p/snail/DPP4 axis. Mol Cancer. 2020;19(1):92. doi:10.1186/s12943-020-01213-6
152. Zhong J, Sun P, Xu N, et al. Canagliflozin inhibits p-gp function and early autophagy and improves the sensitivity to the antitumor effect of doxorubicin. Biochem Pharmacol. 2020;175:113856. doi:10.1016/j.bcp.2020.113856
153. Luo J, Sun P, Zhang X, et al. Canagliflozin modulates hypoxia-induced metastasis, angiogenesis and glycolysis by decreasing HIF-1alpha protein synthesis via AKT/mTOR pathway. Int J Mol Sci. 2021;22(24). doi:10.3390/ijms222413336
154. Abdel-Rafei MK, Thabet NM, Rashed LA, Moustafa EM. Canagliflozin, a SGLT-2 inhibitor, relieves ER stress, modulates autophagy and induces apoptosis in irradiated HepG2 cells: signal transduction between PI3K/AKT/GSK-3beta/mTOR and Wnt/beta-catenin pathways; in vitro. J Cancer Res Ther. 2021;17(6):1404–1418. doi:10.4103/jcrt.JCRT_963_19
155. Carr RM, Ahima RS. Pathophysiology of lipid droplet proteins in liver diseases. Exp Cell Res. 2016;340(2):187–192. doi:10.1016/j.yexcr.2015.10.021
156. Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: getting to the root of fatty liver. Hepatology. 2015;62(3):964–967. doi:10.1002/hep.27839
157. Almeda-Valdes P, Altamirano-Barrera A, Mendez-Sanchez N. Insights in non-alcoholic fatty liver disease pathophysiology with lipidomic analyses. Ann Hepatol. 2015;14(4):567–569.
158. Ress C, Kaser S. Mechanisms of intrahepatic triglyceride accumulation. World J Gastroenterol. 2016;22(4):1664–1673. doi:10.3748/wjg.v22.i4.1664
159. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1062–1079. doi:10.1016/j.metabol.2015.11.006
160. Polyzos SA, Aronis KN, Kountouras J, Raptis DD, Vasiloglou MF, Mantzoros CS. Circulating leptin in non-alcoholic fatty liver disease: a systematic review and meta-analysis. Diabetologia. 2016;59(1):30–43. doi:10.1007/s00125-015-3769-3
161. Haggstrom C, Jonsson H, Bjorge T, et al. Linear age-course effects on the associations between body mass index, triglycerides, and female breast and male liver cancer risk: an internal replication study of 800,000 individuals. Int J Cancer. 2020;146(1):58–67. doi:10.1002/ijc.32240
162. Cho Y, Cho EJ, Yoo JJ, et al. Association between lipid profiles and the incidence of hepatocellular carcinoma: a nationwide population-based study. Cancers. 2021;13(7). doi:10.3390/cancers13071599
163. Yi SW, Kim SH, Han KJ, Yi JJ, Ohrr H. Higher cholesterol levels, not statin use, are associated with a lower risk of hepatocellular carcinoma. Br J Cancer. 2020;122(5):630–633. doi:10.1038/s41416-019-0691-3
164. Sinn DH, Kang D, Cho SJ, et al. Risk of hepatocellular carcinoma in individuals without traditional risk factors: development and validation of a novel risk score. Int J Epidemiol. 2020;49(5):1562–1571. doi:10.1093/ije/dyaa089
165. Nderitu P, Bosco C, Garmo H, et al. The association between individual metabolic syndrome components, primary liver cancer and cirrhosis: a study in the Swedish AMORIS cohort. Int J Cancer. 2017;141(6):1148–1160. doi:10.1002/ijc.30818
166. Lee TY, Wu JC, Yu SH, Lin JT, Wu MS, Wu CY. The occurrence of hepatocellular carcinoma in different risk stratifications of clinically noncirrhotic nonalcoholic fatty liver disease. Int J Cancer. 2017;141(7):1307–1314. doi:10.1002/ijc.30784
167. Goh MJ, Sinn DH, Kim S, et al. Statin use and the risk of hepatocellular carcinoma in patients with chronic hepatitis B. Hepatology. 2020;71(6):2023–2032. doi:10.1002/hep.30973
168. Wu CY, Lin JT, Ho HJ, et al. Association of nucleos(t)ide analogue therapy with reduced risk of hepatocellular carcinoma in patients with chronic hepatitis B: a nationwide cohort study. Gastroenterology. 2014;147(1):143–151 e5. doi:10.1053/j.gastro.2014.03.048
169. Tan Y, Zhang X, Zhang W, et al. The influence of metabolic syndrome on the risk of hepatocellular carcinoma in patients with chronic hepatitis B infection in Mainland China. Cancer Epidemiol Biomarkers Prev. 2019;28(12):2038–2046. doi:10.1158/1055-9965.EPI-19-0303
170. Arain SQ, Talpur FN, Channa NA, Ali MS, Afridi HI. Serum lipid profile as a marker of liver impairment in hepatitis B cirrhosis patients. Lipids Health Dis. 2017;16(1):51. doi:10.1186/s12944-017-0437-2
171. Chrostek L, Supronowicz L, Panasiuk A, Cylwik B, Gruszewska E, Flisiak R. The effect of the severity of liver cirrhosis on the level of lipids and lipoproteins. Clin Exp Med. 2014;14(4):417–421. doi:10.1007/s10238-013-0262-5
172. Bassani L, Fernandes SA, Raimundo FV, Harter DL, Gonzalez MC, Marroni CA. LIPID PROFILE OF CIRRHOTIC PATIENTS AND ITS ASSOCIATION WITH PROGNOSTIC SCORES: a cross-sectional study. Arq Gastroenterol. 2015;52(3):210–215. doi:10.1590/S0004-28032015000300011
173. Tauseef A, Zafar M, Rashid B, et al. Correlation of fasting lipid profile in patients with chronic liver disease: a descriptive cross-sectional study in tertiary care hospital. Cureus. 2020;12(10):e11019. doi:10.7759/cureus.11019
174. Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2012;367(19):1792–1802. doi:10.1056/NEJMoa1201735
175. Tsan YT, Lee CH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis B virus infection. J Clin Oncol. 2012;30(6):623–630. doi:10.1200/JCO.2011.36.0917
176. Hsiang JC, Wong GL, Tse YK, Wong VW, Yip TC, Chan HL. Statin and the risk of hepatocellular carcinoma and death in a hospital-based hepatitis B-infected population: a propensity score landmark analysis. J Hepatol. 2015;63(5):1190–1197. doi:10.1016/j.jhep.2015.07.009
177. Tsan YT, Lee CH, Ho WC, Lin MH, Wang JD, Chen PC. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol. 2013;31(12):1514–1521. doi:10.1200/JCO.2012.44.6831
178. Butt AA, Yan P, Bonilla H, et al. Effect of addition of statins to antiviral therapy in hepatitis C virus-infected persons: results from ERCHIVES. Hepatology. 2015;62(2):365–374. doi:10.1002/hep.27835
179. Mohanty A, Tate JP, Garcia-Tsao G. Statins are associated with a decreased risk of decompensation and death in veterans with hepatitis C-related compensated cirrhosis. Gastroenterology. 2016;150(2):430–40 e1. doi:10.1053/j.gastro.2015.10.007
180. Li X, Sheng L, Liu L, Hu Y, Chen Y, Lou L. Statin and the risk of hepatocellular carcinoma in patients with hepatitis B virus or hepatitis C virus infection: a meta-analysis. BMC Gastroenterol. 2020;20(1):98. doi:10.1186/s12876-020-01222-1
181. Wong YJ, Qiu TY, Ng GK, Zheng Q, Teo EK. Efficacy and safety of statin for hepatocellular carcinoma prevention among chronic liver disease patients: a systematic review and meta-analysis. J Clin Gastroenterol. 2021;55(7):615–623. doi:10.1097/MCG.0000000000001478
182. Friedman GD, Achacoso N, Fireman B, Habel LA. Statins and reduced risk of liver cancer: evidence for confounding. J Natl Cancer Inst. 2016;108(10). doi:10.1093/jnci/djw109
183. Jeong GH, Lee KH, Kim JY, et al. Effect of statin on cancer incidence: an umbrella systematic review and meta-analysis. J Clin Med. 2019;8(6). doi:10.3390/jcm8060819
184. Emberson JR, Kearney PM; Cholesterol Treatment Trialists C. Lack of effect of lowering LDL cholesterol on cancer: meta-analysis of individual data from 175,000 people in 27 randomised trials of statin therapy. PLoS One. 2012;7(1):e29849. doi:10.1371/journal.pone.0029849
185. Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6(7):541–555. doi:10.1038/nrd2221
186. Alipour Talesh G, Trezeguet V, Merched A. Hepatocellular carcinoma and statins. Biochemistry. 2020;59(37):3393–3400. doi:10.1021/acs.biochem.0c00476
187. Sassano A, Katsoulidis E, Antico G, et al. Suppressive effects of statins on acute promyelocytic leukemia cells. Cancer Res. 2007;67(9):4524–4532. doi:10.1158/0008-5472.CAN-06-3686
188. Rao S, Porter DC, Chen X, Herliczek T, Lowe M, Keyomarsi K. Lovastatin-mediated G1 arrest is through inhibition of the proteasome, independent of hydroxymethyl glutaryl-CoA reductase. Proc Natl Acad Sci USA. 1999;96(14):7797–7802. doi:10.1073/pnas.96.14.7797
189. Rombouts K, Kisanga E, Hellemans K, Wielant A, Schuppan D, Geerts A. Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol. 2003;38(5):564–572. doi:10.1016/s0168-8278(03
190. Tatsuta M, Iishi H, Baba M, et al. Suppression by pravastatin, an inhibitor of p21ras isoprenylation, of hepatocarcinogenesis induced by N-nitrosomorpholine in Sprague-Dawley rats. Br J Cancer. 1998;77(4):581–587. doi:10.1038/bjc.1998.94
191. Spampanato C, De Maria S, Sarnataro M, et al. Simvastatin inhibits cancer cell growth by inducing apoptosis correlated to activation of Bax and down-regulation of BCL-2 gene expression. Int J Oncol. 2012;40(4):935–941. doi:10.3892/ijo.2011.1273
192. Kah J, Wustenberg A, Keller AD, et al. Selective induction of apoptosis by HMG-CoA reductase inhibitors in hepatoma cells and dependence on p53 expression. Oncol Rep. 2012;28(3):1077–1083. doi:10.3892/or.2012.1860
193. Weis M, Heeschen C, Glassford AJ, Cooke JP. Statins have biphasic effects on angiogenesis. Circulation. 2002;105(6):739–745. doi:10.1161/hc0602.103393
194. Bayat N, Izadpanah R, Ebrahimi-Barough S, et al. The Anti-angiogenic effect of atorvastatin in glioblastoma spheroids tumor cultured in fibrin gel: in 3D in vitro model. Asian Pac J Cancer Prev. 2018;19(9):2553–2560. doi:10.22034/APJCP.2018.19.9.2553
195. Relja B, Meder F, Wang M, et al. Simvastatin modulates the adhesion and growth of hepatocellular carcinoma cells via decrease of integrin expression and ROCK. Int J Oncol. 2011;38(3):879–885. doi:10.3892/ijo.2010.892
196. Ridruejo E, Romero-Caimi G, Obregon MJ, Kleiman de Pisarev D, Alvarez L. Potential molecular targets of statins in the prevention of hepatocarcinogenesis. Ann Hepatol. 2018;17(3):490–500. doi:10.5604/01.3001.0011.7394
197. Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1alpha/PPAR-gamma/PKM2-mediated glycolysis. J Exp Clin Cancer Res. 2020;39(1):24. doi:10.1186/s13046-020-1528-x
198. Zhou TY, Zhuang LH, Hu Y, et al. Inactivation of hypoxia-induced YAP by statins overcomes hypoxic resistance tosorafenib in hepatocellular carcinoma cells. Sci Rep. 2016;6:30483. doi:10.1038/srep30483
199. Chamoto K, Chowdhury PS, Kumar A, et al. Mitochondrial activation chemicals synergize with surface receptor PD-1 blockade for T cell-dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114(5):E761–E770. doi:10.1073/pnas.1620433114
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.