")
Back to Journals » Cancer Management and Research » Volume 12
Antiproliferative Effects of Telotristat Ethyl in Patients with Neuroendocrine Tumors: The TELEACE Real-World Chart Review Study
Authors Morse MA, Liu E, Joish VN , Huynh L, Cheng M, Duh MS , Seth K, Lapuerta P , Metz DC
Received 6 May 2020
Accepted for publication 13 July 2020
Published 30 July 2020 Volume 2020:12 Pages 6607—6614
DOI https://doi.org/10.2147/CMAR.S261257
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Yong Teng
Michael A Morse,1 Eric Liu,2 Vijay N Joish,3 Lynn Huynh,4 Mu Cheng,4 Mei Sheng Duh,4 Kiernan Seth,3 Pablo Lapuerta,3 David C Metz5
1Duke Cancer Institute, School of Medicine, Duke University, Durham, NC, USA; 2The Neuroendocrine Institute at Rocky Mountain Cancer Centers, Denver, CO, USA; 3Lexicon Pharmaceuticals, Inc., The Woodlands, TX, USA; 4Analysis Group, Boston, MA, USA; 5Neuroendocrine Tumor Program at Penn Medicine, Philadelphia, PA, USA
Correspondence: Michael A Morse, 10 Bryan Searle Drive, Box 3233, Durham, NC 27710, USA
Tel +1 919 681– 3480
Email [email protected]
Purpose: Neuroendocrine tumors (NETs) associated with carcinoid syndrome (CS) overproduce serotonin, mediated by tryptophan hydroxylase-1 (TPH1). The TPH inhibitor telotristat ethyl (TE) reduces peripheral serotonin and relieves CS symptoms. We conducted a real-world clinical practice study to explore the effects of TE on tumor growth in patients with NETs and CS.
Patients and Methods: Single-arm, pre/post chart review study of patients with advanced NETs who received TE for ≥ 6 months and had ≥ 2 radiological scans within 12 months before and ≥ 1 scan after TE initiation. Linear regression and longitudinal analyses assessed changes in tumor size controlling for background NET treatment.
Results: Two hundred patients were enrolled, most (61%) had well-differentiated gastrointestinal NETs (61%) and received TE for an average of 12 months (SD, 7.3). Mean reduction in tumor size after TE initiation was 0.59 cm (p=0.006). Longitudinal analysis showed an 8.5% reduction in tumor size (p=0.045) from pre- to post-TE periods. Documented NET treatment prior to initiating TE and time between scans were not significant predictors of changes in tumor size. Results were consistent in a subgroup of patients with the same documented NET treatment before and after initiating TE.
Conclusion: TE may have antitumor effects consistent with serotonin overproduction in tumor growth.
Keywords: neuroendocrine tumors, carcinoid tumor, malignant carcinoid syndrome, telotristat ethyl, octreotide, lanreotide
Introduction
Secretory neuroendocrine tumors (NETs) that produce the carcinoid syndrome (CS) are associated with overproduction of serotonin, a product of tryptophan metabolism, which causes intestinal motility, excessive bowel movements and carcinoid syndrome diarrhea (CSD).1–3 Approximately 20–35% of patients with well-differentiated NETs (mainly of small bowel origin) experience CS, many of whom (up to 70% in older studies) may develop carcinoid heart disease from serotonin-driven development of endocardial fibrotic plaques in the heart.4–8
In vitro studies have shown proliferative effects of elevated serotonin (5-hydroxytryptamine; 5-HT) on NETs, and elevated serotonin has been associated with increased 1-year mortality for patients with NETs and CS.9–12 Animal models have shown increased levels of tryptophan hydroxylase 1 (TPH1), which synthesizes serotonin, to be correlated with tumor size and growth, and have suggested that inhibition of TPH1 and peripheral serotonin may have antitumor effects.13–16
The primary goals of treatment for advanced NETs are to suppress tumor growth and relieve symptoms. Somatostatin analogs (SSAs) have historically been the initial standard of care for patients with NETs and have demonstrated antiproliferative effects in clinical trials and observational studies.17–19 Pivotal data supporting the antitumor effects of SSAs are from the PROMID and CLARINET randomized controlled trials of octreotide (Sandostatin® LAR Depot, Novartis Pharmaceuticals, East Hanover, New Jersey) and lanreotide (Somatuline® Depot, Ipsen Biopharmaceuticals, Inc., Cambridge, Massachusetts), respectively, including the improved time to tumor progression after 6 months of octreotide versus placebo (14.3 vs 6.0 months) and improved progression-free survival with lanreotide versus placebo (65% vs 33% at 24 months) among other outcomes.17,18 These trials were preceded by several single-arm and comparative prospective clinical practice studies and have since been followed by retrospective database analyses.19–24
The TPH inhibitor telotristat ethyl (TE; Xermelo™, Lexicon Pharmaceuticals, Inc., The Woodlands, Texas) reduces the production of peripheral serotonin and is approved for the treatment of CSD in combination with SSAs.25 In the TELESTAR pivotal trial, TE 250 mg reduced levels of the serotonin marker 5-hydroxyindoleacetic acid (5-HIAA) by a mean of 40.1 mg/24h after 12 weeks compared with an increase with placebo of 11.5 mg/24h.26 While the ability of TE to improve CSD and CS symptoms has been demonstrated in clinical trials and real-world clinical practice studies, the potential antiproliferative effects and associated clinical outcomes of serotonin reduction from TE treatment have not been investigated.26–28 Similar to the early clinical practice studies exploring the potential antiproliferative effects of octreotide,19–24 we conducted a medical chart review study to explore the potential antiproliferative effects of TE on tumor size and growth in patients with advanced NETs and CS.
Materials and Methods
Study Design and Participants
The TELEACE study is a retrospective, single-arm, pre-post physician panel-based chart review of patients who received TE for at least 6 months. Participating physicians were recruited by a professional recruiting organization (Dynata, Plano, TX, USA). Physicians had to have treated ≥ 1 patient with TE who had been diagnosed with advanced (unresectable, locally advanced or metastatic) NET and documented CS according to the patient’s medical record within the past 12 months. Physicians also had to have available records of the tumor size and tumor response before and after TE initiation for each eligible patient. Participating physicians were blinded to the identity of the study sponsor and the study sponsor was blinded to participating physicians. This retrospective chart review maintained the anonymity of patients’ medical records that were abstracted for review and analysis. The New England Independent Review Board® (https://neirb.com) reviewed the study protocol and electronic case report form and determined the study to be exempt from IRB review due to the retrospective observational nature of this medical chart review study, with a waiver of participant consent for records research. The study was conducted in compliance with the principles set forth by the Declaration of Helsinki and further actions were taken to ensure patient privacy and confidentiality. A randomization scheme was implemented during chart abstraction where a random sequence of letters was generated to determine the selection of each medical chart for review; the letters were not retained or recorded. An automatically generated date shift (addition or subtraction of a randomly generated number of days) was assigned to each patient to further preserve the de-identification of collected data.
Adults (≥ 18 years) who received TE treatment after a confirmed advanced NET diagnosis and documented CS in the medical record were eligible for participation. Patients had to have received TE for ≥ 6 months and had to have ≥ 2 radiological scans in the 12 months prior to TE initiation and ≥ 1 scan after TE initiation. Patient records also had to have treatment information related to NET and CS for ≥ 6 months after TE initiation or until death. Eligible patients could not have a histologically poorly differentiated NET based on grade (G3) or Ki67 index > 20%, mixed tumor types according to physician notes, or documented enrollment in any clinical trial during the 6 months following TE initiation.
Outcomes
The primary endpoint was change in tumor size between pre- and post-TE periods, defined as the sum of the longest diameters of target lesions as reported from radiological scans reported on separate dates in the patient’s medical record. Radiological scans were compared directly at the time of the last scan. The scan technique and quality modality were to remain constant; different radiologists may have performed the scans. Tumor size was calculated for patients with ≥ 2 radiological scans during the pre-TE period and ≥ 1 scan during the post-TE period (Figure 1). Target lesion and radiological scan information were based on that provided in the patient’s medical record. Secondary outcomes included physician-assessed tumor response (“improved”, “stayed the same”, or “worsened”). Physician assessment of tumor response based on the radiology report has been shown to be an effective assessment of tumor burden in real-world medical chart review studies where the information required for the Response Evaluation Criteria in Solid Tumors (RECIST) is not typically available.29 Changes in urinary 5-hydroxyindoleacetic acid (5-HIAA) were measured pre-TE treatment and from available post-TE follow-up samples.
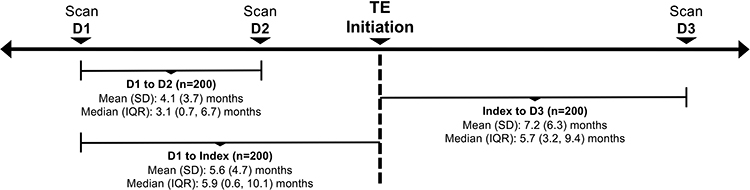 |
Figure 1 Radiological scans during pre- and post-TE treatment periods (overall population). |
Statistical Analysis
A sample size of 95 patients was required based on a 2-sided Wilcoxon signed-rank test with 80% power to detect a 3 percentage point change in tumor size measurements between pre- and post-TE periods. Descriptive statistics were used to summarize the difference in tumor size between the radiological scan performed immediately before TE initiation and the last radiological scan performed after TE initiation, as well as physician-assessed tumor responses and changes in urinary 5-HIAA before and after TE initiation. Linear regression was used to assess change in tumor size after TE initiation, controlling for SSA treatment and additional non-SSA NET treatment (background treatment) before TE initiation. Longitudinal analyses of changes in tumor size used a generalized estimation equation with a log link and unstructured covariance matrix to model the repeated tumor scans in the pre- and post-TE periods, controlling for background NET and CS treatment: log(yij) = 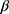 0 +
0 + 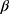 1tij +
1tij + 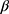 2SSA_preij +
2SSA_preij + 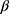 3Additional tx_preij +
3Additional tx_preij + 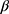 4Post_TEij (where yij = tumor size for patient i at time j; tij = months since first tumor scan for patient i at time j; SSA_preij = indicator for the presence of documented SSA treatment during pre-TE initiation period for patient i at time j; additional tx_preij = indicator for the presence of documented additional non-SSA NET treatment during pre-TE initiation period for patient i at time j; post_TEij = indicator for post-TE period for patient i at time j). Confirmatory analyses were conducted in a subgroup of 65 patients who had the same background NET treatment before and after TE initiation to isolate the effect of TE. All analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
4Post_TEij (where yij = tumor size for patient i at time j; tij = months since first tumor scan for patient i at time j; SSA_preij = indicator for the presence of documented SSA treatment during pre-TE initiation period for patient i at time j; additional tx_preij = indicator for the presence of documented additional non-SSA NET treatment during pre-TE initiation period for patient i at time j; post_TEij = indicator for post-TE period for patient i at time j). Confirmatory analyses were conducted in a subgroup of 65 patients who had the same background NET treatment before and after TE initiation to isolate the effect of TE. All analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Study Population
A total of 114 physicians participated in the study, predominantly from community care settings (62%); nearly all (98%) were oncologists. Medical charts for 200 patients who initiated TE during the study period were included, with a mean of 1.8 charts per physician. The mean age of patients was 61 years and 57% were men; most patients had well-differentiated NETs (61%) with a gastrointestinal primary tumor site (61%; Table 1). Patients received TE for an average of 12 months (SD, 7.3) and a median of 9 months (IQR, 6.8–15.2). Most patients (82%, 163/200) were still receiving TE treatment at the time of data collection. Nearly all patients with recorded SSA treatment information received long-acting octreotide or lanreotide prior to and concurrently with TE treatment (Table 1). A subgroup of 65 patients with the same documented pre- and post-TE NET treatment had a mean duration of TE treatment of 11.4 months (SD, 7.0; median, 7.7) and mean duration of post-TE NET treatment of 12.4 months (SD, 7.9; median 8.1).
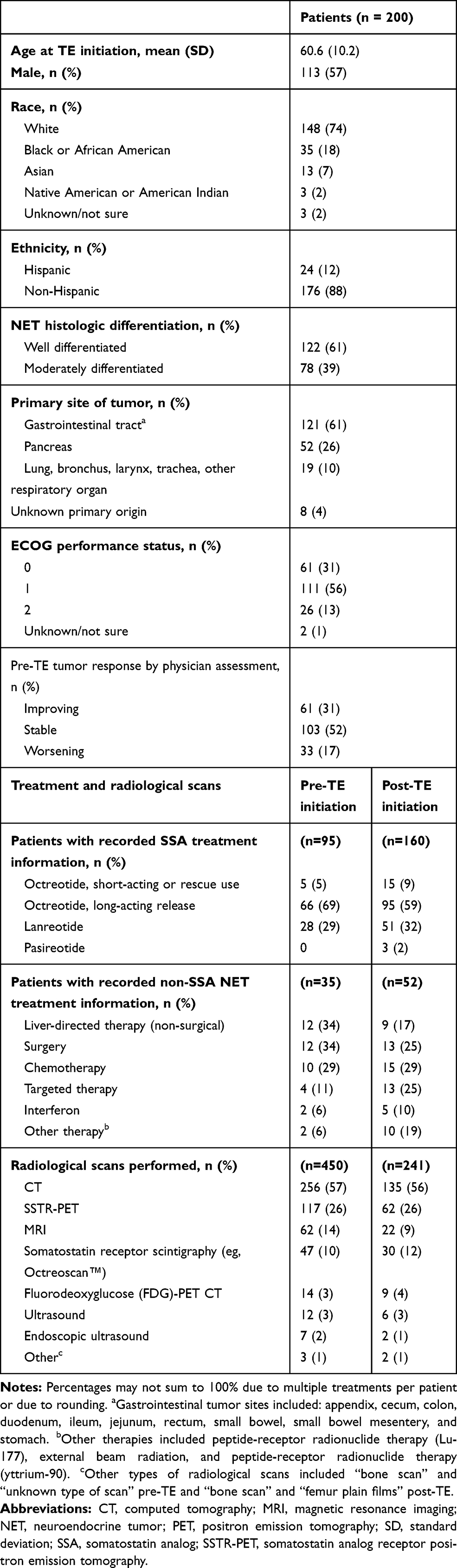 |
Table 1 Patient Demographic and Clinical Characteristics |
Patients had a mean of 2.3 (SD, 0.7) documented radiological scans during the 12 months before TE treatment and 1.2 (SD, 0.6) after TE initiation (median, 2.0 and 1.0, respectively). The mean time from the first scan to TE initiation was 5.6 months (SD, 4.7) and from TE initiation to the last scan was 7.2 months (SD, 6.3; Figure 1). Most scans were based on computed tomography, followed by 68Ga-DOTATOC somatostatin receptor positron emission tomography (SSTR PET), and magnetic resonance imaging (MRI; Table 1).
Change in Tumor Size and Serotonin Levels
In the overall population, mean tumor size was 3.9 cm (SD, 2.2; median, 3.5 cm) during the pre-TE period and decreased by 0.59 cm after TE treatment (95% CI: –1.01, –0.17; P=0.006; Table 2). Regression analysis indicated that additional non-TE NET treatment with or without SSA treatment prior to initiating TE was not a significant predictor of post-TE reduction in tumor size (P=0.26 and P=0.79, respectively). A confirmatory analysis in a subgroup of 65 patients who had the same non-TE NET treatment before and after TE initiation (including SSA and non-SSA treatment) yielded similar results as the primary analysis (mean tumor size decrease of 0.63 cm, 95% CI: –1.24, –0.02; P=0.044).
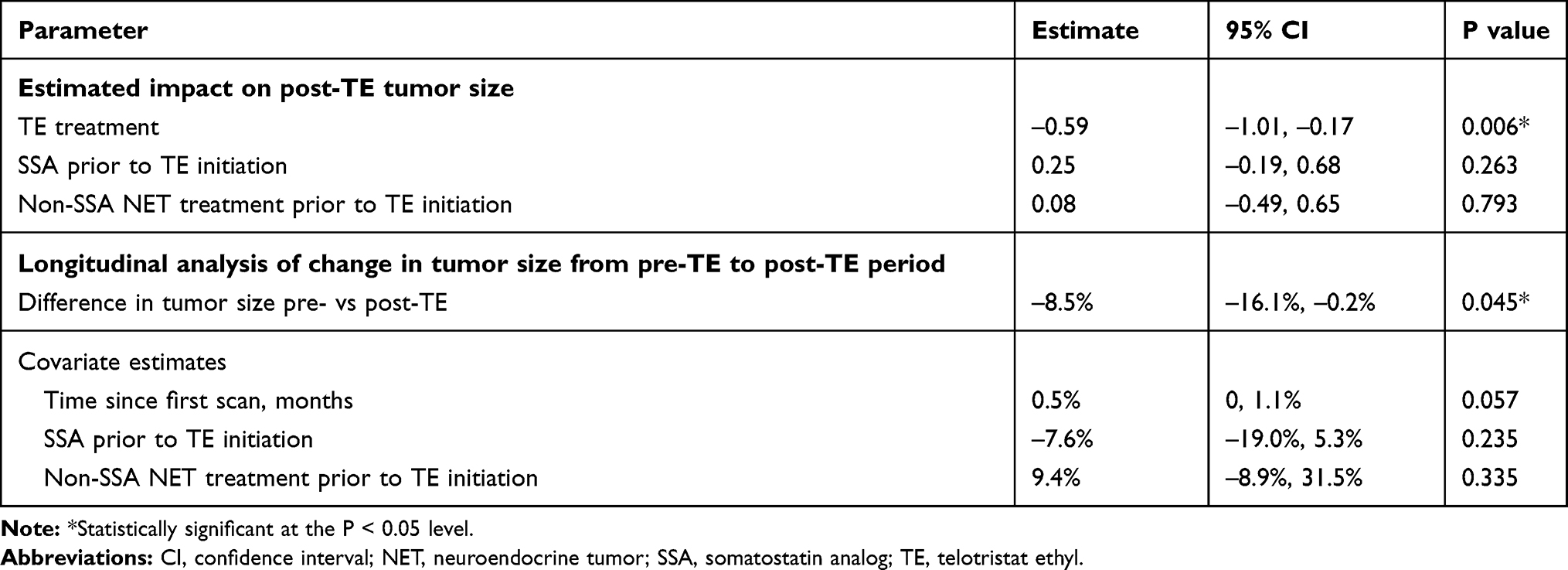 |
Table 2 Change in Tumor Size After TE Initiation (Overall Population) |
In the longitudinal analysis of tumor growth, tumor size decreased 8.5% in the post-TE period (95% CI: –16.1%, –0.2%; P=0.045). Pre-TE SSA treatment, pre-TE non-SSA NET treatment, and time since first radiological scan were not significant predictors of change in tumor size (All P > 0.05). A descriptive posthoc assessment of changes in tumor size by primary tumor site and type of radiological scan was consistent with the primary findings (Supplementary Table S1).
A small sample of patients (n=22) had urinary 5-HIAA values available in both the pre- and post-TE treatment periods. Mean urinary 5-HIAA levels decreased from 72.5 mg/24 hrs (SD, 98.0) before TE initiation to 43.6 mg/24 hrs (SD, 54.7) after TE initiation (P<0.001).
Physician-Assessed Tumor Response
Physician assessment of tumor response was favorable during the TE treatment period (Figure 2). The majority of tumors determined to have worsened in the pre-TE treatment period were improved or stable (81%) after TE initiation. Stable pre-TE tumors were nearly all improved or stable after TE (94%), as were improved pre-TE tumors (97%). Findings were similar in the subgroup of patients with the same SSA or non-SSA NET treatment before and after TE initiation, despite small sample sizes particularly for the group of 6 patients with worsened pre-TE tumors.
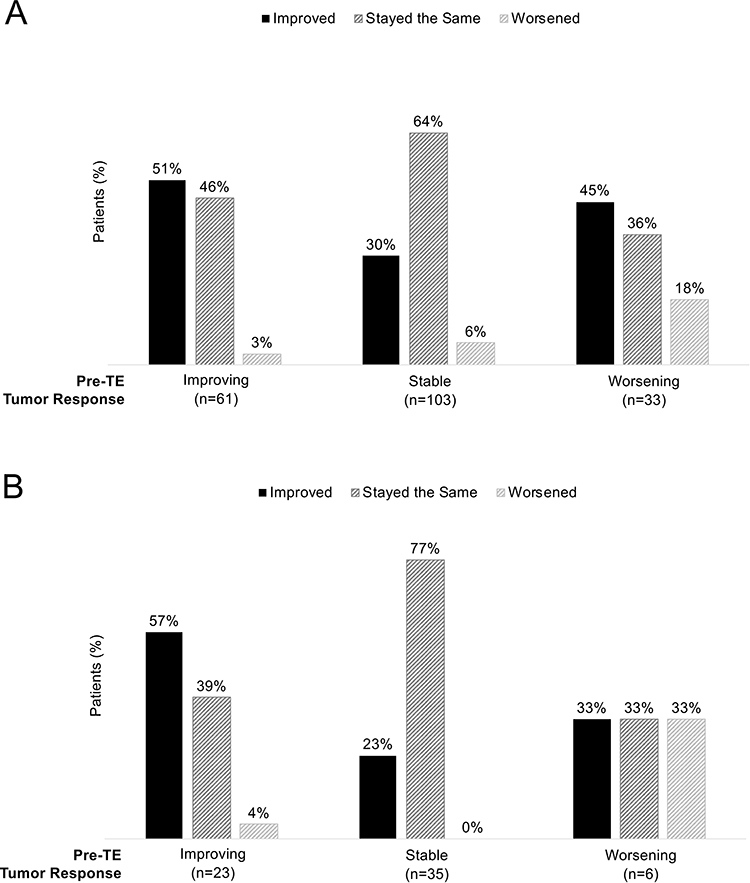 |
Figure 2 Post-TE physician assessment of tumor response by pre-TE assessment. (A) Overall population (n = 200). (B) Same SSA and NET treatment subgroup (n = 65). |
Discussion
This retrospective chart review showed reductions in tumor size among patients with advanced NETs and CS receiving TE in US clinical practice. Post-TE changes in tumor size were not attributable to other NET treatments, including SSAs, or the timing of radiological scans used to assess tumor size. Findings were similar in a subgroup of patients with the same documented SSA or non-SSA NET treatment before and after TE initiation, suggesting a likely independent effect of TE on tumor size.
These findings are consistent with those from the early studies exploring the potential antiproliferative effects of the SSAs octreotide and lanreotide, though the early SSA studies tended to have smaller sample sizes and included many patients with a lower prevalence of CS.19,30-32 Aparicio et al (2001) reported arrested tumor growth in 60% of patients for a median of 11 months among 35 patients treated with octreotide or lanreotide.33 No difference in antitumor effect was reported with higher doses or between SSAs. The single-arm octreotide studies with >50 patients with advanced, well-differentiated NETs reported stable disease in 47–49% of patients treated for a median of 12–14 months,31,34 and in 13% of those treated for ≥ 6 months.35 The median TE treatment duration of 9 months in this study corresponded to a majority of patients showing stable or improved disease after TE initiation across all categories of pre-TE tumor growth assessments (81–97%). This finding remained consistent in the smaller subgroup of 65 patients who had the same pre- and post-TE NET treatment.
The observed reduction in urinary 5-HIAA levels from the pre- to post-TE treatment periods was consistent with the known mechanism of serotonin reduction with TE, though the subgroup with available laboratory values was small. It should be noted that the effect of TE on tumor growth has not been thoroughly evaluated in pancreatic NET cell lines. TE has recently shown a reduction in tumor growth in cell lines derived from colon cancer, liposarcoma, and cholangiocarcinoma.36 Our findings in patients treated in clinical practice are of interest when considering the potential antiproliferative effects of TE in patients with NETs.
This medical chart review of patients in real-world US clinical practice was not limited to the strict patient eligibility criteria of experimental trials, though the observational nature of the study could not account for any potential influence of unmeasured variables. To enhance the external validity of the study and to reduce confounding, a randomization scheme was implemented during the chart abstraction process. The pre-post design, where participants served as their own controls, reduced the potential for confounding and eliminated the challenge of selecting an appropriate control group. Nevertheless, the retrospective, non-randomized design may still have been affected by potential biases including measurement error, non-random missing data, and external validity. The use of TE in clinical practice was observed in a variety of NET types beyond those typically associated with CS. Data extraction from electronic medical records with an average of 1.8 charts per physician did not provide a comprehensive view of clinical practice patterns for a given physician, including the use of SSA or TE, and was limited to charts with documented radiological scan reports. A self-selection bias may have been present among the physicians who chose to participate and responded to the online chart review invitation, and selection bias may have been present as the proportion of eligible charts submitted by each physician was unknown. The assessment of radiological scans and assessment schedules across clinical sites and physicians was heterogeneous, and complete data may not have been available in all medical records. A RECIST-based approach was unavailable for this real-world chart review study, but a clinician-based assessment derived from the medical charts and supported by the radiology report was the most feasible method for characterizing tumor response and has been shown to be effective in assessing tumor burden in real-world chart review studies.29 The sample size was adequate for the primary analysis of tumor size, but sub-analyses included some small subgroups. The longitudinal analysis evaluating the association between TE treatment and change in tumor size utilized all radiological scans, leveraging as much information as possible although the follow-up duration was short. The longitudinal analysis also addressed variability in the time intervals between scans in the pre- and post-TE periods.
This is the first investigation of antiproliferative effects of TE in patients with advanced NETs and CS. While additional prospective clinical studies are needed, these exploratory findings are consistent with the known preclinical mechanisms of serotonin as a potential promoter of tumor growth and the potential antiproliferative effects of TPH inhibition. This real-world clinical practice study showed a potential role for TE treatment to inhibit tumor growth in patients with NETs.
Acknowledgments
This study was sponsored by Lexicon Pharmaceuticals, Inc. Medical writing support was provided by Jeff Frimpter, MPH, funded by Lexicon Pharmaceuticals, Inc.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
This study was funded by Lexicon Pharmaceuticals, Inc., which participated in the design, conduct, analysis and interpretation of findings. DM, MM and EL served as consultants for the study design and conduct and were compensated for their time and effort. LH, MC and MSD are employees of Analysis Group which received funding for data collection and analysis. VNJ, KS and PL are employees of Lexicon Pharmaceuticals, Inc., the study sponsor. Michael A Morse reports personal fees from Lexicon, during the conduct of the study and outside the submitted work. Eric Liu reports personal fees from Lexicon, during the conduct of the study. Vijay N Joish reports employment and stock holdings for Lexicon Pharmaceuticals Inc. Lynn Huynh reports grants from Lexicon, during the conduct of the study, and Novartis, Taiho, Pfizer, and Epizyme, outside the submitted work. Mu Cheng reports grants from Lexicon Pharmaceuticals, during the conduct of the study; and grants from Takeda, Ipsen, Novo Nordisk, Merck, GSK, Pfizer, Epizyme, Bayer, AstraZeneca, CSL Behring, and Sanofi, outside the submitted work. Mei S Duh reports grants from Lexicon, during the conduct of the study. Kiernan Seth reports employment and stock holdings for Lexicon Pharmaceuticals, Inc. Pablo Lapuerta reports employment and stock holdings for Lexicon Pharmaceuticals, Inc. David C Metz reports personal fees from Lexicon, during the conduct of the study; and grants from Lexicon, AAA, Ipsen, and wren, and personal fees from crinetics and curium, outside the submitted work. The authors report no other possible conflicts of interest in this work.
References
1. Gustafsson BI, Hauso O, Drozdov I, Kidd M, Modlin M. Carcinoid heart disease. Int J Cardiol. 2008;129(3):318–324. doi:10.1016/j.ijcard.2008.02.019
2. Zuetenhorst JM, Bonfrer JMGM, Korse CM, Bakker R, van Tinteren TBG, Taal BG. Carcinoid heart disease: the role of urinary 5-hydroxyindoleacetic acid excretion and plasma levels of atrial natriuretic peptide, transforming growth factor-beta and fibroblast growth factor. Cancer. 2003;97(7):1609–1615. doi:10.1002/cncr.11226
3. Pandit S, Bhusal K. Carcinoid syndrome; 2017. StatPearls Publishing: Treasure Island, FL. Available from: https://www.ncbi.nlm.nih.gov/books/NBK448096/.
4. Dasari A, Shen C, Halperin D, et al. Trends in the incidence, prevalence, and survival outcome sin patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017;3(10):1335–1342. doi:10.1001/jamaoncol.2017.0589
5. Turaga KK, Kvols LK. Recent progress in the understanding, diagnosis, and treatment of gastroenteropancreatic neuroendocrine tumors. Can Cancer J Clin. 2011;61(2):113–132. doi:10.3322/caac.20097
6. Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–3072. doi:10.1200/JCO.2007.15.4377
7. Halperin DM, Shen C, Dasari A, et al. Frequency of carcinoid syndrome at neuroendocrine tumour diagnosis: a population-based study. Lancet Oncol. 2017;18(4):525–534. doi:10.1016/S1470-2045(17)30110-9
8. Grozinsky-Glasberg S, Grossman AB, Gross DJ. Carcinoid heart disease: from pathophysiology to treatment – “something in the way it moves”. Neuroendocrinol. 2015;101(4):263–273. doi:10.1159/000381930
9. Siddiqui EJ, Shabbir MA, Mikhailidis DP, Mumtaz FH, Thompson CS. The effect of serotonin and serotonin antagonists on bladder cancer cell proliferation. BJU Int. 2006;97:634–639. doi:10.1111/j.1464-410X.2006.06056.x
10. Ishizuka J, Beauchamp RD, Townsend CM, Greeley GH, Thompson JC. Receptor-mediated autocrine growth-stimulatory effect of 5-hydroxytryptamine on cultured human pancreatic carcinoid cells. J Cell Physiol. 1992;150(1):1–7. doi:10.1002/jcp.1041500102
11. Drozdov I, Kidd M, Gustafsson BI, et al. Auto-regulatory effects of serotonin on proliferation and signaling pathways in lung and small intestine neuroendocrine tumor cell lines. Cancer. 2009;115(21):4934–4945. doi:10.1002/cncr.24533
12. Joish VN, Shah S, Tierce JC, et al. Serotonin levels and 1-year mortality in patients with neuroendocrine tumors: a systematic review and meta-analysis. Future Oncol. 2019;15(12):1397–1406. doi:10.2217/fon-2018-0960
13. Huang L, Mellor AL. Metabolic control of tumor progression and anti-tumor activity. Curr Opin Oncol. 2014;26(1):92–99. doi:10.1097/CCO.0000000000000035
14. Jiang S-H, Li J, Zhang Z-G. Increased serotonin signaling contributes to the Warburg effect in pancreatic tumor cells under metabolic stress and promotes growth of pancreatic tumors in mice. Gastroenterol. 2017;153(1):277–291. doi:10.1053/j.gastro.2017.03.008
15. Gautam J, Banskota S, Regmi SC, et al. Tryptophan hydroxylase 1 and 5-HT7 receptor preferentially expressed in triple-negative breast cancer promote cancer progression through autocrine serotonin signaling. Mol Cancer. 2016;15:75. doi:10.1186/s12943-016-0559-6
16. Matthes S, Bader M. Peripheral serotonin synthesis as a new drug target. Trends Pharmacol Sci. 2018;39(6):560–572. doi:10.1016/j.tips.2018.03.004
17. Rinke A, Müller HH, Schade-Brittinger C, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID study group. J Clin Oncol. 2009;27:4656–4663. doi:10.1200/JCO.2009.22.8510
18. Caplin ME, Pavel M, Ruszniewski P. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371:1556–1557. doi:10.1056/NEJMoa1316158
19. Barrows SM, Cai B, Copley-Merriman C, et al. Systematic literature review of the antitumor effect of octreotide in neuroendocrine tumors. World J Meta-Anal. 2018;6(2):9–20. doi:10.13105/wjma.v6.i2.9
20. Anthony L, Johnson D, Hande K, et al. Somatostatin analogue Phase I trials in neuroendocrine neoplasms. Acta Oncol. 1993;32:217–223. doi:10.3109/02841869309083915
21. Angeletti S, Corleto VD, Schillaci O, et al. Single dose of octreotide stabilize metastatic gastro-entero-pancreatic endocrine tumours. Ital J Gastroenterol Hepatol. 1999;31:23–27.
22. Tomassetti P, Migliori M, Corinaldesi R, Gullo L. Treatment of gastroenteropancreatic neuroendocrine tumours with octreotide LAR. Aliment Pharmacol Ther. 2000;14:557–560. doi:10.1046/j.1365-2036.2000.00738.x
23. Shen C, Shih YC, Xu Y, Yao JC. Octreotide long-acting repeatable use among elderly patients with carcinoid syndrome and survival outcomes: a population-based analysis. Cancer. 2014;120:2039–2049. doi:10.1002/cncr.28653
24. Shen C, Shih YC, Xu Y, Yao JC. Octreotide long-acting repeatable among elderly patients with neuroendocrine tumors: a survival analysis of SEER-medicare data. Cancer Epidemiol Biomarkers Prev. 2015;24:1656–1665. doi:10.1158/1055-9965.EPI-15-0336
25. Lexicon Pharmaceuticals, Inc (2017). Xermelo® (telotristat ethyl) prescribing information. Available from: https://www.xermelo.com/Media/Default/pdfs/Product_Info_telotristat_ethyl.pdf. Accessed March 6, 2020.
26. Kulke MH, Hörsch D, Caplin ME, et al. Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treatment of carcinoid syndrome. J Clin Oncol. 2017;35:14–23. doi:10.1200/JCO.2016.69.2780
27. Pavel ME, Hörsch D, Caplin M, et al. Telotristat etiprate for carcinoid syndrome: a single-arm, multicenter trial. J Clin Endocrinol Metab. 2015;100:1511–1519. doi:10.1210/jc.2014-2247
28. Strosberg J, Joish VN, Giacalone S, et al. TELEPRO: patient-reported carcinoid syndrome symptom improvement following initiation of telotristat ethyl in the real world. Oncologist. 2019;24(11):1446–1452. doi:10.1634/theoncologist.2018-0921
29. Griffith SD, Tucker M, Bowser B, et al. Generating real-world tumor burden endpoints from electronic health record data: comparison of RECIST, radiology-anchored, and clinician-anchored approaches for abstracting real-world progression in non-small cell lung cancer. Adv Ther. 2019;36(8):2122–2136. doi:10.1007/s12325-019-00970-1
30. Shojamanesh H, Gibril F, Louie A, et al. Prospective study of the antitumor efficacy of long-term octreotide treatment in patients with progressive metastatic gastrinoma. Cancer. 2002;94:331–343. doi:10.1002/cncr.10195
31. Di Bartolomeo M, Bajetta E, Buzzoni R, et al. Clinical efficacy of octreotide in the treatment of metastatic neuroendocrine tumors. A study by the Italian trials in medical oncology group. Cancer. 1996;77:402–408. doi:10.1002/(SICI)1097-0142(19960115)77:2<402::AID-CNCR25>3.0.CO;2-4
32. Panzuto F, Di Fonzo M, Iannicelli E, et al. Long-term clinical outcome of somatostatin analogues for treatment of progressive, metastatic, well-differentiated entero-pancreatic endocrine carcinoma. Ann Oncol. 2006;17:461–466. doi:10.1093/annonc/mdj113
33. Aparicio T, Ducreux M, Baudin E, et al. Antitumor activity of somatostatin analogues in progressive metastatic neuroendocrine tumours. Eur J Cancer. 2001;37(8):1014–1019. doi:10.1016/S0959-8049(01)00073-9
34. Janson ET, Oberg K. Long-term management of the carcinoid syndrome. Treatment with octreotide alone and in combination with alpha-interferon. Acta Oncol. 1993;32:225–229. doi:10.3109/02841869309083916
35. Arnold R, Trautmann ME, Creutzfeldt W, et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430–438. doi:10.1136/gut.38.3.430
36. Kliman HJ, Ling IS, Milano KM, et al. Efficacy of telotristat ethyl: a peripheral tryptophan hydroxylase inhibitor that blocks serotonin biosynthesis against cultured liposarcoma, colon cancer, and cholangiocarcinoma cell lines. J Clin Oncol. 2020;38(15_suppl):e15602. doi:10.1200/JCO.2020.38.15_suppl.e15602
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.