")
Back to Journals » OncoTargets and Therapy » Volume 13
Antitumor Actions of Intratumoral Delivery of Membrane-Fused Mitochondria in a Mouse Model of Triple-Negative Breast Cancers
Authors Chang JC , Chang HS, Wu YC, Cheng WL, Lin TT, Chang HJ, Chen ST, Liu CS
Received 11 November 2019
Accepted for publication 23 April 2020
Published 9 June 2020 Volume 2020:13 Pages 5241—5255
DOI https://doi.org/10.2147/OTT.S238143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
Jui-Chih Chang,1 Huei-Shin Chang,1 Yao-Chung Wu,2 Wen-Ling Cheng,1 Ta-Tsung Lin,1 Hui-Ju Chang,1 Shou-Tung Chen,3,4 Chin-San Liu1,5,6
1Vascular and Genomic Center, Changhua Christian Hospital, Changhua 50094, Taiwan; 2Department of Medicine, College of Medicine, China Medical University, Taichung 40447, Taiwan; 3Comprehensive Breast Cancer Center, Changhua Christian Hospital, Changhua 50094, Taiwan; 4Department of Medical Research, Changhua Christian Hospital, Changhua 50094, Taiwan; 5Department of Neurology, Changhua Christian Hospital, Changhua 50094, Taiwan; 6School of Chinese Medicine, Graduate Institute of Chinese Medicine, Graduate Institute of Integrated Medicine, College of Chinese Medicine, Research Center for Chinese Medicine and Acupuncture, China Medical University, Taichung 40447, Taiwan
Correspondence: Chin-San Liu
Changhua Christian Hospital, Department of Neurology, 135 Nanhsiao Street, Changhua 50094, Taiwan, Republic of China
Tel +886 4 7238595 Ext 4751
Fax +886-4-7238595 Ext 4063
Email [email protected]
Shou-Tung Chen
Changhua Christian Hospital, Comprehensive Breast Cancer Center, 135 Nanhsiao Street, Changhua 50094, Taiwan, Republic of China
Tel +886-4-7238595 Ext 4751
Fax +886-4-7238595 Ext 4063
Email [email protected]
Background: The transfer of whole mitochondria has been demonstrated to be beneficial for treating breast cancer because it induces apoptosis and drug sensitivity; however, in vivo evidence of this benefit remains scant. The present study compared the transplantation of mitochondria with instinctive (Mito) and membrane-fused morphologies induced by Pep-1 conjugation (P-Mito) using a mouse model of triple-negative breast cancers.
Materials and Methods: Mice with advanced severe immunodeficiency received orthotopic implantation of MDA-MB-231 human breast cancer cells followed by transplants of 5-bromo-2ʹ-deoxyuridine (BrdU)-labeled Mito or P-Mito (200 μg [10 μg/μL]) through intratumoral injection at multiple points once a week for 4 weeks.
Results: After 1 month of consecutive treatment, 8.2% and 14.2% of the BrdU-labeled mitochondria were preserved in tumors of the Mito and P-Mito groups, respectively. Both Pep-1 and P-Mito treatments reduced tumor weight (21.7% ± 2.43% vs 40.6% ± 2.28%) and led to marked inhibition of Ki67 staining and angiogenesis. However, only the P-Mito group exhibited obvious necrosis and DNA fragmentation accompanied by an altered tumor microenvironment, which included reduced oxidative stress and size of cancer-associated fibroblast populations and enhanced immune cell infiltration. Transmission electron microscopy images further revealed an elongated network of perinuclear mitochondria fused with a few peripheral mitochondria in the nonnecrotic area in the P-Mito group as well as increases in mitochondrial fusion proteins and parkin compared with mitochondrial fission proteins.
Conclusion: In this study, the results of mitochondrial transplantation emphasized that the facilitation of mitochondrial fusion is a critical regulator in breast cancer therapy.
Keywords: mitochondrial transplantation, animal model of breast cancer, Pep-1, MDA-MB-231, tumor growth, mitochondrial fusion
Introduction
Mitochondria play a diverse role in the regulation of breast cancer progression. The mechanism of action of several anticancer drugs involves the destruction of breast cancer cells through mitochondrial dysfunction–induced apoptosis. However, mitochondrial dysfunction also accelerates epithelial-to-mesenchymal transition, thereby promoting deterioration, metastasis, invasion, and drug resistance in cancer cells.1,2 Studies have shown that in contrast to the poorly aggressive and noninvasive cell line MCF-7, triple-negative and highly deteriorated breast cancer cells have a metabolic demand that is more reliant on glycolysis than on aerobic respiration, and their mitochondrial dysfunction is mostly accompanied by abnormal structure and defects.3 Thus, targeting of the mitochondrial function in certain types of breast cancer could be a therapeutic approach;4 however, mitochondria-implicated mechanisms and regulation are still largely elusive and susceptible to unstable microenvironments because of the different structures and accessibilities of nutrients and the heterogeneity of cell populations.4,5
Mitochondria are extremely dynamic organelles. The regulation of their mitochondrial fission/fusion machinery determines their dynamic balance for maintaining cell survival, cell death, and cellular metabolic homeostasis6 in several types of cancer.7 Mitochondrial function is influenced by the coordinative regulation of mitochondrial fusion and fission proteins as well as the triggered removal of damaged mitochondria through autophagy (mitophagy) to control mitochondrial integrity.6 Through the adjustment of dynamic morphological changes between fragmentation and elongation, cancer cells can adapt to changes in nutrient availability and metabolic demand, regulated by the microenvironment, to support cell viability and evade the activation of apoptosis.8 Today, the regulatory role of mitochondrial dynamics in breast cancer therapy remains uncertain and controversial. The mitochondrial fusion process, which is related to the switch from glycolysis toward oxidative phosphorylation supports cancer cell survival under stress conditions;3,9 however, the induction of mitochondrial fusion can inhibit breast cancer cell proliferation.10 In recent studies, pharmacological inactivation of dynamin-related protein 1 (Drp1), the main mitochondrial fission mediator, has been shown to abrogate mitophagy, metabolic reprogramming, and cancer cell viability in breast cancer.11–13 Therefore, compared with various other approaches for targeting cancer metabolism, the manipulation of mitochondrial dynamics may be a more potent strategy for breast cancer therapy.14
Mitochondrial transplantation has been proven to rescue mitochondrial function and reduce oxidative stress in mitochondrial diseases15–18 as well as provide anti–breast-cancer benefits, including reduced cell proliferation, oxidative stress, and drug resistance.19 Although in vivo animal experiments have not produced substantial evidence, our previous study showed that the benefit of mitochondrial transplantation in a breast cancer cell model could be related to the induction of mitochondrial fusion with a consequential decrease in mitochondrial fission.19 Moreover, compared with the transplantation of naked mitochondria (Mito), the transplantation of Pep-1-conjugated mitochondria (P-Mito) increased the internalization and morphological fusion of mitochondria, which may explain the superior performance of P-Mito treatment compared with Mito treatment not only in breast cancer cell lines19 but also in mitochondrial disease cells.16,17 The cell-penetrating peptide Pep-1, apart from being a cargo carrier of intracellular delivery,20 can induce the fusion of two lipid bilayers of isolated mitochondria in a dose-dependent manner,17 which may provide a promising route for exploring the role of the dynamic heterogeneity of mitochondria in breast cancer treatment.
This consecutive study compared the treatment outcomes of Mito and P-Mito transplantation through intratumor injections in mice with advanced severe immunodeficiency (ASID) that had received orthotopic implantation of MDA-MB-231 human breast cancer cells. We found that both P-Mito and Pep-1 treatments reduced tumor growth; however, marked reduction in cancer necrosis was only observed in the P-Mito group, as was an altered tumor microenvironment (TME), which included reduced oxidative stress and cancer-associated fibroblast (CAF) populations and enhanced immune cell infiltration. Furthermore, elongated morphology of the mitochondrial network was only found in solid tumors treated with P-Mito, and mitochondrial dynamics were concomitantly remodeled through increases in mitochondrial fusion proteins (Mitofusin-2 [MFN2] and optic atrophy 1 [OPA1]) and parkin. This resulted in decreased Drp1 phosphorylation and total protein levels compared with those in other groups. Additionally, the mitochondrial cristae density did not differ between treatments, which indicated that the P-Mito-induced machinery of mitochondrial fusion was independent of mitochondrial energy production. Compared with previous in vitro studies, the present study demonstrated a unique facilitation of mitochondrial fusion through the transplantation of P-Mito compared with Mito in mitochondrial transplant breast cancer therapy, and validated that mitochondrial fusion is a critical regulator in breast cancer treatment in vivo.
Materials and Methods
Cell Culture
The human breast adenocarcinoma cell line MDA-MB-231 was purchased from the Bioresource Collection and Research Center (Food Industry Research and Development Institute, Taiwan). Cells were cultured in Leibovitz’s L-15 medium (Gibco/BRL, Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco/BRL) and 1% penicillin-streptomycin (100 U/L penicillin G sodium and 100 mg/L streptomycin sulfate; Gibco/BRL) and incubated at 37°C without CO2. The cells were detached from tissue culture flasks through digestion with 0.25% trypsin and 0.02% EDTA (Gibco/BRL).
Mitochondrial Labeling, Isolation, and Conjugation
Prior to the isolation of donor mitochondria, in vivo bromodeoxyuridine or 5-bromo-2′-deoxyuridine (BrdU; Sigma-Aldrich, St. Louis, MA, USA) labeling of mitochondrial DNA (mtDNA) was performed for the long-term tracking of transplanted mitochondria.21 Male C57BL/6 mice, the mitochondrial donors, received an intraperitoneal injection of BrdU at 0.5 mg/g of body weight. After 36 h of BrdU incorporation, the mice were sacrificed; their livers were collected to isolate mitochondria following the procedure described in a previous study.22 In brief, fresh liver tissue (approximate weight = 0.25 g) was cut into small pieces of less than 5 mm. Samples were transferred to a 2-mL tube containing 5 mL of ice-cold homogenizing buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris-HCl, and 1 mM EDTA; pH = 7.4). The tissues were homogenized using a Precellys homogenizer (Bertin Technologies, Saint-Quentin-en-Yvelines Cedex, France) to dissociate the mitochondria at a speed of 5000 rpm for 15 s at 4°C with a 2-s rest interval for homogenization. Proteinase K (0.05 mg/mL; Sigma-Aldrich) was added to the homogenate and mixed well through inversion. Subsequently, the homogenate was incubated on ice for 10 min. Nuclear debris was removed through centrifugation of the solution at 750× g for 10 min, and the collected supernatants were added to freshly prepared bovine serum albumin (BSA; 0.05 mg/mL; Sigma-Aldrich) and mixed well by inversion. The homogenates were centrifuged at 3000× g for 10 min, and the supernatants were filtered sequentially using 20-µm- and 5-µm-mesh filters (PluriStrainer; pluriSelect, Leipzig, Germany) on ice. After centrifugation at 9000× g for 10 min at 4°C, mitochondrial pellets were collected and placed in an ice-cold MiR05 respiration buffer (0.5 mM EGTA, 3 mM MgCl2, 60 mM lactobionic acid, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM D-sucrose, and 0.1% w/v BSA) for use. The concentration of freshly isolated mitochondria was determined using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL, USA). Isolated mitochondria were conjugated with Pep-1 peptide (1:0.57 weight ratio; Anaspec, San Jose, CA, USA), which was P-Mito or not (Mito), through gentle mixing and then allowed to stand at room temperature for 10 min to form the P-Mito complex, as described in previous studies.15,16 Mito and P-Mito were applied immediately after they had been prepared.
Mouse Orthotropic Breast Tumor Model and Mitochondrial Transplantation
All animal treatment protocols were approved by the Institutional Animal Care and Use Committee of Changhua Christian Hospital (CCH-AE-105-011) and followed the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines for mice including appropriate preoperative/postoperative care, asepsis, minimum suffering and sacrifice. Seven-week-old female ASID mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/YckNarl) were purchased from the National Laboratory Animal Center (NLAC), NARLabs, Taiwan. The mice were housed in ventilated cages with autoclaved chow, water, and bedding and maintained in an appropriate environment with a 12-h light/dark cycle, temperature of approximately 26°C, and relative humidity of 40–60% with ad libitum access to food and water. Following 1 week of acclimation, the mice were ready for cancer cell transplantation. In total, 1 × 106 MDA-MB231 cells were suspended in 100 μL of Dulbecco’s phosphate-buffered saline (PBS) and injected unilaterally into the right-side fat pads of the #4 mammary glands of the 8-week-old female ASID mice. For the mitochondrial transplantation study, intratumoral multipoint injection of Mito or P-Mito (200 μg suspended in 20 μL of MiR05 respiration buffer) for four once-weekly treatments was administered to each mouse starting from when their tumors became palpable (1.5–2 mm in diameter). A control cohort received injections of the vehicle (MiR05 respiration buffer) and Pep-1 (1.9 mM). Each group had at least six transplant replicates. After 25 days of treatment, tumorigenesis was evaluated by analyzing the volumes of subcutaneous breast tumors in the mice by using a 3D laser scanning device (TumorImager; Biopticon Corporation, Princeton, NJ, USA) and measuring tumor weights.
In vivo Tracking of BrdU-Labeled Mitochondria
Imaging and quantitative determination of BrdU-labeled mitochondria in breast tumors were performed using a tomographic near-infrared imaging system (FMT; PerkinElmer, Waltham, MA, USA). BrdU antibody without BSA and azide at a concentration of 1 mg/mL (BU20a clone; MyBioSource, San Diego, CA, USA) was fluorescently conjugated using an Alexa Fluor® 647 Protein Labeling Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions, and 62.5 μM conjugated BrdU antibody was diluted with 100 μL of saline and injected into the tail veins of the mice. An FMT 2500 fluorescence molecular tomography imaging system (Perkin Elmer Inc., Norwalk, CT, USA) was used to perform whole-body imaging of the mice 24 h after they were injected with the BrdU antibody. The animals were anesthetized using a mixture of isoflurane and oxygen, placed in an imaging cassette, and maintained under anesthesia in a thermally regulated (37°C) chamber during the imaging session. To quantify the concentration of the BrdU antibody, the known concentration of the conjugated antibody (2.5 μM) was used as a reference dye for simultaneous detection.
Flow Cytometry
To estimate the expression of BrdU-labeled mitochondria after their in vivo transplantation, the mitochondrion-binding affinity of the Alexa Fluor 647 dye-labeling monoclonal BrdU antibody (Clone: BU20a; MyBioSource, San Diego, CA, USA), used for in vivo tracking, was analyzed in vitro through the co-staining of mitochondria with mitochondrial dye (MitoTracker Green, 500 nM) using flow cytometry. To avoid the error of unsaturated staining caused by the low antibody concentration, we diluted the antibody at 1/100 (32.5μ) according to the concentration recommended for manual flow cytometry (100–500 dilution). First, 0.6 mg of BrdU-labeled mitochondria were suspended in 1 mL of PBS containing MitoTracker dye and the antibody, and then they were incubated in darkness at room temperature for 1 h. After mitochondria had been washed, their fluorescent expression was analyzed using flow cytometry through the collection of 10,000 events.
Histology Analysis
The animals were sacrificed when their tumors reached 10% of their body weight, and primary tumors were harvested, embedded in paraffin, sectioned, and subjected to hematoxylin-eosin (H&E) staining or antibodies against BrdU (1:100 dilution; MyBioSource), Ki67 (1:100 dilution; Abcam, Cambridge, MA, USA), CD31 (1:300 dilution; Abcam), alpha-smooth muscle actin (α-SMA, 1:300 dilution; Abcam), CD4 (1:400 dilution; Abcam), or CD8 (1:100 dilution, Abcam). The color was developed using a horseradish peroxidase (HRP)-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, Bar Harbour, ME, USA) and a 3,3ʹ-diaminobenzidine tetrahydrochloride (DAB; Abcam) substrate. Images on slides were visualized using an Olympus BX40 light microscope equipped with a computer-controlled digital camera (DP71; Olympus Center Valley, PA, USA).
Terminal Transferase-Mediated dUTP Nick End Labeling (TUNEL) Staining
Tumor tissues were embedded in paraffin and sectioned (thickness = 5 µm). Tissues were stained using a DeadEnd™ Colorimetric Apoptosis Detection System (Promega, Madison, WI, USA) to detect DNA fragmentation in the tumor sections in accordance with the manufacturer’s protocol. In brief, the slides were incubated with equilibration buffer for 10 min before being treated with 20 µg/mL of proteinase K solution for 10 min. After being washed in PBS, sections were incubated with terminal deoxynucleotidyl transferase (TdT) enzyme at 37°C for 1 h in a humidified chamber to incorporate biotinylated nucleotides at the 3′ hydroxyl ends of the DNA. The slides were incubated in HRP-labeled streptavidin to bind the biotinylated nucleotides and then detected with a stable DAB chromagen. Dark-brown cytoplasmic staining was employed to identify apoptotic cells.
Immunohistochemistry of 8-Hydroxydeoxyguanosine
Mice were sacrificed when their tumors reached 10% of their body weight. Primary tumors and organs were harvested, embedded in paraffin, sectioned, and stained with 8-hydroxydeoxyguanosine (8-OHdG) antibody (Abcam, 1:200 dilution) overnight at 4°C. After being washed in PBS, slides were incubated for secondary antibody for 1 h and examined using an Olympus optical microscope. The immunohistochemical staining results in each group were quantified by three observers.
Transmission Electron Microscopy
Tumor tissues were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH = 7.2) for 4 h at room temperature, dehydrated in graded solutions of ethanol for 20 min, and then infiltrated and embedded into resin for ultrathin 70-nm sections to be cut using a Leica EM UC7 ultramicrotome (Leica Microsystems, Wetzlar, Germany). The sections were assessed using transmission electron microscopy (TEM; Hitachi H-7000, Japan). The diameters of the mitochondria were calculated using ImageJ 1.41 software (ImageJ, NIH, USA) to measure the population of the longest axis (maximum diameter) within 0.5–1 μm or greater than 1 μm. The mitochondrial cristae density of each mitochondrion was calculated by normalizing the cristae numbers to the maximum diameter of the mitochondria (mitochondrial length).
mtDNA Copy Numbers
An aliquot of 50 ng of DNA was subjected to a quantitative polymerase chain reaction (PCR) using a LightCycler-FastStart DNA Master SYBR Green I kit (Roche Applied Science, Indianapolis, IN, USA). DNA fragments of the nicotinamide adenine dinucleotide dehydrogenase subunit 1 (ND1) gene (mt-DNA–encoded) and the βactin gene (nuclear DNA–encoded, used as an internal control) were amplified with specific primer pairs. The relative mtDNA copy number was determined by normalizing the crossing points on the quantitative PCR curves between the ND1 and βactin genes by using RelQuant software (Roche Applied Science).
Western Blot Analysis
Randomly selected tumor tissues were homogenized using a Precellys homogenizer (Bertin Technologies) in Pierce IP lysis buffer (Thermo Fisher Scientific) containing proteinase inhibitors (Thermo Fisher Scientific). The protein concentrations were determined using a Pierce BCA Protein Assay kit. Subsequently, the samples were denatured, and approximately 30 μg of the protein was subjected to electrophoresis on 10% or 12% Mini-PROTEAN TGX stain-free gels (Bio-Rad, Hercules, CA, USA). Next, the proteins were transferred to polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo RTA Mini PVDF transfer kit (Bio-Rad) and Trans-Blot Turbo Blotting System (Bio-Rad) in accordance with the manufacturer’s instructions. The membranes were then blocked in BlockPRO™ Blocking Buffer (Visual Protein Biotechnology, Taipei, Taiwan) for 1 h at room temperature and probed with monoclonal antibodies of mitochondrial dynamic proteins, including OPA1 (Novus Biologicals, Littleton, CO, USA), MFN2 (Sigma-Aldrich), Drp1 (Novus Biologicals), Phospho-DRP1 (Ser616) (Cell Signaling Technology, Danvers, MA, USA), PTEN-induced putative kinase 1 (PINK1; Abcam), and parkin (Abcam). The membranes probed with HRP-conjugated secondary antibodies were visualized using a chemiluminescence Western HRP substrate (EMD Millipore, Billerica, MA, USA) and quantified using an image acquisition system (FUSION SL; Viber Lourmat, Marne-la-Vallee, France).
Statistical Analysis
Data were analyzed and visualized using GraphPad Prism version 4.0 (GraphPad Software, San Diego, CA, USA). Data were expressed as the mean ± standard deviation or mean ± standard error of the mean (tumor volume/weight). Statistical differences were determined using Student’s t tests and a two-way analysis of variance; p < 0.05 was considered statistically significant. Each experiment was performed independently a minimum of three times.
Results
Presence of BrdU-Labeled Mitochondria in Tumors After Intratumoral Transplantation
To confirm the intactness of the intratumorally injected mitochondria, a morphological examination using TEM analysis was performed on liver-derived mitochondria immediately after they were isolated prior to Pep-1 conjugation (P-Mito). TEM images showed that most mitochondria had intact mitochondrial architecture of the inner membrane with cristae and crista junctions as well as highly variable crista morphologies (Figure 1A). Moreover, more than 95% of the isolated mitochondria successfully expressed MitoTracker dye-positive signals (Q1 area), but only 49.9 ± 2.05% coexpressed a BrdU signal (Q2 area). This indicated that the mitochondrion-binding affinity of BrdU antibody under a working concentration of 32.5 μM was 15.3 μg of mitochondria/μM BrdU (Figure 1B). Imaging and quantitation of BrdU-labeled mitochondria in breast tumors were performed 25 days after transplantation (Figure 1C and D). In vivo fluorescence tomography imaging revealed the presence of BrdU-positive signals in both mitochondria-treated groups compared with the sham group 36 h after 62.5 μM BrdU antibody labeled with the dye Alexa Fluor 647 was injected into tail veins (Figure 1C). The concentration of the remaining dye was converted using the total fluorescent expression of a known concentration of the dye (Ref. dye). The BrdU concentration that remained in the P-Mito group (0.7 ± 0.06 μM) was significantly higher than that in the Mito group (0.4 ± 0.05 μM; Figure 1C, right panel). Additionally, the mitochondrial amount was calculated using the binding efficiency of the BrdU antibody to BrdU-labeled mitochondria (15.3 μg mitochondria/μM BrdU) tested in vitro, as previously mentioned (b). The Mito and P-Mito groups had 65.6 ± 8.09 μg and 113.9 ± 5.58 μg of exogenous mitochondria, respectively, which are equivalent to 8.2% ± 1.01% and 14.2% ± 0.07% of the total transplant amount (approximately 800 μg of mitochondria), respectively (Figure 1C). The mitochondrial internalization was further confirmed by anti-BrdU immunohistochemical staining and it consistently revealed that BrdU expression (brown color) was more obvious in the P-Mito group than in the Mito group (Figure 1D). Moreover, to observe the internal state of the injected mitochondria, a between-group comparison of BrdU distribution was performed on magnified images of breast tumor cells, with sharp cytoplasmic boundaries marked as dashed circles in the lower panel of Figure 1D. The comparison revealed that the BrdU expression in the tumor cells’ cytoplasm was higher in the P-Mito group than in the Mito group, whereas the Mito group had a relatively obvious expression in cellular stroma (indicated by arrows, Figure 1D). This meant that the injected mitochondria in the P-Mito group had higher permeability because more mitochondria remained inside the cells.
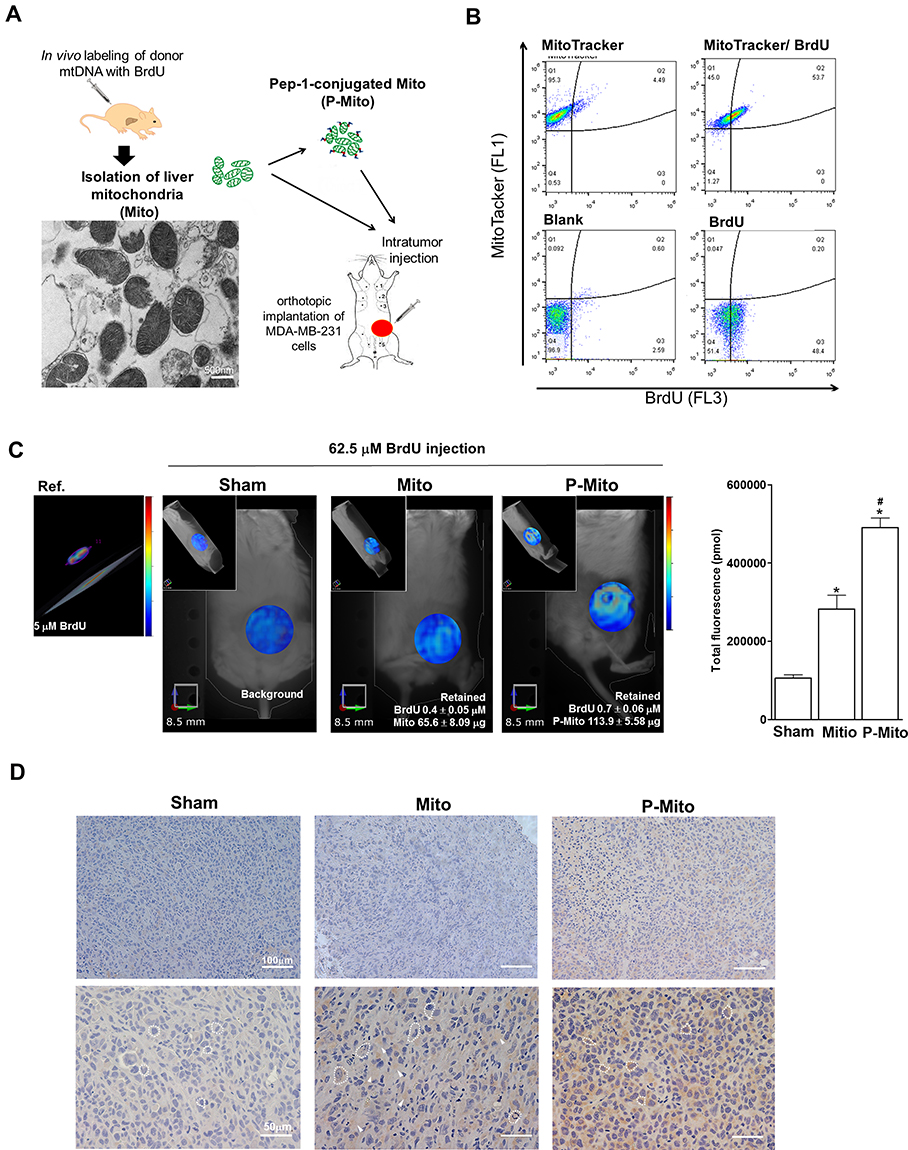 |
Figure 1 Presence of foreign mitochondria revealed by mitochondrial DNA (mtDNA) labeling with (BrdU) in ASID mice bearing breast tumors after 25 days of consecutive treatment. (A) Isolation of donor mitochondria with BrdU-labeled mtDNA from livers was performed after 36 h of in vivo BrdU incorporation; the mitochondrial morphology after fresh isolation was analyzed using TEM before Pep-1 conjugation (P-Mito). (B) The antibody binding affinity of BrdU was analyzed by co-staining isolated mitochondria (1 mg/mL) with BrdU antibody tagged with Alexa Fluor 647 dye (32.5 μM) and MitoTracker Green (500 nM), a mitochondria-specific dye, using flow cytometry. (C) Imaging and quantification of transplanted mitochondria were performed through in vivo fluorescence tomography of mice 36 h after they had received tail vein injections of 62.5 μM BrdU antibody labeled with Alexa Fluor 647 dye. The concentration of the remaining dye and mitochondrial amount were converted using the total fluorescent intensity of a known concentration of the reference dye (Ref., 2.5 μM) and calculated using the binding efficiency of the BrdU antibody with BrdU-labeled mitochondria (15.3 μg mitochondria/μM BrdU), respectively. (D) Immunohistochemical staining of thin slices with the anti-BrdU antibody demonstrated the performance of BrdU-positive expression (brown color) in solid tumors. *p < 0.05, difference relative to the sham group. #p < 0.05, difference between the Mito and P-Mito groups. |
Transplantation of Pep-1-Conjugated Mitochondria Arrested Tumor Growth, Enhanced Cell Necrosis, and Altered the Tumor Microenvironment in Breast Cancer
The ASID mice with MDA-MB-231–cell tumors in their unilateral mammary fat pads were treated once a week for 4 weeks with an MiR05 respiration buffer (sham group) or a buffer containing 1.9 mM Pep-1, 200 μg of Mito, or 200 μg of P-Mito through multipoint intratumoral injections. Photographs of the excised tumors provided clear evidence of the remarkable tumor-inhibiting effect of P-Mito and Pep-1 transplantation (Figure 2A). Quantification of tumor growth was assessed by measuring the weight of the breast tumor tissues, as shown in Figure 2B, and by determining the subcutaneous tumor volume using a noninvasive 3D scanner (Figure 2C). Pep-1 and P-Mito moderately reduced primary tumor weights by 21.7% ± 2.43% and 40.6% ± 2.28%, respectively, compared with the control group after 25 days of constitutive treatment (Figure 2B); however, simultaneous reductions in tumor volume with time were only observed in the P-Mito group after 16 (reduction of 32.0% ± 0.58%) and 20 days of treatment (reduction of 40.2% ± 0.05%; Figure 2C). No differences in tumor growth were observed between the sham and Mito groups compared with the control group (Figure 2).
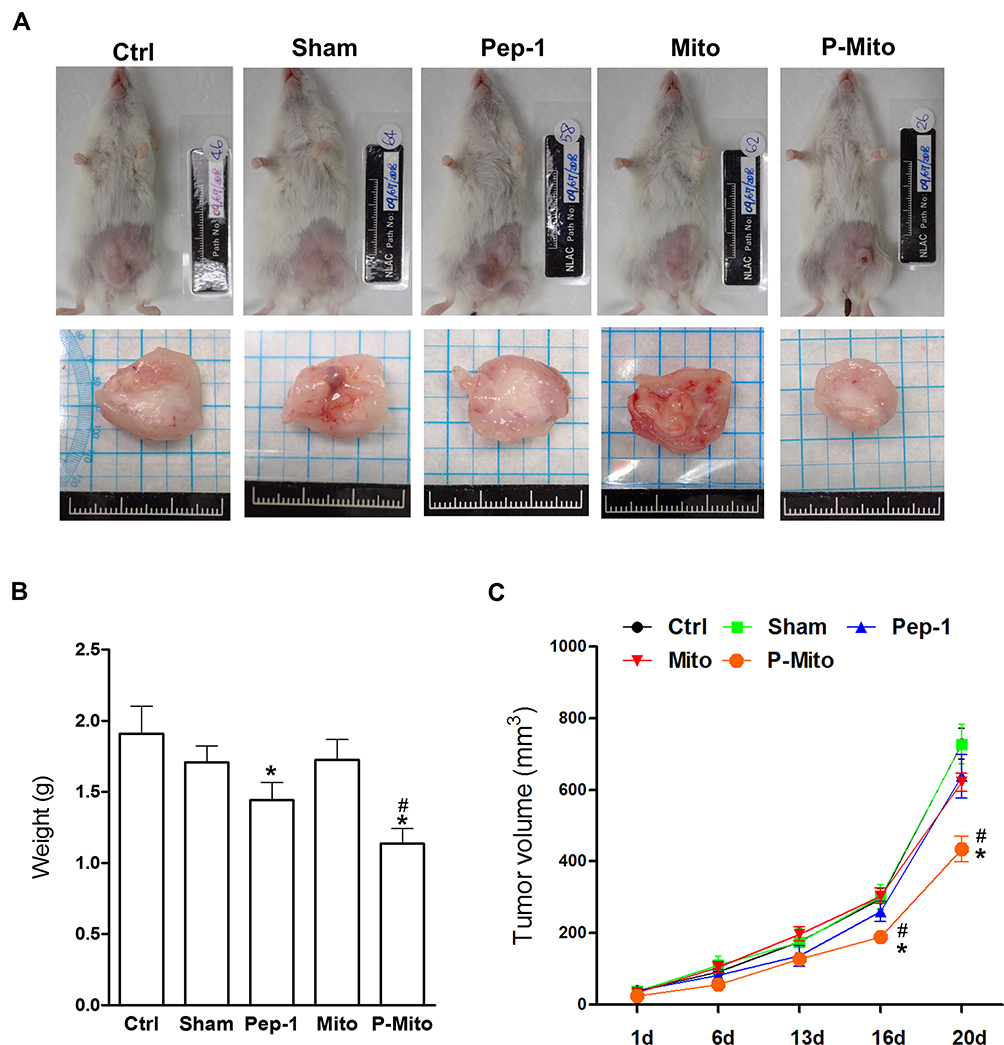 |
Figure 2 Growth of breast tumors in the mouse orthotopic breast cancer model after consecutive treatments. (A) Breast tumors in each group were imaged 1 month after treatments. (B) Changes in tumor weight were evaluated after 25 days. (C) The tumor volumes over time in each group were evaluated after treatments. *p < 0.05, difference relative to the control group. #p < 0.05, difference relative to the Pep-1 group. |
Immunohistochemical determination of Ki67 (cell proliferation), CD31 (endothelial cells), and TUNEL (apoptosis; Figure 3A) further revealed the stronger tumor growth inhibition ability in the Pep-1 and P-Mito groups compared with the control group, including significant inhibition of Ki67-positive cells and vascular density (Figure 3B). However, only the P-Mito group exhibited a significant increase in the total number of TUNEL-positive apoptotic cells compared with the Pep-1 group (Figure 3B). This finding implied that the antitumor efficacy of P-Mito transplantation was greater than that of Pep-1 treatment in the breast cancer mouse model. Similarly, H&E staining confirmed that the prominent induction of necrosis (N; necrotic area) was only observed in the P-Mito group (Figure 4A). P-Mito also reduced the size of major CAF populations, as indicated by the results of immunohistochemistry (IHC) of alpha-smooth muscle actin (alpha-SMA) in the TME. Moreover, IHC revealed the rare expression of CD4+ and CD8+ T cells in the tumors because of the xenograft model of immunodeficient mice; however, a distinction between the control and P-Mito groups showed that P-Mito still noticeably increased the migration of CD4+ and CD8+ T cells into the tumor margin (as indicated by the arrow in Figure 4B), which promoted stromal infiltration. The TME being formed by P-Mito was further supported by an assessment of oxidative stress using IHC of 8-hydroxydeoxyguanosine (8-OHdG), a biomarker of oxidative DNA damage (Figure 5), because the increase in oxidative stress also translated to the TME. In the P-Mito group, 8-OHdG expression was significantly decreased compared with the control group (Figure 5A and B). This also meant that P-Mito-induced necrosis occurred independently of oxidative damage.
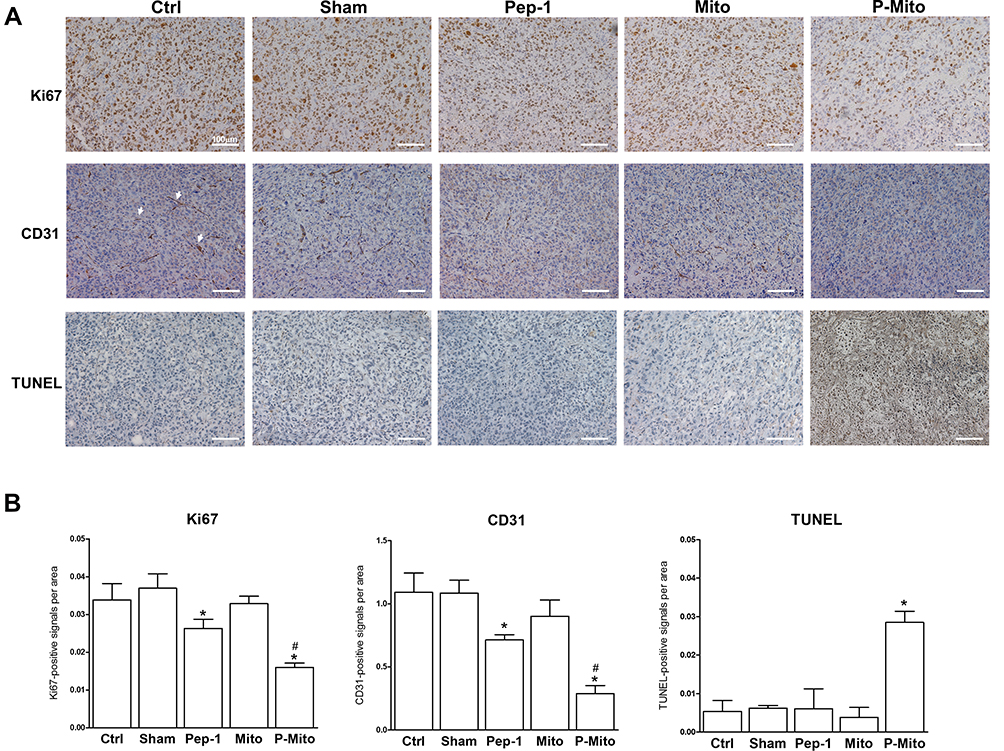 |
Figure 3 Analysis of cell proliferation, angiogenesis, and apoptosis following 1 month of treatment in mouse breast tumors. (A) Imaging and (B) quantification of tumors from different treatment groups through immunohistochemical analysis for the expression of Ki-67 (cell proliferation), CD31 (microvessel density), and TUNEL (apoptosis). *p < 0.05, difference compared with the control group. #p < 0.05, difference compared with the Pep-1 group. |
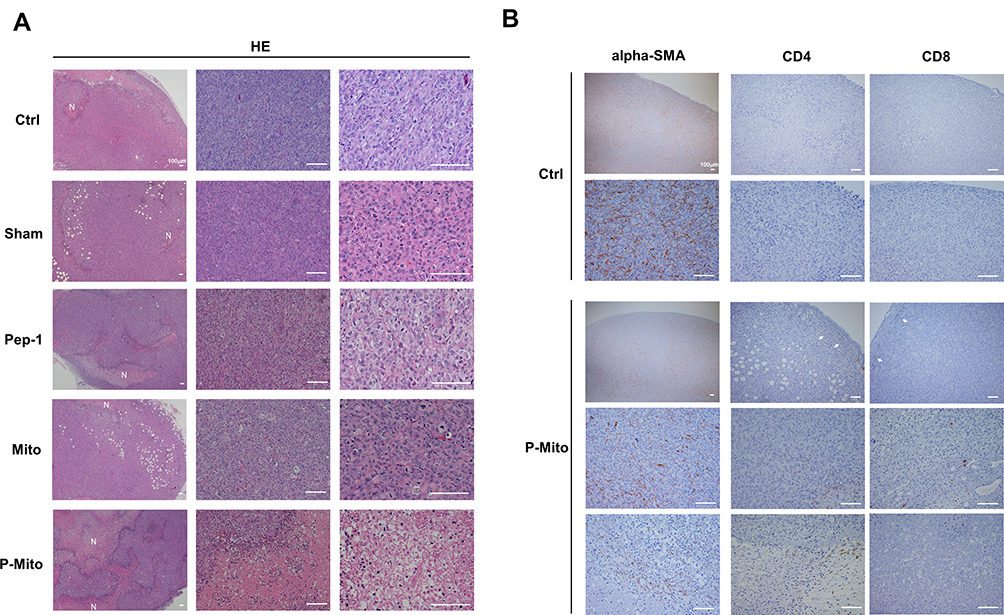 |
Figure 4 Histological evaluation of tumor morphology, fibroblasts, and immune cells in the tumor microenvironment (TME). (A) Representative H&E staining revealing tumor necrosis (necrotic area, N) with different magnifications. (B) Alpha-smooth muscle actin (alpha-SMA) and CD4, CD8-positive immunohistochemical staining revealed the population change of CAFs and the distribution of tumor-infiltrating T lymphocytes in P-Mito-treated breast tumors compared with the control (Ctrl). CD4+ and CD8+ T lymphocytes were observed at the intratumor and tumor margin, as indicated by the arrows. |
 |
Figure 5 Expression of 8-hydroxydeoxyguanosine (8-OHdG) in breast tumors. (A) Immunohistochemical staining was used to evaluate and (B) quantify the levels of 8-OHdG, a biomarker of oxidative damage in breast tumors. *p < 0.05, difference compared with the other groups. |
Pep-1-Conjugated Mitochondrial Transplantation Induced Mitochondrial Fusion (but Not Mitochondrial Cristae Density or mtDNA Copy Number) in Breast Tumors
A comparison using TEM of the mitochondrial ultrastructures and morphologies of the nonnecrotic area in each group (Figure 6) revealed that a dramatic elongated phenotype was only present in the P-Mito group (Figure 6A). Furthermore, the population with mitochondrial lengths > 1.0 μm in diameter was 34% ± 9.7% of the total mitochondrial population (Figure 6B). These mitochondria exhibited relatively complete structures of the crista junction connecting the mitochondrial inner-boundary membrane through a tubular structure of relatively uniform size (indicated by the arrows in Figure 6A). Furthermore, the cristae were more regular and arranged in a discernible pattern in the P-Mito group compared with those in the other groups, which exhibited partial disorganization of the cristae, as indicated by the star in Figure 6A. Moreover, despite a relatively large mitochondria size (diameter) in the P-Mito group, no statistical differences in cristae intensity (measured using the ratio of the cristae number to the mitochondrial diameter (Figure 6C) and mtDNA copy number (Figure 6D) were found between groups in terms of the common population of mitochondria).
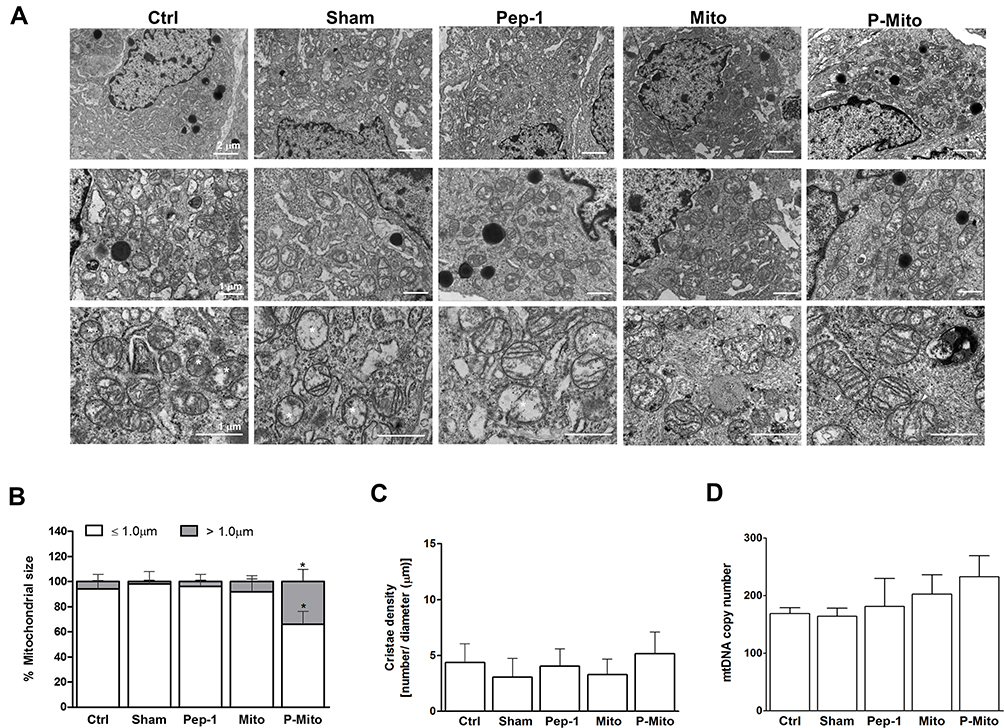 |
Figure 6 Ultrastructural analysis of mitochondria and mitochondrial DNA (mtDNA) copy number in mouse breast tumors. (A) Ultrastructural morphology was visualized using TEM with different magnifications. (B) Mitochondrial size was evaluated by measuring the proration of the mitochondrial maximal diameter and classifying mitochondria as ≤1 μm or >1 μm. (C) The cristae density (number of cristae/mitochondrial length) was quantitatively analyzed simultaneously. (D) The performance of the mitochondrial DNA (mtDNA) copy number against the control β-actin in breast tumors was determined through a PCR. *p < 0.05, difference compared with the other groups. |
Western blot analysis of the mitochondrial dynamics and mitophagy-related proteins revealed that only P-Mito treatment simultaneously enhanced the levels of the mitochondrial fusion proteins OPA1 and MFN2 as well as the levels of the mitophagy protein PINK1-short form (PINK1-S, cleavage form of PINK1) and parkin compared with the control group; however, a consistent decrease in the mitochondrial fission protein Drp-1 and its phosphorylated form at serine 616 (p-Drp1) was observed for both P-Mito and Pep-1 treatment (Figure 7A and B). The opposite effect of significantly downregulated PINK1-S and parkin was found in the Pep-1 group compared with the P-Mito group (Figure 7B). This reflected the facilitation of mitophagy, and increased mitochondrial integrity was indicated by decreased full-length PINK accumulation (enhanced PINK-1 cleavage) in the outer membrane of damaged mitochondria in the P-Mito group compared with the Pep-1 group. As shown in the TEM images in Figure 6, the mitochondrial morphology in the P-Mito group elongated to maintain mitochondrial integrity. Thus, the regulatory benefit of P-Mito was related to the simultaneous induction of mitochondrial fusion and mitophagy for improving mitochondrial homeostasis rather than just the reduction of Drp1 mediates mitochondrial network fragmentation.
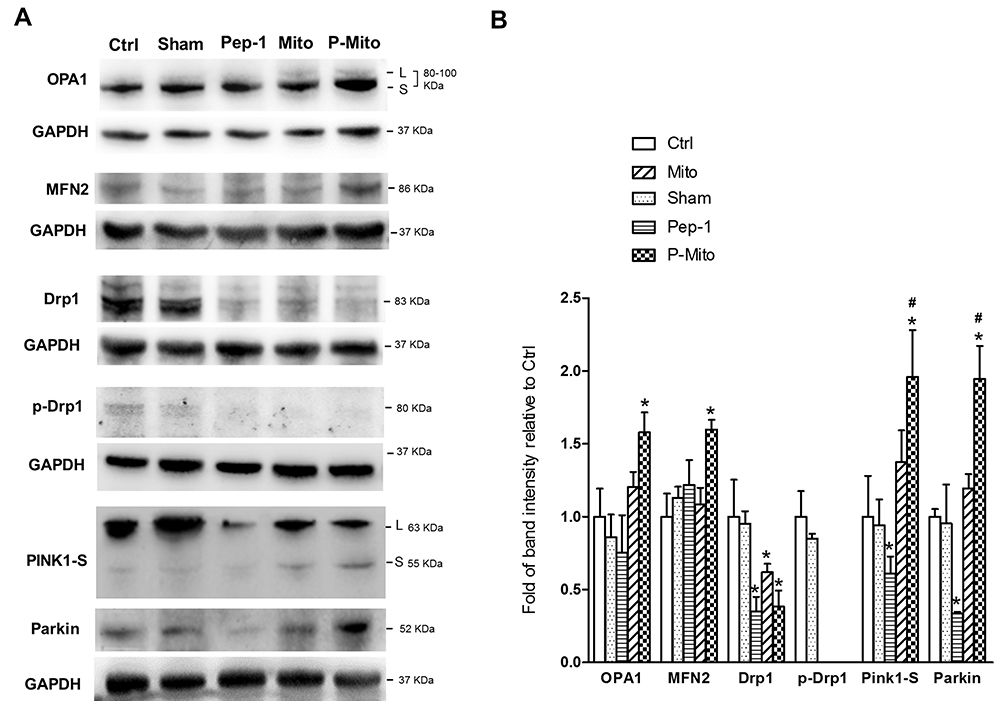 |
Figure 7 Expression of mitochondrial dynamic-related proteins in mouse breast tumors. (A) Levels of the mitochondrial dynamic-related proteins OPA1, MFN2, and Drp1, the phosphorylated form (serine 616) of Drp1 (p-Drp1), and mitophagy-related proteins, namely PINK1-S and parkin, were analyzed and (B) quantified using normalization to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). *p < 0.05, difference compared with the control group. #p < 0.05, difference compared with the Pep-1 group. |
Discussion
Recently, mitochondrial transplantation has been indicated to be a potential treatment for breast cancer, and we further validated the efficacy of mitochondrial transplantation in an orthotropic mice model of human breast cancer. Compared with a cell study based on a two-dimensional monoculture, mouse models of breast cancer were optimal for reflecting the actual complex interaction between cancer cells and their microenvironments.23 By comparing the transplants of mitochondria with intrinsic (Mito) and fused morphologies induced by Pep-1 modification (P-Mito), this study validated the critical mechanism of increased mitochondrial fusion and parkin protein in breast cancer treatment with mitochondrial transplantation and supported the regulatory relation between mitochondrial fusion in the inhibition of cell proliferation10 and reduced chemotherapy resistance in breast cancer.10,24 Notably, only treatment with P-Mito transplantation, not that of Mito, was validated. This may be related to the weaker penetrative ability of the transplanted Mito in the complex and a disorganized TME because Pep-1 modification can increase the incorporation efficiency of mitochondria in specific diseased cells.17,18 This may have prevented mitochondrial inactivation caused by prolonged exposure to the extracellular environment25 and hindered mitochondrial adherence to the cell outer membrane, which would lead to less uptake.19,26 In fact, the strategy of fusion with Pep-1 has been implemented to enhance the transmembrane movement of protein-derived cosmetics or drugs in vitro and in vivo.27,28 In addition, regarding the facilitation of outer membrane fusion of isolated mitochondria through Pep-1 modification,17 we cannot rule out the effect of delivering “membrane-fused mitochondria,” P-Mito, because Mito treatment with random structures under the same conditions has not been proven to be useful. Mitochondrial network remodeling is critical in tumorigenesis and increases the mitochondrial proportion of fused networks through genetic or drug manipulation.29 This was shown to impair cancer cell growth and metastasis.30
To track the performance of the transplanted mitochondria, donor mtDNA was labeled using in vivo incorporation of BrdU instead of mitochondrial protein targeting of recombinant GFP17,18 because of the availability of long-term tracking.31 However, we found that only 49.9% ± 2.05% of isolated mitochondria could be successfully labeled with BrdU following a regular protocol of 36-h in vivo incorporation.32 Through the conversion of the known concentration of injected mitochondria, the remaining amount of transplanted mitochondria estimated after 1 month of consecutive treatment was significantly higher in the P-Mito group than in the Mito group. However, we cannot rule out the error in the number assessment of remaining mitochondria caused by a barrier using a cell-impermeable anti-BrdU antibody. The high BrdU signal in P-Mito group was due to the antibody infiltrating into the necrotic/disrupted cells induced by P-Mito. Therefore, to confirm mitochondrial internalization in tumor cells, the result of BrdU IHC showed an increase in the mitochondrial penetration capacity through Pep-1 modification. We further suggested the maintenance of transplanted mitochondria could be related with the escape of fused mitochondria from mitophagy degradation.33 Although P-Mito treatment enhanced parkin production, it still reduced the amount of parkin recruited to degrade mitochondria, which manifested as an increasing cleavage form of PINK1.34 Parkin is a key protein in mitophagy and was initially identified in Parkinson disease;35 accumulating evidence suggests that the function of parkin is diverse and complex in multiple cell types, extending beyond mitophagy regulation. Additionally, parkin is a tumor suppressive gene in several common cancers, namely breast, lung, colorectal, and ovarian cancers.35 Thus, the benefit of parkin upregulation may not be related to the mitophagy-dependent method in P-Mito treatment of breast cancer. Therefore, further studies are warranted.
The findings of the present study regarding the regulation of Pep-1 in tumor-bearing mice contradicts those reported for Pep-1 regulation in cell models.19 Pep-1 treatment reportedly increases cancer cell proliferation and in vivo tumorgenesis of treated cells;19 however, for an already-formed breast tumor, Pep-1 treatment through intratumoral injection only reduced tumor weight, cell proliferation, and angiogenesis in the present study. Its anticancer efficacy was not as significant as that of P-Mito treatment, including the promotion of apoptosis and tumor necrosis. This may explain why Pep-1 treatment was unable to also reduce tumor volume. The cause remains unclear; however, we excluded the regulatory correlation of Pep-1 with mitochondrial dynamic balance and mtDNA replication in consideration of no influence on those expressions. The outcome was consistent with the results of Meloni et al.36 The therapeutic efficacy of Pep-1 is diversified and dependent on various diseases, as observed previously; for example, Pep-1 is an invalidated treatment for neuroprotection in Parkinson disease;18 however, it sustained cell survival in patients with oxidative damage from mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke syndrome.17
Consistent with the results of our previous study,19 mitochondrial cristae density consistently indicated the upregulation of mitochondrial fusion in a manner independent of mitochondrial respiratory capacity under P-Mito treatment. No direct correlation was demonstrated between mitochondrial dynamics and the mitochondrial membrane potential in MDA-MB-231 breast cancer cells from the overexpression of Drp1 and MFN1 or silencing of MFN1 and MFN2.14 Moreover, this highlights an available approach through the exclusive intervention of mitochondrial dynamics to adjust the sensitivity of breast cancer cells against environment-induced stress.14 Increasing mitochondrial fusion relieves mitochondrial oxidative damage through the complementation of damaged content and contributes to the maintenance of mitochondrial homeostasis.30 However, decreased mitochondrial oxidative stress in the CAF population in the TME reportedly suppresses tumor progression and metastasis in cancers.37 This illuminates how P-Mito remodels the tumorigenic TME, including through reductions in the CAF population and oxidative stress, as revealed by a significant reduction in the level of 8-OHdG, an oxidized nucleoside of DNA. The tumor-promoting and immunosuppressive role of CAF and its derivations of the extracellular matrix, enzymes, and chemical factors have generally been recognized in most recent reports.38,39 Notably, in prostate cancer, cancer cells hijack functional mitochondria derived from CAF through mitochondrial transfer between cells to promote malignancy.40 Thus, we suggest that P-Mito–induced reductions in the CAF population not only reduce the frequency of mitochondrial transfer between CAF and cancer cells but also block CAF-mediated immunosuppression in the tumor stroma, thereby enhancing tumor T cell infiltration in the TME to help fight breast cancer. Although mitochondrial transplantation is a milder treatment, it is not as effective as other drastic breast cancer treatments, with a reduction of >80% in tumor formation;41 however, it alters the dynamic properties of mitochondria and remodels the tumorigenic TME of breast cancer cells accompanied by the reactive oxygen species–induced malignant progression of breast cancer.42 Because of the highly positive correlations of oxidative stress with tumor recurrence in residual cancer cells,43 anticancer drug resistance44 and the risk of metastasis in patients with breast cancer,45 research on the antitumor efficacy of chemotherapy combined with mitochondrial transplantation for improving chemoresistance and side-effects caused by high doses of anticancer drugs is ongoing.
Conclusion
Mitochondrial transplantation, which is the artificial manipulation of mitochondrial transfer, altered mitochondrial dynamic properties in tumor cells and remodeled the TME, including by reducing oxidative stress and the CAF population and enhancing immune cell infiltration. This emphasizes that facilitation of mitochondrial fusion is a critical regulator in breast cancer therapy.
Acknowledgments
We would like to thank the NLAC, NARLabs, Taiwan, for their technical support in the contract breeding of ASID mice and animal transplantation. This study was supported by grants from the National Science Council (MOST 106-2314-B-371-004-; MOST 107-2314-B-371-005-) and Changhua Christian Hospital (107-CCH-MST-012).
Disclosure
The authors declare that they have no conflicts of interest. All data generated or analyzed during this study have been included in this article.
References
1. Guerra F, Guaragnella N, Arbini AA, et al. Mitochondrial dysfunction: a novel potential driver of epithelial-to-mesenchymal transition in cancer. Front Oncol. 2017;7:295. doi:10.3389/fonc.2017.00295
2. Han SY, Jeong YJ, Choi Y, et al. Mitochondrial dysfunction induces the invasive phenotype, and cell migration and invasion, through the induction of AKT and AMPK pathways in lung cancer cells. Int J Mol Med. 2018;42:1644–1652. doi:10.3892/ijmm.2018.3733
3. Avagliano A, Ruocco MR, Aliotta F, et al. Mitochondrial flexibility of breast cancers: a growth advantage and a therapeutic opportunity. Cells. 2019;8(5):401. doi:10.3390/cells8050401
4. Deus CM, Coelho AR, Serafim TL, et al. Targeting mitochondrial function for the treatment of breast cancer. Future Med Chem. 2014;6:1499–1513. doi:10.4155/fmc.14.100
5. Giannattasio S, Mirisola MG, Mazzoni C. Cell stress, metabolic reprogramming and cancer. Front Oncol. 2018;8:236. doi:10.3389/fonc.2018.00236
6. Ni H-M, Williams JA, Ding W-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015;4:6–13. doi:10.1016/j.redox.2014.11.006
7. Srinivasan S, Guha M, Kashina A, et al. Mitochondrial dysfunction and mitochondrial dynamics-the cancer connection. Biochim Biophys Acta Bioenerg. 2017;1858:602–614. doi:10.1016/j.bbabio.2017.01.004
8. Maycotte P, Marin-Hernandez A, Goyri-Aguirre M, et al. Mitochondrial dynamics and cancer. Tumor Biol. 2017;39:1010428317698391. doi:10.1177/1010428317698391
9. Li J, Huang Q, Long X, et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene. 2017;36(34):4901. doi:10.1038/onc.2017.98
10. Braganza A, Quesnelle K, Bickta J, et al. Myoglobin induces mitochondrial fusion, thereby inhibiting breast cancer cell proliferation. J Biol Chem. 2019;294:7269–7282. doi:10.1074/jbc.RA118.006673
11. Zou P, Liu L, Zheng LD, et al. Coordinated upregulation of mitochondrial biogenesis and autophagy in breast cancer cells: the role of dynamin related protein-1 and implication for breast cancer treatment. Oxid Med Cell Longev. 2016;2016:1–10. doi:10.1155/2016/4085727
12. Lucantoni F, Prehn JH. Metabolic targeting of breast cancer cells with the 2-Deoxy-D-glucose and the mitochondrial bioenergetics inhibitor Mdivi-1. Front Cell Dev Biol. 2018;6:113. doi:10.3389/fcell.2018.00113
13. Han Y, Cho U, Kim S, et al. Tumour microenvironment on mitochondrial dynamics and chemoresistance in cancer. Free Radic Res. 2018;52(11–12):1271–1287. doi:10.1080/10715762.2018.1459594
14. Zhao J, Zhang J, Yu M, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32(40):4814. doi:10.1038/onc.2012.494
15. Chang J-C, Liu K-H, Li Y-C, et al. Functional recovery of human cells harbouring the mitochondrial DNA mutation MERRF A8344G via peptide-mediated mitochondrial delivery. Neurosignals. 2013;21(3–4):160–173. doi:10.1159/000341981
16. Chang J-C, Liu K-H, Chuang C-S, et al. Treatment of human cells derived from MERRF syndrome by peptide-mediated mitochondrial delivery. Cytotherapy. 2013;15(12):1580–1596. doi:10.1016/j.jcyt.2013.06.008
17. Chang J-C, Hoel F, Liu K-H, et al. Peptide-mediated delivery of donor mitochondria improves mitochondrial function and cell viability in human cybrid cells with the melas A3243g mutation. Sci Rep. 2017;7(1):10710. doi:10.1038/s41598-017-10870-5
18. Chang J-C, Wu S-L, Liu K-H, et al. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine–induced neurotoxicity. Transl Res. 2016;170:40–56. e43. doi:10.1016/j.trsl.2015.12.003
19. Chang J-C, Chang H-S, Wu Y-C, et al. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J Exp Clin Cancer Res. 2019;38(1):30. doi:10.1186/s13046-019-1028-z
20. Divita G, Deshayes S, Konate K, et al. Cell penetrating peptides for intracellular delivery of molecules. Google Patents. 2018.
21. Battersby BJ, Shoubridge EA. Selection of a mtDNA sequence variant in hepatocytes of heteroplasmic mice is not due to differences in respiratory chain function or efficiency of replication. Hum Mol Genet. 2001;10:2469–2479. doi:10.1093/hmg/10.22.2469
22. Preble JM, Pacak CA, Kondo H, et al. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J Vis Exp. 2014;91:e51682.
23. Breslin S, O’Driscoll L. The relevance of using 3d cell cultures, in addition to 2d monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget. 2016;7(29):45745. doi:10.18632/oncotarget.9935
24. Anderson GR, Wardell SE, Cakir M, et al. Dysregulation of mitochondrial dynamics proteins are a targetable feature of human tumors. Nat Commun. 2018;9(1):1–13. doi:10.1038/s41467-018-04033-x
25. Gollihue JL, Patel SP, Rabchevsky AG. Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma. Neural Regen Res. 2018;13(2):194. doi:10.4103/1673-5374.226382
26. Elliott R, Jiang X, Head J. Mitochondria organelle transplantation: introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res Treat. 2012;136(2):347–354. doi:10.1007/s10549-012-2283-2
27. Luo X-G, Ma D-Y, Wang Y, et al. Fusion with Pep-1, a cell-penetrating peptide, enhances the transmembrane ability of human epidermal growth factor. Biosci Biotechnol Biochem. 2016;80:584–590. doi:10.1080/09168451.2015.1091714
28. Bolhassani A, Jafarzade BS, Mardani G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides. 2017;87:50–63. doi:10.1016/j.peptides.2016.11.011
29. Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166(3):555–566. doi:10.1016/j.cell.2016.07.002
30. Senft D, Ronai ZA. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 2016;39:43–52. doi:10.1016/j.ceb.2016.02.001
31. Huang P-J, Kuo -C-C, Lee H-C, et al. Transferring xenogenic mitochondria provides neural protection against ischemic stress in ischemic rat brains. Cell Transplant. 2016;25:913–927. doi:10.3727/096368915X689785
32. Calkins MJ, Reddy PH. Assessment of newly synthesized mitochondrial DNA using BrdU labeling in primary neurons from alzheimer’s disease mice: implications for impaired mitochondrial biogenesis and synaptic damage. Biochim Biophys Acta Mol Basis Dis. 2011;1812:1182–1189. doi:10.1016/j.bbadis.2011.04.006
33. Rambold AS, Kostelecky B, Lippincott-Schwartz J. Together we are stronger: fusion protects mitochondria from autophagosomal degradation. Autophagy. 2011;7(12):1568–1569. doi:10.4161/auto.7.12.17992
34. Matsuda N, Sato S, Shiba K, et al. Pink1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol. 2010;189(2):211–221. doi:10.1083/jcb.200910140
35. Liu J, Zhang C, Hu W, et al. Parkinson’s disease-associated protein parkin: an unusual player in cancer. Cancer Commun. 2018;38:40. doi:10.1186/s40880-018-0314-z
36. Meloni BP, Craig AJ, Milech N, et al. The neuroprotective efficacy of cell-penetrating peptides tat, penetratin, Arg-9, and Pep-1 in glutamic acid, kainic acid, and in vitro ischemia injury models using primary cortical neuronal cultures. Cell Mol Neurobiol. 2014;34:173–181. doi:10.1007/s10571-013-9999-3
37. Balliet RM, Capparelli C, Guido C, et al. Mitochondrial oxidative stress in cancer-associated fibroblasts drives lactate production, promoting breast cancer tumor growth: understanding the aging and cancer connection. Cell Cycle. 2011;10(23):4065–4073. doi:10.4161/cc.10.23.18254
38. Liu T, Han C, Wang S, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. 2019;12(1):1–15. doi:10.1186/s13045-019-0770-1
39. Liu T, Zhou L, Li D, et al. Cancer-associated fibroblasts build and secure the tumor microenvironment. Front Cell Dev Biol. 2019;7. doi:10.3389/fcell.2019.00060
40. Ippolito L, Morandi A, Taddei ML, et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene. 2019;38(27):5339–5355. doi:10.1038/s41388-019-0805-7
41. Ouhtit A, Gaur RL, Abdraboh M, et al. Simultaneous inhibition of cell-cycle, proliferation, survival, metastatic pathways and induction of apoptosis in breast cancer cells by a phytochemical super-cocktail: genes that underpin its mode of action. J Cancer. 2013;4(9):703. doi:10.7150/jca.7235
42. Ma J, Zhang Q, Chen S, et al. Mitochondrial dysfunction promotes breast cancer cell migration and invasion through hif1α accumulation via increased production of reactive oxygen species. PLoS One. 2013;8(7):e69485. doi:10.1371/journal.pone.0069485
43. Havas KM, Milchevskaya V, Radic K, et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J Clin Invest. 2017;127(6):2091–2105. doi:10.1172/JCI89914
44. Okon IS, Zou M-H. Mitochondrial ros and cancer drug resistance: implications for therapy. Pharmacol Res. 2015;100:170–174. doi:10.1016/j.phrs.2015.06.013
45. Gill JG, Piskounova E, Morrison SJ. Cancer, oxidative stress, and metastasis. Cold Spring Harbor Symp Quant Biol. 2016;81:163–175. doi:10.1101/sqb.2016.81.030791
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.