Back to Journals » Clinical Epidemiology » Volume 9
Antinuclear antibody prevalence in a general pediatric cohort from Mexico City: discordance between immunofluorescence and multiplex assays
Authors Somers EC ![]() , Monrad SU, Warren JS, Solano M, Schnaas L, Hernandez-Avila M, Tellez-Rojo MM, Hu H
, Monrad SU, Warren JS, Solano M, Schnaas L, Hernandez-Avila M, Tellez-Rojo MM, Hu H
Received 6 September 2016
Accepted for publication 18 October 2016
Published 20 December 2016 Volume 2017:9 Pages 1—8
DOI https://doi.org/10.2147/CLEP.S121632
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor H Sorensen
Emily C Somers,1–3 Seetha U Monrad,1 Jeffrey S Warren,4 Maritsa Solano,5 Lourdes Schnaas,6 Mauricio Hernandez-Avila,5 Martha Maria Tellez-Rojo,5 Howard Hu7
1Divison of Rheumatology, Department of Internal Medicine, 2Department of Environmental Health Sciences, 3Department of Obstetrics & Gynecology, 4Division of Clinical Pathology, Department of Pathology, University of Michigan, Ann Arbor, MI, USA; 5Center for Nutrition and Health Research, National Institute of Public Health, Cuernavaca, Morelos, 6Department of Developmental Neurobiology, National Institute of Perinatology, Mexico City, Mexico; 7Occupational and Environmental Health, Dalla Lana School of Public Health, University of Toronto, Toronto, ON, Canada
Objective: To characterize antinuclear antibody (ANA) prevalence according to distinct assay methodologies in a pediatric cohort from Mexico City, and to further examine associations with age and sex.
Methods: Serum ANA were measured by indirect immunofluorescence assay (IFA) and multiplex immunoassay in 114 children aged 9–17 years. IFA was considered positive at a cutoff titer of ≥1:80. Agreement between assay methods was assessed by kappa statistic. Sensitivity, specificity, and 95% confidence intervals (CIs) of the multiplex were computed with IFA as the reference standard.
Results: Of the 114 children (mean age 14.7 [standard deviation 2.1] years; 54 [47%] female), 18 of 114 (15.8%) were ANA positive by IFA, and 11 of 114 (9.6%) by 11-antigen multiplex assay. ANA prevalence was higher in females compared with males by both of the methods (ratios 1.6–1.9 to 1). Agreement between tests was classified as slight by kappa (κ=0.177 [95% CI −0.051, 0.406]). The multiplex immunoassay had sensitivity of 22.2% (95% CI 6.4, 47.6) and specificity of 92.7% (95% CI 85.6, 97.0), and failed to capture 3 of 4 (75%) of the high-titer (≥1:1280) IFA-positives.
Conclusion: Up to 15% of children in this general population cohort were ANA positive, with a higher rate of positivity among females according to both assay methods. Substantial discordance in ANA results was found between IFA and multiplex methods, even for high-titer IFA positives. These findings underscore the need to sufficiently account for assay characteristics when interpreting ANA test results, and support IFA as the more appropriate assay for studies of subclinical autoimmunity.
Keywords: Autoreactivity, biomarker, immune dysfunction, preclinical, subclinical autoimmunity, autoantibodies, pediatric, epidemiology
Introduction
Antinuclear antibodies (ANAs), increasingly referred to by the more technically correct term “autoantibodies to cellular antigens”,1 are associated with numerous autoimmune diseases, such as systemic lupus erythematosus (SLE), systemic sclerosis, Sjögren’s syndrome, and idiopathic inflammatory myopathies. ANAs are highly sensitive for some of these disorders, and are considered the serologic hallmark of SLE, with 95% of SLE patients being ANA-positive.2
Numerous studies have demonstrated that circulating autoantibodies can be detected several years in advance of clinical autoimmune diagnoses. A seminal study of 130 SLE patients found that 88% had at least one SLE-associated autoantibody up to several years prior to diagnosis.3 The existence of autoantibodies in asymptomatic individuals who subsequently develop disease has been reported in a variety of other autoimmune conditions, including rheumatoid arthritis4–6 and type 1 diabetes.7 The terms “preclinical”, “subclinical”, and even “benign” autoimmunity have been used to describe the presence of autoantibodies or other markers of autoreactivity or altered immune response in the absence of tissue injury or clinical manifestations. We favor the term “subclinical autoimmunity” in prospective settings, as a reflection that not all individuals with markers of autoimmunity will progress to overt autoimmune disease. A conceptual framework has emerged, whereby a subclinical autoimmunity phase of variable duration is thought to represent an important early stage in the development of clinical autoimmune disease.8–12 Accurate identification of which individuals will progress from subclinical autoimmunity to a pathologic state, and identification of underlying risk factors, depends on the ability to reliably measure autoantibodies in the epidemiologic setting.
Approximately 13% of the general population demonstrates ANA positivity according to standard immunofluorescence methodology (at a 1:80 cutoff titer).13,14 While pediatric populations have been less well studied, a US-NHANES study reported ANA prevalence rates among 12–19-year-olds of approximately 9.5% for non-Hispanic whites, 12.5% for Mexican Americans, and 14% for non-Hispanic blacks.14 Similar rates were reported in two other large series of children: 12.3% in an Italian study of 261 children aged 8–13 years15 and 14.6% among 151 persons aged 5–20 years in a Brazilian study.16
Traditionally, autoantibody detection has been performed by indirect immunofluorescence assay (IFA), which remains the reference standard of choice by professional bodies.1,17 In recent years, semiquantitative solid-phase assays utilizing multiplex bead technology have been FDA-approved, and have become commonplace due to their automated, high-throughput nature. However, discordance of ANA results between these assay types has been demonstrated, at variable degrees across racial and ethnic groups.18,19 A pediatric series found discordance between IFA and multiplex results among juvenile inflammatory arthritis (JIA) patients, with all 10 JIA patients ANA positive by IFA but negative by multiplex.20 Thus, it is essential to learn how the test characteristics of different assays may vary across population subsets, as has been called for by international recommendations on ANA assessment.1
We performed this study to characterize ANA prevalence according to distinct assay methodologies in a pediatric cohort from Mexico City, and to further examine associations with age and sex.
Methods
Study population
The study population was comprised of offspring born to mothers who had previously enrolled in a birth cohort in Mexico City – the Early Life Exposures in Mexico to Environmental Toxicants (ELEMENT) study – during 1994–2003. As described in detail elsewhere,21–24 the original ELEMENT study enrolled low-to-moderate income, pregnant women from the National Institute of Perinatology, Hospital General Dr. Manuel Gea Gonzalez, or clinics affiliated with the Mexican Social Security Institute. Eight hundred twenty-eight children of these mothers were enrolled during 2007–2011 into a follow-up study. For the present study, 114 offspring with banked serum from follow-up visits in 2010–2011 were included. Ethics approval was obtained from the respective institutional review boards (National Institute of Public Health of Mexico, Harvard School of Public Health, University of Michigan, and participating hospitals). Maternal written informed consent and written assent from the children were obtained.
ANA testing
ANA assays were performed in the CLIA (Clinical Laboratory Improvement Amendments)-certified Clinical Immunopathology Laboratory at the University of Michigan Health System using stored serum from the ELEMENT biobank. Specimens were initially collected in standard gold SST (serum separator tube) vacutainers and allowed to clot at room temperature before centrifugation at 3,500 rpm for 10 minutes. Serum was then aliquoted into cryovials and stored at −70°C prior to analysis.
Indirect IFA
Serum was applied to HEp-2 cell slides (Fluorescent HEp-2 ANA; Immuno Concepts, Sacramento, CA, USA) after dilution in 0.15 M NaCl and 10 mM phosphate buffered saline (PBS), pH 7.4, and incubated for 30 minutes at room temperature in a moist chamber. After washing twice in PBS for 10 minutes, cells were incubated with fluorescein isothiocyanate-conjugated goat anti-human Ig (IgG heavy and light chains) for 30 minutes in the dark. After two further washes, slides were assembled with buffered glycerol, pH 9.5, and coverslipped. ANA titer was determined by testing successive twofold dilutions of the serum to endpoint. PBS buffer and negative human control serum supplied with the commercial kit (Immuno Concepts) were used as negative controls. Analysis was performed by two independent expert observers using a Zeiss Rx 100 microscope under 400× magnification. Samples were classified as ANA-HEp-2 positive if a well-defined indirect immunofluorescence pattern was identified (≥3+/4) at 1:80 dilution by both observers. Patterns were determined based on standard classification criteria.25 Fluorescence photomicrographs from our laboratory with representative images are included in Figure 1A–D. Expert consensus is lacking as to the titer to consider positive for children, and some have used a dilution of 1:40.1 We utilized a cutoff of 1:80, as it is our standard institutional positive/negative cutoff based on a laboratory validation study in which the false-positive rate was set at ≤5% (J Warren, unpublished data, 2007).
 | Figure 1 Representative photomicrographs of antinuclear antibody (ANA) patterns at 400x magnification. (A) negative ANA; (B) positive/homogeneous pattern; (C) positive/speckled pattern; (D) positive/nucleolar pattern. |
Multiplex immunoassay
We used the ANA multiplex immunoassay (BioPlex 2200 ANA Screen; BioRad, Hercules, CA, USA), an automated two-color fluorescence microbead flow system in which recombinant or purified native antigen has been linked to beads. Specific antigen beads were incubated with serum, followed by incubation with fluorochrome-linked antibody directed against human immunoglobulin G. This test detects autoantibodies to 11 antigens with clinical relevance to autoimmune/rheumatic diseases (double-stranded DNA [dsDNA], SS-A, SS-B, Sm, RNP, SmRNP, Scl-70, Jo-1, centromere B, ribosomal P, chromatin). A negative result indicates that the serum shows no reactivity for these antibodies.
Statistics
Characteristics of the study population were compared between groups using two-sample t-tests or Wilcoxon rank-sum tests for continuous data, or chi-squared tests for categorical data. Agreement between assays was assessed by the kappa statistic. Kappa statistic interpretation was classified as follows: <0, less than chance; 0.01–0.20, slight; 0.21–0.40, fair; 0.41–0.60, moderate; 0.61–0.80, substantial; 0.81–0.99, almost perfect.26 Sensitivity and specificity, with exact binomial 95% confidence intervals (CIs), of the multiplex test were calculated, with IFA as the “gold standard”. Stata v.13 software was used (StataCorp LP, College Station, TX, USA).
Results
Of the 114 children in this study, 54 (47%) were female. Ages at follow-up ranged from 9 to 17 years (mean [SD] 14.7 [2.1]), and were similar between males and females (female 14.5 [0.30]; male 15.0 [0.26]; p=not signficant).
ANA prevalence
ANA results according to assay methodology are presented in Table 1. ANA positivity was found in 18 of 114 (15.8%) by IFA and 11 of 114 (9.6%) by multiplex. Three of the children who were multiplex positive had only dsDNA positivity among the 11 antigens tested. The presence of only anti-dsDNA by multiplex, in the absence of other relevant findings, is considered by some experts to represent a false-positive, often due to anti-single-stranded DNA (ssDNA) antibodies.27 When these three children were classified as negative by multiplex, the multiplex-positive proportion decreased to 8 of 114 (7%) overall, 5 of 54 for females (9.3%), and 3 of 60 for males (5%). Regardless of assay methodology, in this pediatric population, a higher proportion of females compared with males tested positive for ANAs, though not reaching statistical significance, with female-to-male ratios for ANA positivity ranging from 1.6–1.9 to 1. Speckled immunofluorescence patterns predominated at lower titers, and the homogeneous pattern at higher titers (Figure 2A). One mitotic spindle pattern was found, which is considered an atypical pattern and thus classified as ANA-negative at our institution; this person also was negative by multiplex. No other atypical patterns were observed in our cohort, including antibodies to dense fine speckles (anti-DFS70) which have been increasingly noted in healthy populations.28 From the multiplex-11 panel, 4 specificities did not yield any positives (ribosomal P, SS-A, centromere B, and Jo-1); distribution shown in Figure 2B.
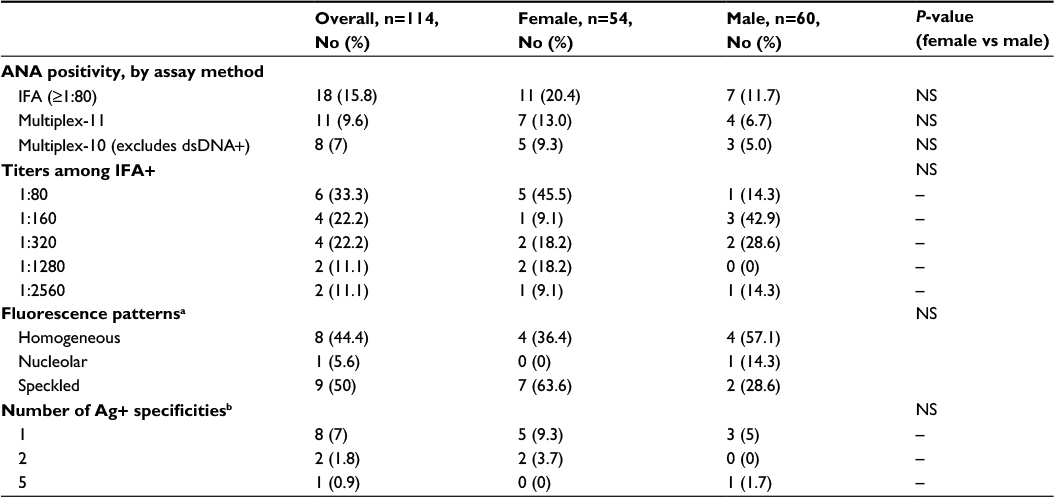 | Table 1 ANA positivity according to assay method in a pediatric cohort (aged 9–17 years) from Mexico City Notes: aAmong IFA positives, bamong multiplex-11 positives. Abbreviations: Ag, antigen; ANA, antinuclear antibody; dsDNA, double-stranded DNA; IFA, immunofluorescence assay; NS, not significant. |
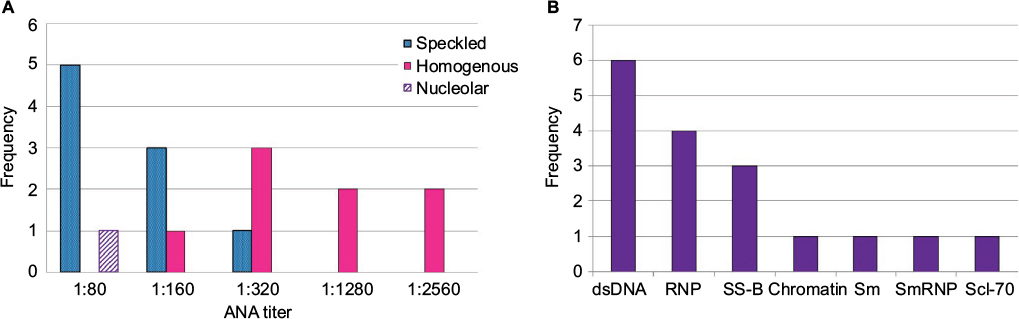 | Figure 2 (A) ANA titers and patterns, by indirect IFA. (B) Autoantibody specificities by multiplex-11 assay. Four specificities did not yield any positives (ribosomal P, SS-A, centromere B, and Jo-1). Abbreviations: ANA, antinuclear antibody; dsDNA, double-stranded DNA; IFA, immunofluorescence assay. |
Age patterns
According to IFA, mean age was similar between those who screened positive (14.7 years [SD 2.2]) versus negative (15.2 years [1.9]); p=not significant. By multiplex, there was a trend toward older age among multiplex-positives (15.8 years [1.3]) compared with multiplex-negatives (14.6 years [2.2]); p=0.07 by Wilcoxon rank-sum test. In general, the IFA screen captured positives from a broader age distribution, with the multiplex predominantly capturing positives among those aged >15 years (Figure 3).
 | Figure 3 Age distributions according to ANA screening status by indirect IFA and multiplex-11 assays. Abbreviations: ANA, antinuclear antibody; IFA, immunofluorescence assay. |
Agreement between assays
The observed agreement between IFA and multiplex-11 in the overall study population was 81.6%, compared to expected agreement by chance of 77.6%, yielding a kappa coefficient of κ=0.177 (95% CI −0.051, 0.406); by conventional interpretation, this indicates “slight” agreement. Agreement was higher for boys (κ=0.305 [95% CI −0.075, 0.684]) than for girls (κ=0.076 [95% CI −0.208, 0.360]). The extent of overlap between tests is presented Figure 4. Of the 25 children with a positive ANA by either test, only 4 (16%) were dual-positive (by both IFA and multiplex); the lower overall prevalence captured by multiplex did not simply reflect a subset of the IFA positives. All four dual-positives occurred in children older than 15 years. With IFA (≥1:80) as the reference standard, multiplex sensitivity was 22.2% for both the 11- and 10-antigen panels; specificity was 92.7% and 95.8%, respectively. All three of the multiplex dsDNA-only positives were negative by IFA, lending support for exclusion of the dsDNA antigen from this test. There was discordance across the range of IFA titers (Table 2), and the multiplex failed to capture 3 of 4 (75%) of the high-titer IFAs (≥1:1280).
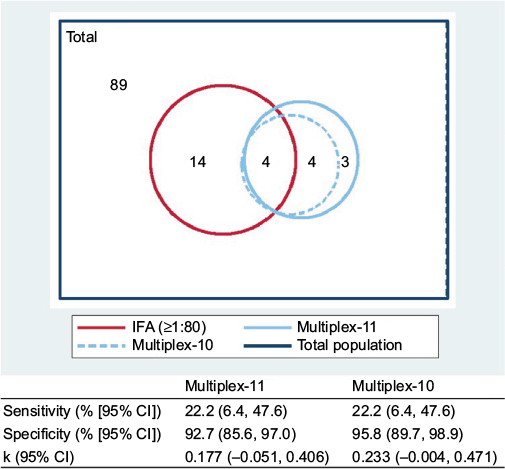 | Figure 4 Proportional Venn diagram displaying overlap in positivity for ANA assay methods (n=114). Corresponding sensitivity, specificity, and kappa estimates are presented, for multiplex compared to IFA (≥1:80) as the “gold standard”. Multiplex-10 excludes the dsDNA antigen from the full multiplex-11 panel. Abbreviations: ANA, antinuclear antibody; dsDNA, double-stranded DNA; IFA, immunofluorescence assay. |
 | Table 2 Comparison of IFA titers and number of multiplex-positive antigens Abbreviations: Ag, antigen; IFA, immunofluorescence assay; dsDNA, double-stranded DNA; neg, negative. |
Discussion
We have described ANA prevalence according to competing assay methodologies in a pediatric cohort from the general Mexico City population. Understanding serologic patterns associated with subclinical autoimmunity in various population subsets is an important component of immunoepidemiology. As ANA results are highly dependent upon assay type, appreciation of methodologic differences and their implications is essential. Further, as significant health disparities in the incidence, severity and age of onset have been documented for autoimmune diseases such as SLE,29–32 it follows that subclinical autoimmunity should be specifically characterized in various population demographics. Among the 9–17-year-olds in our study, 15.8% were positive by IFA and 9.6% by multiplex. This rate of ANA-IFA positivity extends the high end of the range of approximately 9.5%–14.6% reported in other pediatric populations using IFA at a 1:80 cutoff,14–16 and is compatible with epidemiologic data indicating that risk of SLE for Hispanics is higher than for non-Hispanic whites, yet lower than for blacks.33,34 In our study, the proportion of positive females was close to double that of males for each assay method. It is well-recognized that autoimmune diseases disproportionately affect females, with female-to-male prevalence ratios of up to 9:1.35 Our data add to accumulating evidence that the phenomenon of subclinical autoimmunity is likewise more prevalent among females, even in childhood and adolescence.36
We found that ANA positivity was lower based on the multiplex methodology compared to IFA, which is consistent with findings from patients with clinical autoimmune diseases such as SLE and other ethnic groups.19 This is in part due to the fact that solid-phase methods such as multiplex assays test for a restricted range of approximately 8–11 antigens, compared with the broader range of 100–150 possible autoantigens identified by IFA.37 However, we found that the lower positivity rate for the multiplex assay was not simply a reflection of a smaller subset of the IFA positives being captured by multiplex, as would have been theoretically expected. Rather, discordance occurred bidirectionally (ie, multiplex positive/IFA negative and vice versa); only 4 specimens among the 114 tested were concordant positives (yielding an ANA positivity rate of 3.5% for specimens classified as positive by both methods). Such discordance may be due to recombinant antigens, such as those used in the multiplex assay, displaying different epitopes than those on native antigens, such as are used in the IFA.38 Further, autoantigen profiles have been shown to vary between racial and ethnic groups;39,40 thus, the defined set of antigens on the multiplex may not be uniformly applicable across groups. In our study, the discordance between multiplex and IFA was particularly notable at high titers of IFA positivity, with three quarters of IFA positives at titers of 1:1280 or higher being identified as negative by multiplex.
Moreover, our data demonstrate that the performance characteristics of the 2 ANA assays varied between boys and girls, with lower agreement among girls, where the coefficient of agreement was classified as “slight”. This may indicate that there is a broader diversity of autoantigens among girls that is captured by the IFA (which detects all autoantibodies binding to Hep-2 cell nuclei) but not by multiplex (with its restricted range of available epitopes). The lower agreement among girls is concerning given the substantially higher risk of autoimmune disease among females. We also found better agreement among older children.
While we could not directly examine differences across race or ethnicity in this Mexican population, other studies have found the relative performance of ANA assay methods to vary according to race. Bruner et al reported that in both SLE cases and controls from a multiethnic population including African-, Mexican-, and European-Americans, ANA prevalence according to IFA vs multiplex was most similar for African Americans, and least similar for European Americans; among controls, both tests yielded a prevalence rate of ~20% among African Americans, whereas prevalence was 1.3- and 1.9-fold higher for IFA compared to multiplex among Mexican Americans and European Americans, respectively.19 Further, they found the false-positive rate for multiplex (ie, multiplex positive but IFA negative) among SLE cases to range from 2.1% for African Americans to 7.6% for Mexican Americans, whereas among controls, false positive rates were more similar between groups (11.4%–13.2%).19 The differential levels of agreement between tests across demographic groups need to be taken into consideration when evaluating choice of screening tool and interpreting results.
Based on the high discordance between tests, and particularly that the multiplex failed to capture the majority of high titer IFA positives in our study population, we would argue that the IFA remains the test of choice for screening, including in the context of epidemiologic research related to subclinical autoimmunity. Despite existing guidelines maintaining support for IFA as the reference standard,1,17 the multiplex assay has become increasingly popular due to its automated nature and lack of reliance on specialized technicians, potentially lower cost, and focus on a subset of antigens clinically relevant to autoimmune rheumatic diseases. These attributes are likewise attractive for epidemiologic research. However, we contend that given the low sensitivity (22%) of the multiplex, it is poorly suited as a first-stage or screening test, as it would miss a high proportion of “true positive” cases for whom a positive ANA would prompt further evaluation in the clinical setting, or for whom a signal of immune dysfunction would be missed in the research setting. Conversely, “false positives” would result in the wrong subset of persons being identified as having evidence of autoimmunity. Thus, the multiplex may not ultimately be cost-effective if it triggers unnecessary evaluation or if it misses opportunities for early intervention. Perhaps the multiplex is better suited as a follow-up to a positive IFA screen when identification of antigen specificity is a primary concern.
Understanding patterns of subclinical autoimmunity in the population is important for the elucidation of factors involved in initial perturbation of the immune system and subsequent progression toward a clinical state. While conventional wisdom has been that ANA positivity in children may often be due to infection, Allen et al did not find historical or intercurrent viral infections to be associated with an increased risk of ANA positivity.41 Studies have begun to explore the association between environmental toxicants and subclinical autoimmunity. We recently reported that in the general US population, low-level methylmercury exposure was associated with higher risk of ANA positivity (by IFA) among females of reproductive age.42 Further research into the role of environmental exposures in the development of autoimmunity relies on assays with favorable reliability and with broad applicability to various population subsets.
Our study clearly demonstrates that ANA testing based on multiplex and IFA assays yields not only divergent overall prevalence rates but differential agreement across demographic subsets. By simultaneously testing the same specimens by both methods, discordance in results cannot be ascribed to temporal physiologic changes, as might be contemplated in the clinical setting when serial testing yields opposing results. While the majority of data related to ANA testing stem from clinical series of persons with known or suspected autoimmune disease, we have extended the literature by focusing on a general population cohort of children. Given the homogeneous nature of our study population, the generalizability of results to adults or other racial and ethnic groups is unknown. However, we have focused on an ethnic group with heightened risk of SLE and potentially other autoimmune diseases compared to the non-Hispanic white population,33,34 and provide enhanced characterization of ANA testing in this group.
Conclusion
We have demonstrated substantial variation in the performance of competing assays for ANA detection in a pediatric cohort. Our data support a growing body of evidence demonstrating that, despite these two tests often being used interchangeably, they are in fact disparate assays. As in the clinical setting, this should be taken into consideration in research studies of subclinical autoimmunity.
Acknowledgments
This work was supported by NIH/NIEHS grants K01ES019909, P30ES017885, R01ES021446, R01ES007821, P01ES022844 and the National Institute of Public Health/Ministry of Health of Mexico. The American British Cowdray Hospital provided facilities used for subject recruitment and sample collection. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Disclosure
The authors report no conflicts of interest in this work.
References
Agmon-Levin N, Damoiseaux J, Kallenberg C, et al. International recommendations for the assessment of autoantibodies to cellular antigens referred to as anti-nuclear antibodies. Ann Rheum Dis. 2014;73(1):17–23. | ||
Kavanaugh A, Tomar R, Reveille J, Solomon DH, Homburger HA. Guidelines for clinical use of the antinuclear antibody test and tests for specific autoantibodies to nuclear antigens. American College of Pathologists. Arch Pathol Lab Med. 2000;124(1):71–81. | ||
Arbuckle MR, McClain MT, Rubertone M V, Scofield RH, Dennis GJ, James JA, Harley JB. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003;349(16):1526–1533. | ||
Rantapää-Dahlqvist S, de Jong B a W, Berglin E, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003;48(10):2741–2749. | ||
Nielen MMJ, van Schaardenburg D, Reesink HW, et al. Simultaneous development of acute phase response and autoantibodies in preclinical rheumatoid arthritis. Ann Rheum Dis. 2006;65(4):535–537. | ||
Brink M, Hansson M, Mathsson L, et al. Multiplex analyses of antibodies against citrullinated peptides in individuals prior to development of rheumatoid arthritis. Arthritis Rheum. 2013;65(4):899–910. | ||
Verge CF, Gianani R, Kawasaki E, Yu L, Pietropaolo M, Chase HP, Eisenbarth GS. Number of autoantibodies (against insulin, GAD or ICA512/IA2) rather than particular autoantibody specificities determines risk of type I diabetes. J Autoimmun. 1996;9(3):379–383. | ||
Bizzaro N. Autoantibodies as predictors of disease: the clinical and experimental evidence. Autoimmun Rev. 2007;6(6):325–333. | ||
Cooper GS. Unraveling the etiology of systemic autoimmune diseases: peering into the preclinical phase of disease. J Rheumatol. 2009;36(9):1853–1855. | ||
NIAMS. NIAMS preclinical disease phase of skin and rheumatic autoimmune diseases roundtable summary; 2010. Available from: http://www.niams.nih.gov/News_and_Events/Meetings_and_Events/Roundtables/2010/preclinical_autoimmunity_roundtable.asp. Accessed October 15, 2014. | ||
Deane KD, Norris JM, Holers VM. Preclinical rheumatoid arthritis: identification, evaluation, and future directions for investigation. Rheum Dis Clin North Am. 2010;36(2):213–241. | ||
Deane KD, El-Gabalawy H. Pathogenesis and prevention of rheumatic disease: focus on preclinical RA and SLE. Nat Rev Rheumatol. 2014;10(4):212–228. | ||
Tan EM, Feltkamp TE, Smolen JS, et al. Range of antinuclear antibodies in “healthy” individuals. Arthritis Rheum. 1997;40(9):1601–1611. | ||
Satoh M, Chan EKL, Ho LA, et al. Prevalence and sociodemographic correlates of antinuclear antibodies in the United States. Arthritis Rheum. 2012;64(7):2319–2327. | ||
Sperotto F, Cuffaro G, Brachi S, Seguso M, Zulian F. Prevalence of antinuclear antibodies in schoolchildren during puberty and possible relationship with musculoskeletal pain: a longitudinal study. J Rheumatol. 2014;41(7):1405–1408. | ||
Hilário MOE, Len CA, Roja SC, Terreri MT, Almeida G, Andrade LEC. Frequency of antinuclear antibodies in healthy children and adolescents. Clin Pediatr (Phila). 2004;43(7):637–642. | ||
Meroni PL, Schur PH. ANA screening: an old test with new recommendations. Ann Rheum Dis. 2010;69(8):1420–1422. | ||
Shanmugam VK, Switowski DR, Saddic N, Wang H, Steen VD. Comparison of indirect immunofluorescence and multiplex antinuclear antibody screening in systemic sclerosis. Clin Rheumatol. 2011;30(10):1363–1368. | ||
Bruner BF, Guthridge JM, Lu R, et al. Comparison of autoantibody specificities between traditional and bead-based assays in a large, diverse collection of patients with systemic lupus erythematosus and family members. Arthritis Rheum. 2012;64(11):3677–3686. | ||
Xu M, Roberts BB, Busby BA, et al. Evaluation of multiplex antinuclear antibody assay in pediatric patients. Lab Med. 2007;38(11):671–675. | ||
González-Cossío T, Peterson KE, Sanín LH, et al. Decrease in birth weight in relation to maternal bone-lead burden. Pediatrics. 1997;100(5):856–862. | ||
Hernandez-Avila M, Peterson KE, Gonzalez-Cossio T, Sanin LH, Aro A, Schnaas L, Hu H. Effect of maternal bone lead on length and head circumference of newborns and 1-month-old infants. Arch Environ Health. 2002;57(5):482–488. | ||
Ettinger AS, Lamadrid-Figueroa H, Téllez-Rojo MM, et al. Effect of calcium supplementation on blood lead levels in pregnancy: a randomized placebo-controlled trial. Environ Health Perspect. 2009;117(1):26–31. | ||
Braun JM, Hoffman E, Schwartz J, et al. Assessing windows of susceptibility to lead-induced cognitive deficits in Mexican children. Neurotoxicology. 2012;33(5):1040–1047. | ||
Bradwell A, Hughes R, Harden E. Atlas of Hep-2 Patterns. 2nd ed. Birmingham, UK: The Binding Site; 2003. | ||
Viera AJ, Garrett JM. Understanding interobserver agreement: the kappa statistic. Fam Med. 2005;37(5):360–363. | ||
Ghirardello A, Villalta D, Morozzi G, et al. Evaluation of current methods for the measurement of serum anti double-stranded DNA antibodies. Ann N Y Acad Sci. 2007;1109(1):401–406. | ||
Schmeling H, Mahler M, Levy DM, et al. Autoantibodies to dense fine speckles in pediatric diseases and controls. J Rheumatol. 2015;42(12):2419–2426. | ||
Somers EC, Marder W, Cagnoli P, et al. Population-based incidence and prevalence of systemic lupus erythematosus: the Michigan Lupus Epidemiology and Surveillance Program. Arthritis Rheumatol. 2014;66(2):369–378. | ||
Lim SS, Bayakly AR, Helmick CG, Gordon C, Easley KA, Drenkard C. The incidence and prevalence of systemic lupus erythematosus, 2002–2004: the Georgia Lupus Registry. Arthritis Rheumatol. 2014;66(2):357–368. | ||
Housey M, DeGuire P, Lyon-Callo S, et al. Incidence and prevalence of systemic lupus erythematosus among Arab and Chaldean Americans in southeastern Michigan: the Michigan Lupus Epidemiology and Surveillance Program. Am J Public Health. 2015;105(5):e74–e79. | ||
Somers EC, Thomas SL, Smeeth L, Schoonen WM, Hall AJ. Incidence of systemic lupus erythematosus in the United Kingdom, 1990–1999. Arthritis Rheum. 2007;57(4):612–618. | ||
Dall’Era MC, Snipes K, Cisternas M, Gordon C, Helmick CG. Preliminary population-based incidence and prevalence estimates of systemic lupus erythematosus: the California Lupus Surveillance Project. Arthritis Res Ther. 2014;16(Suppl 1):A38. | ||
Izmirly P, Wan I, Sahl S, et al. Preliminary population-based incidence and prevalence estimates of systemic lupus erythematosus from the Manhattan Lupus Surveillance Program. Arthritis Rheumatol. 2015;67(Suppl 10). | ||
Cooper GS, Bynum ML, Somers EC. Recent insights in the epidemiology of autoimmune diseases: improved prevalence estimates and understanding of clustering of diseases. J Autoimmun. 2009;33(3–4):197–207. | ||
Li Q-Z, Karp DR, Quan J, et al. Risk factors for ANA positivity in healthy persons. Arthritis Res Ther. 2011;13(2):R38. | ||
American college of rheumatology. Position statement: methodology of testing for antinuclear antibodies; 2011. Available from: https://www.rheumatology.org/Practice/Clinical/Position/Position_Statements/. Accessed February 4, 2015. | ||
Satoh M, Tanaka S, Chan EK. The uses and misuses of multiplex autoantibody assays in systemic autoimmune rheumatic diseases. Front Immunol. 2015;6(April):1–4. | ||
Shapira Y, Poratkatz B-S, Gilburd B, et al. Geographical differences in autoantibodies and anti-infectious agents antibodies among healthy adults. Clin Rev Allergy Immunol. 2012;42(2):154–163. | ||
Jurencák R, Fritzler M, Tyrrell P, Hiraki L, Benseler S, Silverman E. Autoantibodies in pediatric systemic lupus erythematosus: ethnic grouping, cluster analysis, and clinical correlations. J Rheumatol. 2009;36(2):416–421. | ||
Allen RC, Dewez P, Stuart L, Gatenby PA, Sturgess A. Antinuclear antibodies using HEp-2 cells in normal children and in children with common infections. J Paediatr Child Health. 1991;27(1):39–42. | ||
Somers EC, Ganser MA, Warren JS, Basu N, Wang L, Zick SM, Park SK. Mercury exposure and antinuclear antibodies among females of reproductive age in the United States: NHANES. Environ Health Perspect. 2015;123(8):792–98. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.