Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Antioxidant activity of pomegranate juice reduces emphysematous changes and injury secondary to cigarette smoke in an animal model and human alveolar cells
Authors Husari A, Hashem Y, Bitar H, Dbaibo G, Zaatari G, Sabban M
Received 24 September 2015
Accepted for publication 26 November 2015
Published 3 February 2016 Volume 2016:11(1) Pages 227—237
DOI https://doi.org/10.2147/COPD.S97027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Ahmad Husari,1,* Yasmine Hashem,1 Hala Bitar,1 Ghassan Dbaibo,2,3 Ghazi Zaatari,4 Marwan El Sabban5,*
1Division of Pulmonary and Critical Care Medicine, Department of Internal Medicine, 2Department of Pediatrics and Adolescent Medicine, Division of Pediatric Infectious Diseases, 3Department of Biochemistry and Molecular Genetics, 4Department of Pathology and Laboratory Medicine, 5Department of Anatomy, Cell Biology and Physiological Sciences, Faculty of Medicine, American University of Beirut, Beirut, Lebanon
*These authors contributed equally to this work
Background: Cigarette smoke (CS) increases oxidative stress (OS) in the lungs. Pomegranate juice (PJ) possesses potent antioxidant activities, attributed to its polyphenols. This study investigates the effects of PJ on the damaging effects of CS in an animal model and on cultured human alveolar cells (A549).
Methods: Male C57BL/6J mice were divided into the following groups: Control, CS, CS + PJ, and PJ. Acute CS exposure was for 3 days, while chronic exposure was for 1 and 3 months (5 days of exposure/week). PJ groups received daily 80 µmol/kg via bottle, while other groups received distilled water. At the end of the experiments, different parameters were studied: 1) expression levels of inflammatory markers, 2) apoptosis, 3) OS, and 4) histopathological changes. In vitro, A549 cells were pretreated for 48 hours with either PJ (0.5 µM) or vehicle. Cells were then exposed to increasing concentrations of CS extracted from collected filters. Cell viability was assessed by counting of live and dead cells with trypan blue staining.
Results: Acutely, a significant increase in interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α expression, apoptosis, and OS was noted in CS when compared to Control. PJ significantly attenuated the expression of inflammatory mediators, apoptosis, and OS. Chronically (at 1 and 3 months), increased expression of TNF-α was observed, and lung sections demonstrated emphysematous changes when compared to Control. PJ supplementation to CS animals attenuated the increased expression of TNF-α and normalized lung cytoarchitecture. At the cellular level, CS extract reduced cellular proliferation and triggered cellular death. Pretreatment with PJ attenuated the damaging effects of CS extract on cultured human alveolar cells.
Conclusion: The expression of inflammatory mediators associated with CS exposure and the emphysematous changes noted with chronic CS exposure were reduced with PJ supplementation. In vitro, PJ attenuated the damaging effects of CS extract on cultured human alveolar cells.
Keywords: reactive oxygen species, antioxidants, acute lung injury, emphysema, pomegranate extract, cigarette smoke, inflammatory mediators
Introduction
Cigarette smoke (CS) is a major risk factor for chronic bronchitis, asthma, and lung cancer. It also remains the main culprit in the pathogenesis of chronic obstructive pulmonary disease (COPD).1–3 CS exposure is known for tipping the oxidative balance in the respiratory system, culminating in an oxidative stress (OS) status.4 CS contains >5,000 different chemicals, many of which are oxidants. It is estimated that each puff may contain >1015 free radicals that deplete endogenous antioxidants and tilt the delicate balance in favor of an OS.4–6 Increased OS is well described in patients with COPD;7 increased levels of H2O2 and 8-isoprostane were demonstrated in the exhaled breaths of COPD patients, and increased markers of OS were also present in lung cells of COPD patients.8–10 An increase in the levels of nitrotyrosine, an indicator of cellular damage secondary to free radicals, has also been noted in sputum leukocytes and lung tissue of COPD patients when compared to healthy subjects.11,12
Punica granatum L. (Punicaceae) is usually consumed as pomegranate juice (PJ). Polyphenols, present in PJ, possess potent antioxidants, which may contribute to its antiatherosclerotic and anti-inflammatory properties.13–16 Dietary supplementation of polyphenols may potentially play a therapeutic role in protecting against CS-induced OS.17 Polyphenols act as scavengers of oxygen radical and hydroxyl radical molecules and increase the levels of the antioxidant glutathione by the induction of glutamate cysteine ligase.4 The combination of polyphenols with other phytochemicals such as ellagic acid synergistically enhances the superior antioxidant properties of PJ.18 In addition, there is the added benefit of the anti-inflammatory properties of polyphenols due to the inhibition of nuclear factor kappa B (NF-κB) expression/activation, interleukin (IL)-8 release, cyclooxygenase-2, and heme oxygenase-1.19,20
This study examined whether supplementation of PJ attenuates the damaging effects of acute and chronic CS exposure both in the lungs of an animal model and at the cellular level.
Methods
In vivo study
The Institutional Animal Care and Use Committee of the American University of Beirut approved this study. Four-month-old adult male C57BL/6J mice (22–25 g body weight) were subjected to a 12-hour dark/light cycle. Temperatures of the room and chambers were maintained at 22°C–24°C, and animals were allowed unlimited access to water and standard rodent chow except when animals were placed in the exposure apparatus. The CS exposure apparatus (ONARES; CH Technologies, Westwood, NJ, USA) consisted of a smoke generator, mixing/conditioning chamber, and a 12-port “nose–only” rodent exposure carousel. One port of the carousel was dedicated for sampling analysis and the remaining eleven ports were used for animal exposure. Animals were divided into four groups: Control, CS, CS + PJ, and PJ. Each group consisted of eleven animals, and all animals were acclimated to retainers for 1 week before initiating exposure to laboratory air or CS. Mice were then positioned in retainers and placed into the holes of the carousel. Animals received a continuous flow of CS or room air into the airways via the “nose–only” delivery system. CS was generated from 3R4F cigarettes (University of Kentucky, Lexington, KY, USA) with 0.9 mg total particulate matter (TPM), 9.4 mg tar, and 0.726 mg nicotine per cigarette. As described previously, the machine was set at one puff every minute, with duration of 2 seconds per puff and a volume of 35 mL per puff.21
The sampling system consisted of a vacuum pump attached to one port of the carousel, which drew the diluted aerosol at 1 L/min (controlled by a critical orifice) through a 47 mm fiberglass filter disk (CH Technologies). Filters were replaced every 30 minutes. The TPM per cubic meter (ΔW) was determined gravimetrically by weighing each filter before CS exposure using an analytical balance. TPM concentration was calculated by dividing ΔW over the time and air sampling rate (1 L/min).
The study was performed at three different time points. The acute exposure time setup was deduced from the literature and was set for 3 consecutive days.22 Animals were exposed to CS or laboratory air twice daily (9 am and 2 pm) for 3 hours each. The calculated TPM for the acute exposure was approximately 100 TPM/m3. The chronic exposure setup consisted of two time points (1 and 3 months). Animals were exposed for two sessions of CS (9 am and 2 pm) per day for 5 days/week. Each session, however, lasted for 1 hour and the calculated TPM was approximately 100 TPM/m3.
At the conclusion of the experiment, animals were anesthetized and exsanguinated by severing the aorta. The diaphragm was dissected to allow free lung expansion. The lower lobe of left lung was excised for pulmonary water content evaluation. The left upper lobe was fixed in formalin for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay, OS measurements, and pathology examination. The remaining right lung lobes were frozen for RNA extraction.
Pomegranate juice (PJ) processing and administration
PJ concentrate (Wonderful variety; POM Wonderful, Los Angeles, CA, USA), utilized in this study, was administered via bottle to the CS + PJ and PJ groups. PJ was initiated 1 week before CS or room air exposure and was maintained throughout the experiment. Animals received 80 μmol/kg/day of PJ, while the Control and CS groups received free water. The dose of PJ supplementation was deduced from previous studies.23
Wet-to-dry lung weight
The left lower lobe was weighed and then placed in a 95°C oven to dry for 2 days. The dry tissue was weighed, and the wet-to-dry (W/D) ratio was then calculated.
Transcription expression of IL-1, IL-6, and tumor necrosis factor-α
Changes in the inflammatory mediators’ transcriptional levels were assessed using the reverse transcriptase–polymerase chain reaction (PCR) method. RNA was extracted using the TRIzol method (Invitrogen, Carlsbad, CA, USA) as described before.24 Briefly, 1 mL of TRIzol reagent was used per 50–100 mg of tissue sample, followed by chloroform extraction. RNA samples were precipitated and stored at -80°C. RNA was quantified using the 260/280 nm absorbance ratio method. Total RNA (5 μg) was reverse-transcribed into first–strand complementary DNA (cDNA). Real-time-PCR was performed using the iCycler (Bio-Rad Laboratories, Hercules, CA, USA) with SYBR Green. Specific primers (Tib-Molbiol, Berlin, Germany) were used to assess the expression of the inflammatory mediators in these tissues (IL-1β: Forward CACCTCTCAAGCAGAGCACAG, Reverse GGGTTCCATGGTGAAGTCAAC; IL-6: Forward TCCTACCCCAACTTCCAATGCTC, Reverse TTGGATGGTCTTGGTCCTTAGCC; tumor necrosis factor-α [TNF-α]: Forward AATGGGCTCCCTCTCATCAGTTC, Reverse TCTGCTTGGTGGTTTGCTACGAC). PCR products and their corresponding melting temperatures were analyzed using the iQ5 Optical System Software (Bio-Rad Laboratories). Correction for loading was achieved by subtracting for local background and normalizing against the cDNA levels of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene (GAPDH: Forward GTATTGGGCGCCTGGTCACC, Reverse CGCTCCTGGAAGATGGTGATGG).
Assessment of oxidative stress
Dihydroethidium (DHE) (Molecular Probes; Thermo Fisher Scientific, Waltham, MA, USA) (10 μmol/L dissolved in dimethyl sulfoxide) was applied to lung sections and incubated in a light-protected humidified chamber at 37°C for 15 minutes. Fluorescent images of ethidium-stained tissue were scanned for signal with a scanning confocal microscope (Zeiss, Oberkochen, Germany). Ethidium bromide was excited at 488 nm and emission fluorescence was detected at 560 nm.
Assessment of apoptosis
TUNEL assay was used to monitor the extent of DNA fragmentation.23 Fluorescein-conjugated dUTP incorporated in nucleotide polymers was detected and analyzed using fluorescence microscopy (LSM 410; Zeiss). Positive and negative controls were used to verify the specificity of the TUNEL assay. TUNEL-positive nuclei were distinguished from the TUNEL-negative nuclei by counterstaining with Hoechst 33258.
Lung histology
The upper lobe of the left lung was fixed in 10% buffered formalin, embedded in paraffin, serially sectioned, and stained with hematoxylin and eosin (H&E). A board-certified pathologist, blinded to the different animal groups, evaluated the histopathologic findings under light microscopy (Axio Observer; Zeiss) and determined the degree of lung injury based on degree of inflammatory cell infiltration, alveolar edema, and emphysema.
Quantification of emphysema
Pulmonary mean linear intercept (Lm), an indicator of air space size, was calculated for each sample based on random fields observed at a total magnification of ×200 using a crossline.24 The total length of the crossline divided by the number of the alveolar walls intersecting the test lines was defined as Lm.
In vitro study
Preparation of CS extract
CS extract (CSE) was obtained from filters collected during the animal studies. On the basis of the weight of the TPM collected by the filter, Dulbecco’s Modified Eagle’s Medium (DMEM) incomplete medium was added to yield a final concentration of 10 mg/mL. All recovered media were then mixed together and sterilized using 0.22 μm filters (Costar; Corning, NY, USA).
Cell culture
A549 cells were grown in DMEM high glucose (4.5 g/L) culture media and supplemented with penicillin-G 100 U/mL, streptomycin 100 μg/mL (Gibco-BRL, Paisley, UK), and 10% fetal bovine serum (Sigma-Aldrich Co, St Louis, MO, USA). Cells were seeded in 24-well plates at a density of 60,000 cells per well. Cells receiving PJ supplementation were incubated with 0.5 μM of PJ for 48 hours before exposure to CSE or placebo sterile water. Exposure to CSE for 24 hours was initiated by mixing CSE (which was prepared as stock solutions at concentration of 10 mg/mL) and complete media to the desired final concentration (0.5, 1, 2, 4, and 8 mg/mL). Images were then taken using a light microscope (Axio Observer; Zeiss). Cells were then counted using trypan blue dye to differentiate between dead and live cells. The Institutional Review Board of the American University of Beirut did not require approval be sought for the use of the cell lines in this study, as they are available commercially.
Measurement of ROS
A549 cells were seeded in 24-well plates containing cover slips at a density of 60,000 cells per well. Cells receiving PJ supplementation were incubated with 0.5 μM of PJ for 48 hours before exposure to CSE or placebo sterile water. CSE stock solutions were prepared at a concentration of 10 mg/mL and exposure to CSE for 24 hours was initiated by mixing CSE and complete media to the desired final concentration (0.5, 1, 2, 4, and 8 mg/mL). Cells were then washed with 1× phosphate-buffered saline, pH =7.4. Cells were incubated with 300 μL of DHE (10 μmol/L dissolved in dimethyl sulfoxide and incubated in light-protected humidified chamber at 37°C) for 15 minutes. DHE was then removed and 4% formaldehyde was added for 30 minutes. The cover slips were then mounted on glass slides using ProLong Antifade. Images were taken using a laser scanning confocal microscope (LSM-710; Zeiss).
Results
Acute CS exposure
There was no significant change in the weight of animals between the different groups throughout the different experiments (acute and chronic). There was no difference in W/D ratio, the expression of inflammatory mediators, apoptotic activity, or OS between Control and PJ. A statistically significant increase in mean W/D in CS was noted when compared to Control (P=1.1×10−7). PJ supplementation attenuated the increase in W/D observed in CS (P=1×10−6) (Figure 1A). The expression of IL-6, IL-1β, and TNF-α was significantly increased in CS when compared to Control (P=0.04, 0.01, and 0.004, respectively) and PJ supplementation to CS animals significantly reduced the expression of inflammatory mediators noted in CS (Figure 1B–D). The lungs of CS animals demonstrated a significant increase in OS and in the number of TUNEL-positive apoptotic nuclei when compared to Control (Figure 2A and B). Again, PJ supplementation significantly attenuated the increased OS and apoptotic activity noted in CS.
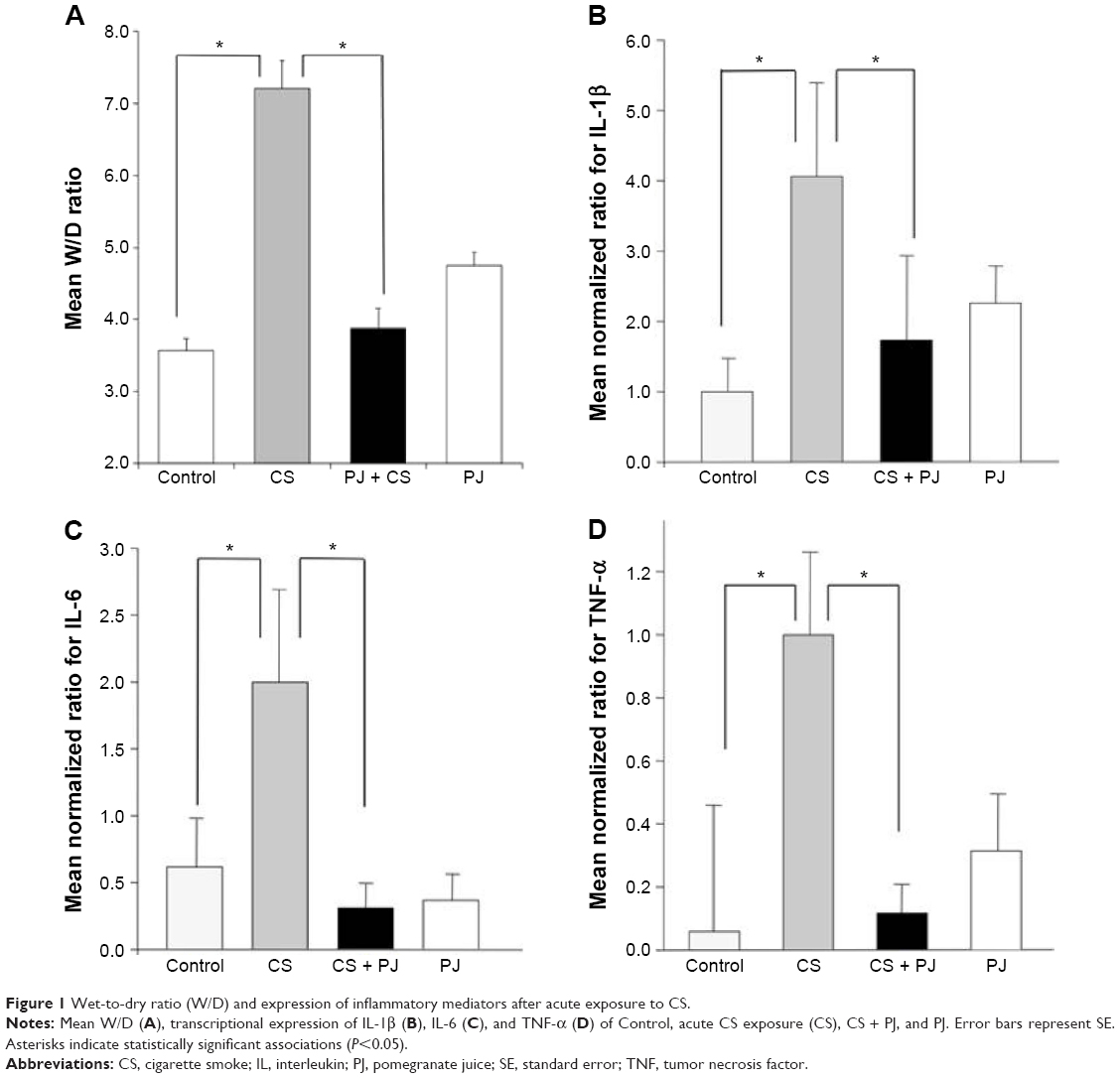 | Figure 1 Wet-to-dry ratio (W/D) and expression of inflammatory mediators after acute exposure to CS. |
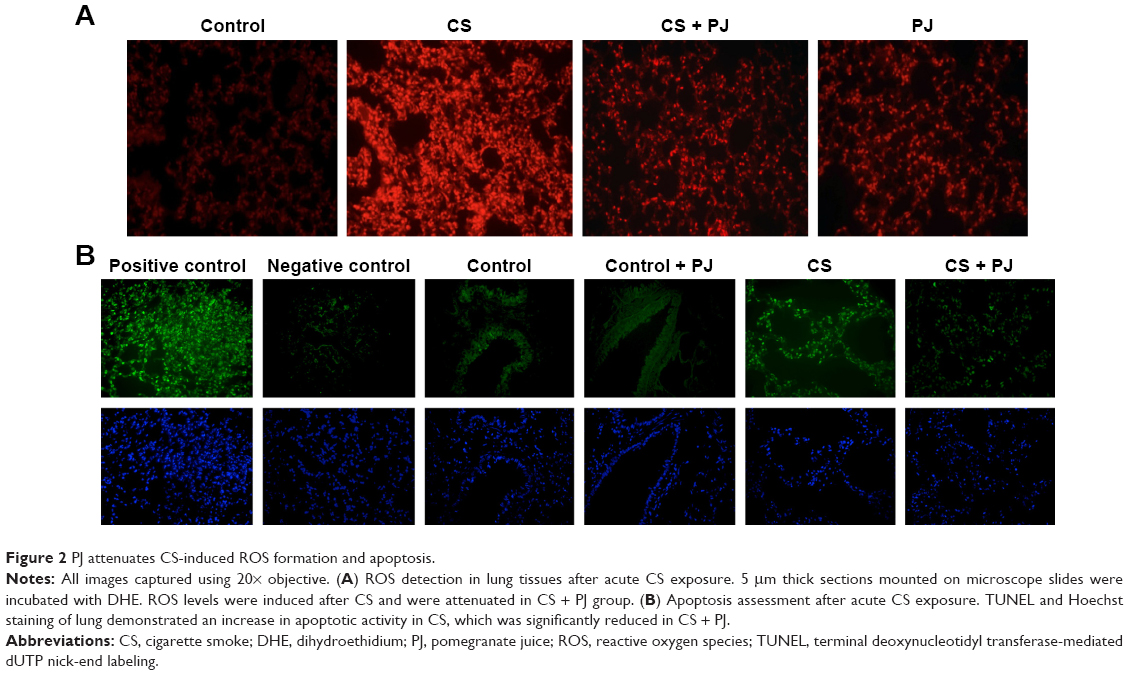 | Figure 2 PJ attenuates CS-induced ROS formation and apoptosis. |
H&E staining of lung sections of CS animals revealed the increased presence of inflammatory cells (macrophages and lymphocytes) across the bronchioles and in lung parenchyma. Edematous and thickened alveolar walls were also observed. CS animals treated with PJ displayed normal alveolar structure with minimal infiltration of inflammatory cells and swelling of the alveoli (Figure 3).
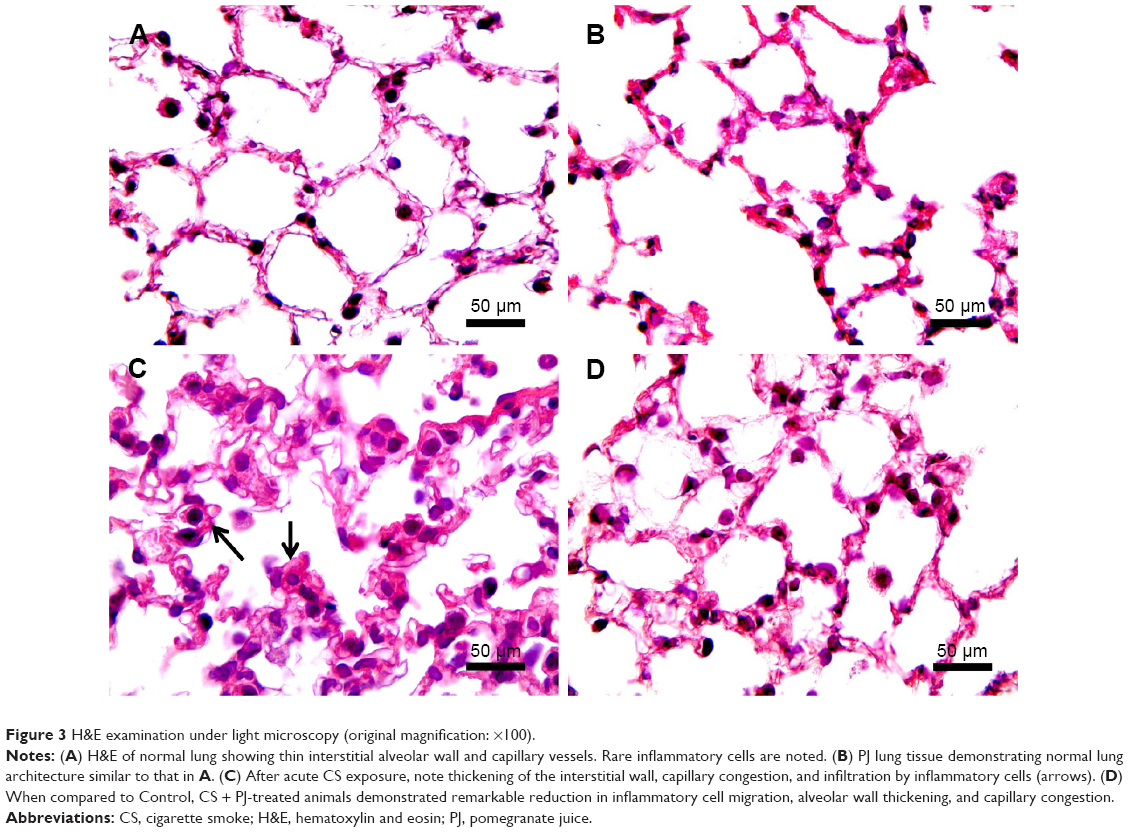 | Figure 3 H&E examination under light microscopy (original magnification: ×100). |
One-month CS exposure
After 1 month of CS exposure, only TNF-α demonstrated persistent increased expression in CS when compared to Control (P=0.04) (Figure 4A). PJ supplementation in the CS + PJ group normalized the increased expression of TNF-α observed in CS. Histologically, subtle changes suggestive of early enlargement of the airspaces, accompanied with limited destruction of the normal alveolar architecture (increases of airspace enlargement), were noted in CS. In contrast, CS treated with PJ displayed normal alveolar structure (Figure 5).
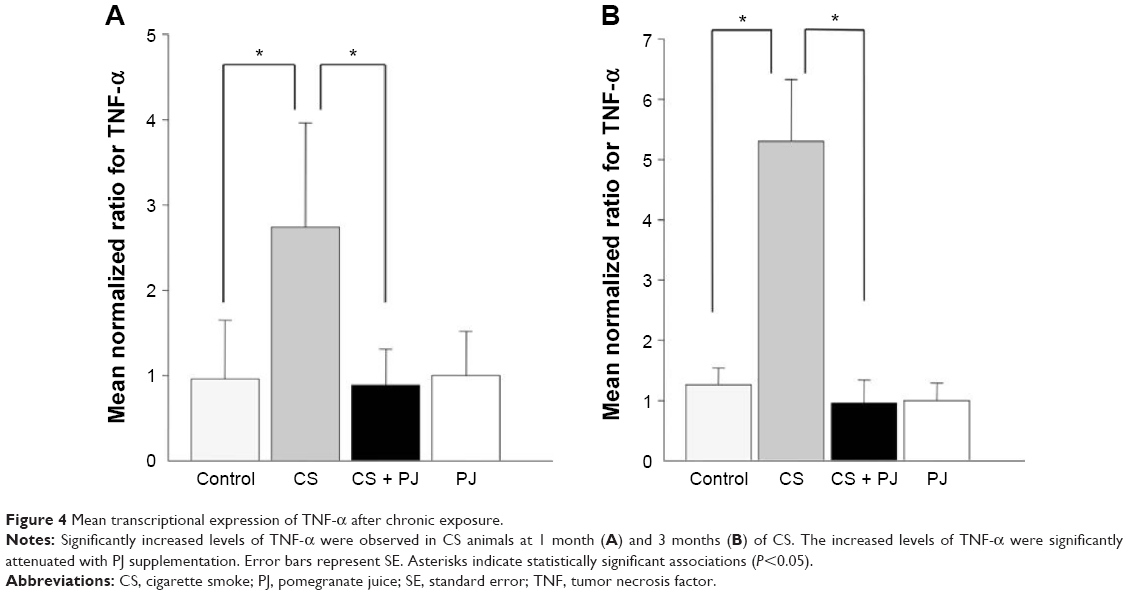 | Figure 4 Mean transcriptional expression of TNF-α after chronic exposure. |
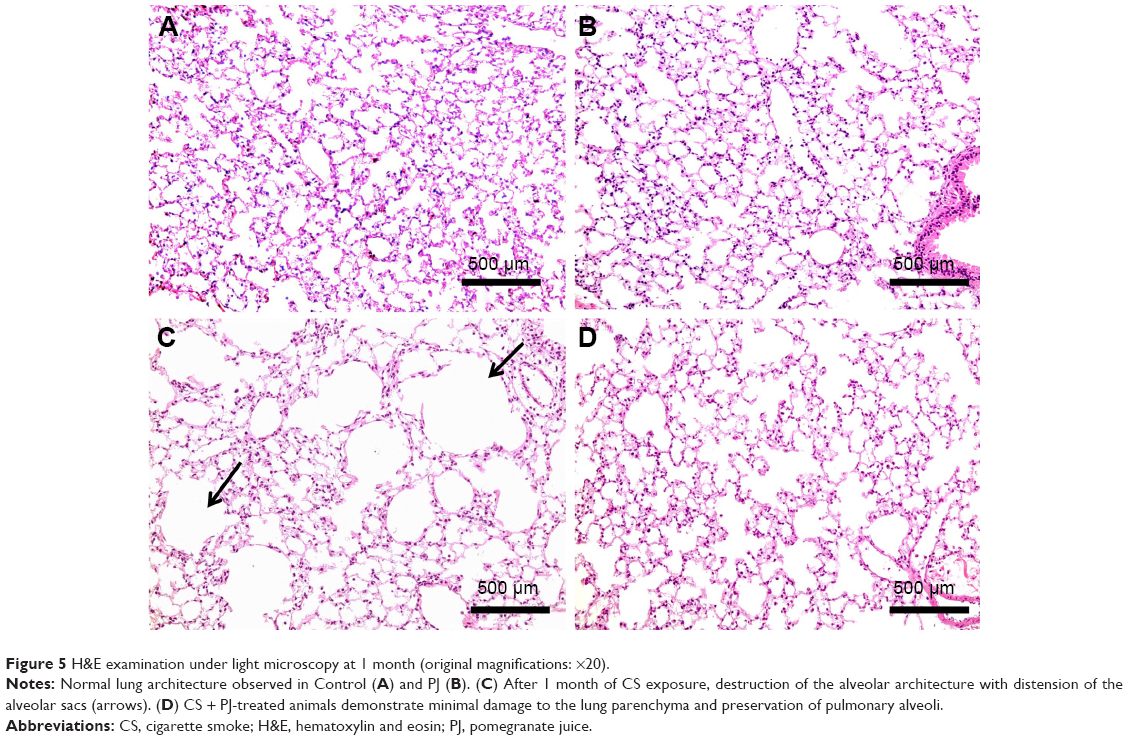 | Figure 5 H&E examination under light microscopy at 1 month (original magnifications: ×20). |
Three-month CS exposure
Similar to the results of 1-month CS exposure, increased expression of TNF-α was also observed in CS, which was attenuated with PJ supplementation (P=0.001) (Figure 4B). Histological evaluation after 3 months of CS, however, revealed significant emphysematous changes with enlargement of the airspaces, accompanied by the destruction of the normal alveolar architecture in CS (Figure 6). Linear intercept data confirmed the significant increase in airspace size in CS lungs compared to Control lungs (P<0.0001). Chronic supplementation with PJ reversed the emphysematous changes noted histologically and attenuated the increase in Lm distance observed in the CS group (P<0.0001).
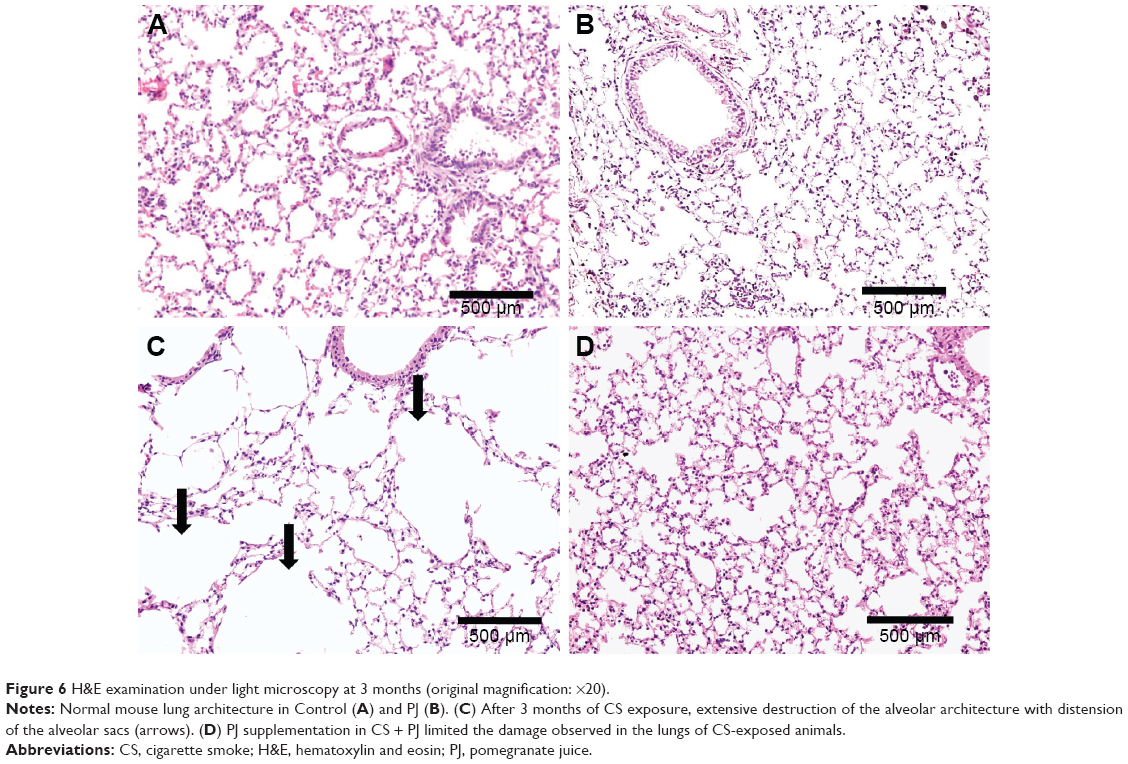 | Figure 6 H&E examination under light microscopy at 3 months (original magnification: ×20). |
In vitro study
CSE reduced cellular proliferation, when compared to Control, in a dose-dependent manner. At 2 mg/mL of CSE exposure, cellular proliferation was completely arrested and cellular death was observed. Human alveolar cells, pretreated with PJ at a dose of 0.5 μM, demonstrated significant resistance to the effects of CSE and shifted cellular inhibition and death to higher doses of CSE. Arrest of cellular growth and cellular death were seen in CSE-only exposure at 2 mg/mL of CSE, whereas cellular proliferation was nearly unaffected and cells appeared healthy in PJ + CSE at the same concentration (Figure 7). Similarly, a significant increase in OS in A549 cells was observed with increased concentrations of CSE. Again, PJ supplementation suppressed CS-induced reactive oxygen species (ROS) activity in A549 cells (Figure 8).
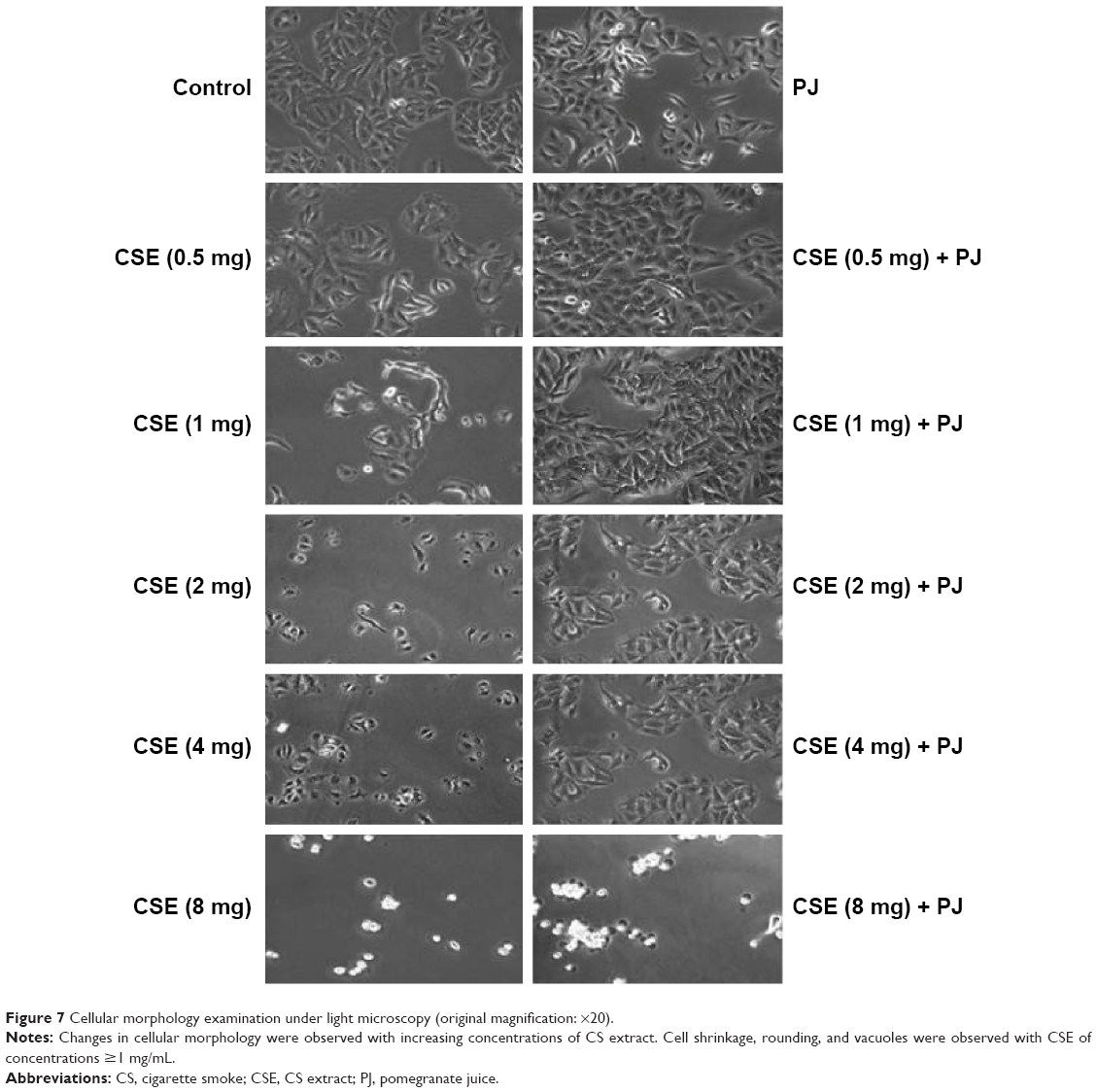 | Figure 7 Cellular morphology examination under light microscopy (original magnification: ×20). |
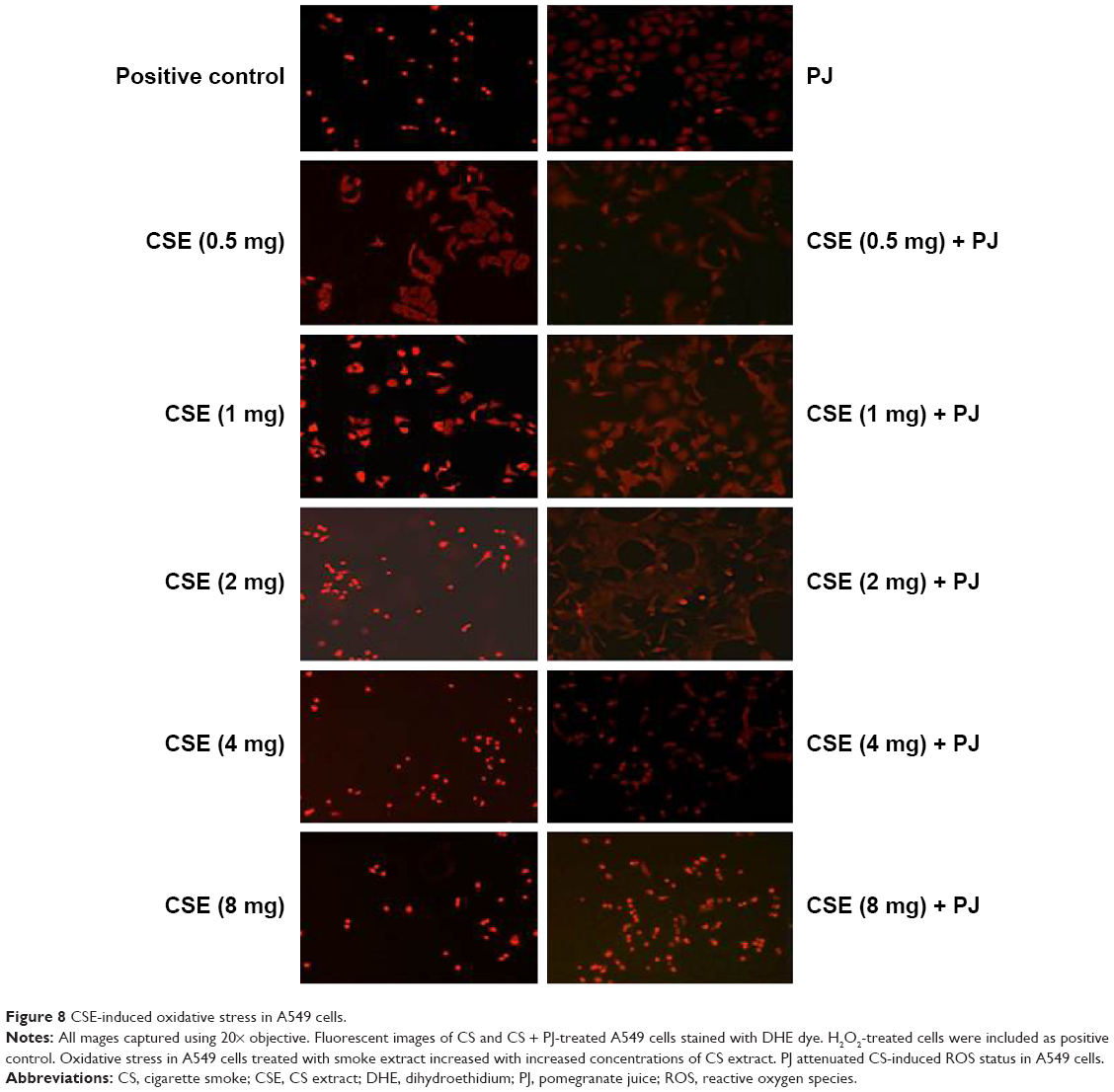 | Figure 8 CSE-induced oxidative stress in A549 cells. |
Discussion
This study examined the damaging effects of CS in an animal model at different time points (acute and chronic) and in human alveolar cells. The “nose–only” delivery system, utilized in the animal study, delivered CS directly into the airways in a continuous, precise, and timely manner. This eliminated inconsistent CS exposure associated with whole-body exposure. As such, accelerated lung injury and early emphysematous changes were observed after 1 month of CS exposure.25 Acute CS exposure was associated with a significant increase in W/D ratio, OS, cellular death, and a surge in inflammatory mediators, in association with infiltration by inflammatory cells.26–28 After 1 or 3 months of CS exposure, only the expression of TNF-α remained persistently elevated, and emphysematous changes with loss of alveolar sacs were noted at 1 month but were clearly evident at 3 months. The findings of persistent increased expression of TNF-α are consistent with the detrimental role of TNF-α in CS. TNF-α is known to prime neutrophils, on exposure to CS, resulting in an increase in its oxidative burst expression, leading to augmented lung damage. In mouse animal models similar to our model, TNF-α was noted to be central in CS-induced loss of alveoli and emphysema.28–30 At the cellular level, CSE, in a dose-dependent manner, inhibited cellular growth and induced cellular death of human alveolar cell cultures.
The study then examined the role of antioxidants in attenuating lung injury secondary to CS. This role is questioned due to recent conflicting, and rather disappointing, animal and human studies that described limited – and possibly adverse – effects associated with exogenous antioxidant supplementation.31–33 Excessive administration of antioxidants suppresses total endogenous ROS formation and diminishes the capability of the recipient to kill bacteria and eliminate damaged or precancerous cells.34 The results of this study supported the role of pomegranate as a powerful antioxidant in protecting the lungs from CS exposure. In vivo, PJ reversed and attenuated all the damaging effects of CS. Acutely, PJ supplementation attenuated the increase in W/D ratio, OS, apoptosis, and the surge of all inflammatory mediators associated with CS. Chronically, PJ attenuated the elevated expression of TNF-α and the emphysematous changes observed histologically with the increase in airspaces and the loss of alveoli. Finally, at the cellular level, PJ created significant resistance to the effects of CSE and higher doses of CSE concentration were needed to elicit similar results as in CSE-only cell cultures.
The success in achieving therapeutic outcomes with antioxidant supplementation is dependent on the choice, the dose of the antioxidant, and the timing of administration.34,35 PJ, utilized in this study, is an important source of powerful antioxidants that include anthocyanins, pelargonidin, and polyphenols. PJ possesses the added value of anti-inflammatory properties as well.19,20 As for the precise dose of daily PJ supplementation, based on previous studies, the daily dose of PJ was set at 80 μmol/kg/day.23 Finally, the timing of administration is vital. In this study, PJ supplementation was initiated 1 week before the exposure to CS, priming animals with supplementary antioxidants needed to neutralize ROS generated from CS.
Emphysema is the hallmark of CS-induced lung injury.3 This study demonstrated the favorable effects of antioxidant supplementation, represented by PJ, in limiting the damaging effects of CS and preventing the formation of emphysematous changes in the lung in an animal model. Further animal and human studies are needed to explore the beneficial role of antioxidants, with clear emphasis on the design of the study, and these need to precisely determine the dosing and timing of antioxidant administration in order to elicit a positive protective effect.
Acknowledgments
This study was supported by the Mikati Foundation, Beirut, Lebanon, and the Medical Practice Plan and the University Research Board at the American University of Beirut, Beirut, Lebanon.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir Med. 2012;106(3):319–328. | ||
U.S. Department of Health and Human Services. The Health Consequences of Smoking – 50 Years of Progress: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2015. | ||
U.S. Department of Health and Human Services. How Tobacco Smoke Causes Disease: What It Means to You. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2015. | ||
Pasupathi P, Saravanan G, Farook J. Oxidative stress bio markers and antioxidant status in cigarette smokers compared to nonsmokers. J Pharm Sci Res. 2009;1(2):55–62. | ||
Forni LG, Mora-Arellano VO, Packer JE, et al. Nitrogen dioxide and related free radicals: electron-transfer reactions with organic compounds in solution containing nitrite or nitrate. J Chem Soc Perkin Trans. 1986;2:1–6. | ||
Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J. 2006;28(1):219–242. | ||
Bowler RP, Barnes PJ, Crapo JD. The role of oxidative stress in chronic obstructive pulmonary disease. COPD. 2004;1(2):255–277. | ||
Antczak A, Ciebiada M, Pietras T, et al. Exhaled eicosanoids and biomarkers of oxidative stress in exacerbation of chronic obstructive pulmonary disease. Arch Med Sci. 2012;8(2):277–285. | ||
Foschino MP, Carpagnano GE, Spanevello A, et al. Inflammation, oxidative stress and systemic effects in mild chronic obstructive pulmonary disease. Int J Immunopathol Pharmacol. 2007;20(4):753–763. | ||
Kostikas K, Papatheodorou G, Psathakis K, et al. Oxidative stress in expired breath condensate of patients with COPD. Chest. 2003;124(4):1373–1380. | ||
Lisette IZ, Kunz JB, Simona EB. Smoking status and anti-inflammatory macrophages in bronchoalveolar lavage and induced sputum in COPD. Respir Res. 2011;12:34. | ||
Ichinose M, Sugiura H, Yamagata S, et al. Increase in reactive nitrogen species production in chronic obstructive pulmonary disease airways. Am J Respir Crit Care Med. 2000;162(2 pt 1):701–706. | ||
Langley P. Why a pomegranate? BMJ. 2000;321(7629):1153–1154. | ||
El-Nemr SE, Ismail IA, Ragab M. Chemical composition of juice and seeds of pomegranate fruit. Nahrung. 1990;7:601–606. | ||
Gil MI, Tomas-Barberan FA, Hess-Pierce B, et al. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J Agric Food Chem. 2000;48(10):4581–4589. | ||
Filomena N, Sharon W, Lilach OL, et al. Beneficial effects of pomegranate juice on oxidation-sensitive genes and endothelial nitric oxide synthase activity at sites of perturbed shear stress. Proc Natl Acad Sci U S A. 2001;102(13):4896–4901. | ||
Arts IC, Hollman PC. Polyphenols and disease risk in epidemiologic studies. Am J Clin Nutr. 2005;81(1 suppl):317S–325S. | ||
Sreekumar S, Sithul H, Muraleedharan P, et al. Pomegranate fruit as a rich source of biologically active compounds. Biomed Res Int. 2014;2014:686921. | ||
Biswas SK, McClure D, Jimenez LA, et al. Curcumin induces glutathione biosynthesis and inhibits NF-κB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7(1–2):32–41. | ||
Yun N, Kang JW, Lee SM. Protective effects of chlorogenic acid against ischemia/reperfusion injury in rat liver: molecular evidence of its antioxidant and anti-inflammatory properties. J Nutr Biochem. 2012;23(10):1249–1255. | ||
Coggins CR. A review of chronic inhalation studies with mainstream cigarette smoke, in hamsters, dogs, and nonhuman primates. Toxicol Pathol. 2001;29(5):550–557. | ||
Bond JA, Chen BT, Griffith WC, et al. Inhaled cigarette smoke induces the formation of DNA adducts in lungs of rats. Toxicol Appl Pharmacol. 1989;99(1):161–172. | ||
Husari A, Khayat A, Bitar H, et al. Antioxidant activity of pomegranate juice reduces acute lung injury secondary to hyperoxia in an animal model. BMC Res Notes. 2014;21(7):664. | ||
Husari A, Khayat A, Awdeh H, et al. Activated protein C attenuates acute lung injury and apoptosis in a hyperoxic animal model. Shock. 2010;33(5):467–472. | ||
Beckett EL, Stevens RL, Jarnicki AG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol. 2013;131(3):752–762. | ||
Takubo Y, Guerassimov A, Ghezzo H, et al. Alpha1-antitrypsin determines the pattern of emphysema and function in tobacco smoke-exposed mice: parallels with human disease. Am J Respir Crit Care Med. 2002;166(12 pt 1):1596–1603. | ||
Kamiide Y, Furuya M, Inomata N, et al. Chronic exposure to cigarette smoke causes extrapulmonary abnormalities in rats. Environ Toxicol Pharmacol. 2015;39(2):864–870. | ||
Churg A, Wang RD, Tai H, et al. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170(5):492–498. | ||
Friedrichs B, Neumann U, Schüller J, et al. Cigarette-smoke-induced priming of neutrophils from smokers and non-smokers for increased oxidative burst response is mediated by TNF-α. Toxicol In Vitro. 2014;28(7):1249–1258. | ||
Li YT, He B, Wang YZ. Exposure to cigarette smoke upregulates AP-1 activity and induces TNF-alpha overexpression in mouse lungs. Inhal Toxicol. 2009;21(7):641–647. | ||
Bjelakovic G, Nikolova D, Gluud LL, et al. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev. 2012;14:3. | ||
Albanes D. Beta-carotene and lung cancer: a case study. Am J Clin Nutr. 1999;69(6):1345–1350. | ||
Weissig V, Guzman-Villanueva D. Nanocarrier-based antioxidant therapy: promise or delusion? Expert Opin Drug Deliv. 2015;12(11):1783–1790. | ||
Jain M, Chandel NS. Rethinking antioxidants in the intensive care unit. Am J Respir Crit Care Med. 2013;188(11):1283–1285. | ||
Poljsak B, Šuput D, Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxid Med Cell Longev. 2013;2013:956792. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.