Back to Journals » Infection and Drug Resistance » Volume 12
Antimicrobial resistance, virulence genes profiling and molecular relatedness of methicillin-resistant Staphylococcus aureus strains isolated from hospitalized patients in Guangdong Province, China
Authors Liang Y, Tu C, Tan C ![]() , El-Sayed Ahmed MAEG
, El-Sayed Ahmed MAEG ![]() , Dai M, Xia Y, Liu Y, Zhong LL
, Dai M, Xia Y, Liu Y, Zhong LL ![]() , Shen C
, Shen C ![]() , Chen G, Tian GB, Liu J
, Chen G, Tian GB, Liu J ![]() , Zheng X
, Zheng X ![]()
Received 29 October 2018
Accepted for publication 20 December 2018
Published 25 February 2019 Volume 2019:12 Pages 447—459
DOI https://doi.org/10.2147/IDR.S192611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Yingjian Liang,1,* Changli Tu,1,* Cuiyan Tan,1 Mohamed Abd El-Gawad El-Sayed Ahmed,2,3,4 Min Dai,5 Yong Xia,6 Yan Liu,7 Lan-Lan Zhong,2,3 Cong Shen,2,3 Guanping Chen,2,3 Guo-Bao Tian,2,3 Jing Liu,1 Xiaobin Zheng1
1Department of Respiratory Medicine, The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, China; 2Department of Immunology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, China; 3Key Laboratory of Tropical Diseases Control, Sun Yat-sen University, Ministry of Education, Guangzhou, China; 4Department of Microbiology and Immunology, Faculty of Pharmacy, Misr University for Science and Technology (MUST), 6th of October City, Egypt; 5School of Laboratory Medicine, Chengdu Medical College, Chengdu, China; 6Department of Clinical Laboratory Medicine, Third Affiliated Hospital of Guangzhou Medical University, Guangzhou, China; 7Clinical laboratory, The Fifth Affiliated Hospital of Sun Yat-Sen University, Zhuhai, China
*These authors contributed equally to this work
Purpose: The main objective of this study was to decipher the prevalence, antimicrobial resistance, major virulence genes and the molecular characteristics of methicillin-resistant Staphylococcus aureus (MRSA) isolated from different clinical sources in southern China.
Materials and methods: The present study was performed on 187 non-duplicate S. aureus clinical isolates collected from three tertiary hospitals in Guangdong Province, China, 2010–2016. Antimicrobial susceptibility testing was performed by the disk diffusion method and by measuring the minimum inhibitory concentration. Screening for resistance and virulence genes was performed. Clonal relatedness was determined using various molecular typing methods such as multilocus sequence typing, spa and staphylococcal chromosomal cassette mec (SCCmec) typing. Whole genome sequencing was performed for three selected isolates.
Results: Out of 187 isolates, 103 (55%) were identified as MRSA. The highest prevalence rate was found among the skin and soft tissue infection (SSTI) samples (58/103), followed by sputum samples (25/103), blood stream infection samples (15/103) and others (5/103). Antimicrobial susceptibility results revealed high resistance rates for erythromycin (64.1%), clindamycin (48.5%), gentamicin (36.9%) and ciprofloxacin (33.98%). All isolates were susceptible to vancomycin. Resistance genes and mutation detected were as follows: aac(6’)-aph(2”) (24.3%), dfrG (10.7%), rpoB (21.4%), cfr (0%), fexA (1.94%), gyrA (35.92%), gyrB (0.97%), grlA (20.4%), grlB (10.68%), ermA (21.4%), ermB (18.44%), ermC (21.4%) and lnuA (18.44%). Profiling of virulence genes revealed the following: sea (11.7%), seb (21.4%), sec (0.97%), sed (0.97%), hla (86.41%), hlb (17.48%), hlg (10.68%), hld (53.4%), Tsst-1 (3.9%) and pvl (27.2%). Clonal relatedness showed that ST239-SCCmecA III-t37 clone was the most prevalent clone.
Conclusion: Our study elucidated the prevalence, antibiotic resistance, pathogenicity and molecular characteristics of MRSA isolated from various clinical sources in Guangdong, China. We found that the infectious rate of MRSA was higher among SSTI than other sources. The most predominant genotype was ST239-SCCmecA III-t37 clone, indicating that ST239-t30 clone which was previously predominant had been replaced by a new clone.
Keywords: MRSA, antibiotic resistance, resistance genes, virulence factors, molecular typing, whole genome sequencing
Introduction
Staphylococcus aureus is a highly virulent opportunistic pathogen capable of causing a variety of infections, including skin and soft tissue infection (SSTI), blood stream infection (BSI) and toxin-mediated syndromes as well as life-threatening diseases.1-3 S. aureus, as one of the “ESKAPE” organisms, is a growing threat worldwide, as it can cause a variety of serious nosocomial infections.4 Infections caused by S. aureus are divided into two types: methicillin-sensitive S. aureus (MSSA) and methicillin-resistant Staphylococcus aureus (MRSA), which is also known as oxacillin-resistant S. aureus.5
Recently, MRSA strains have become responsible for about 25%–50% of clinical infections caused by S. aureus in many countries.6 Commonly, the MSSA becomes MRSA via acquisition of large and potentially transmissible genomic islands called staphylococcal chromosomal cassette mec (SCCmec), which harbor mecA/mecC genes. Awareness of the different antibiotic resistance patterns and the molecular characteristics of MRSA can effectively help in the treatment of MRSA infections as well as slowing the epidemic spread of it. MRSA has acquired multiple resistance to a wide range of antibiotics including aminoglycosides. Practically, MRSA was found to be resistant to all available β-lactam antibiotics.3,7
Several reports about the prevalence, molecular characterization and antimicrobial resistance pattern of MRSA in China are available since China represents a country of high prevalence rate of MRSA, especially in Guangdong Province, one of the largest southern coastal provinces which has a large population and is adjoining Hong Kong and Taiwan. However, a large proportion of these reports are mainly focusing on a single source of MRSA infection, ignoring the possibility of MRSA infection from multiple sources. Therefore, our study aims to focus on MRSA isolates collected from different clinical sources.8-16
Materials and methods
Clinical isolates
This cross-sectional study was conducted on a total of 187 non-repetitive S. aureus isolates collected from different clinical specimens in three tertiary hospitals in Guangdong Province, China, denoted as H1, H2 and H3, in the year 2010–2016. All the MRSA isolates were collected from the routine laboratory work of these hospitals. Specimens were collected from different wards including medical ward, surgical ward, intensive care unit and pediatrics department and were recovered from different clinical sources including SSTI, blood, sputum and others (inflamed joint fluid and ascitic fluid). The isolates were identified as S. aureus by using the BD PhoenixTM100 microbial identification system (bioMérieux, Marcy l’Etoile, France).
For detection of MRSA, all the identified S. aureus were tested for their antimicrobial susceptibility by disk diffusion technique using cefoxitin (FOX; 30 µg) according to the Clinical and Laboratory Standards Institute (CLSI, 2017) guidelines. Then mecA gene was detected by PCR as previously described.17 The samples positive for the mecA gene and resistant to FOX were identified as MRSA.
DNA extraction
All the samples were cultured on Mueller–Hinton agar plates at 33°C overnight. Then, a single bacterial colony was suspended onto 3 mL of sterile lysogeny broth medium (Oxoid, Hampshire, UK) and subsequently incubated at 33°C with vigorous shaking for 8 hours. Then, DNA was extracted using Hipure bacterial DNA kit (Magen, Guangzhou, China) according to the manufacturer’s instructions.
Antimicrobial susceptibility and minimum inhibitory concentration (MIC) determination
In vitro antimicrobial susceptibility of MRSA isolates was determined by modified Kirby–Bauer disk diffusion method according to the CLSI 2017 guidelines. Antimicrobial drugs tested included: penicillin (P), FOX, rifampicin (RFD), quinupristin/dalfopristin (Q/D) and trimethoprim–sulfamethoxazole (SXT). MIC was detected by broth microdilution method and interpreted according to the CLSI 2017 guidelines for erythromycin (ERY), clindamycin (CLI), gentamicin (GEN), ciprofloxacin (CIP), chloramphenicol (CHL), nitrofurantoin (NIT) and vancomycin (VAN). Tigecycline (TGC) was evaluated using MICs according to the European Committee on Susceptibility Testing guidelines (version 1.3, 2010; Basel, Switzerland).
Molecular detection of antibiotic resistance genes
Isolates were screened for the production of the common resistance genes and mutation for different antimicrobial classes by PCR as follows: RFD (rpoB), CHL (cfr and fexA), aminoglycosides (acc(6′)-aph(2″)), macrolides (ermA, ermB and ermC), lincosamides (lnuA), SXT (dfrG) and fluoroquinolones (gyrA, gyrB, grlA and grlB).14,18–22 Purified positive PCR products were sequenced using BigDye terminator chemistry on an automated ABI 3130 sequencer (PE Applied Biosystems, Foster City, CA, USA) based on Sanger’s sequencing method. Gene sequences were corrected using CodonCode Aligner version 7.1.2. sequencing analysis software and then analyzed using BlastN and BlastP against the National Center for Biotechnology Information database (www.ncbi.nlm.nih.gov).
Detection of virulence factors
Detection of virulence genes was performed among MRSA isolates. Ten exotoxins coding gene, including four enterotoxins (sea, seb, sec and sed), four hemolysin toxins (hla, hlb, hld and hlg), toxic shock syndrome toxin (tsst-1) and Panton–Valentine leukocidin (pvl) were amplified by multiplex PCR as previously described.2
Molecular typing of MRSA
All the MRSA strains were typed by three different molecular characterization methods as follows: SCCmec, spa type and multilocus sequence typing (MLST).
In the SCCmec typing, MRSA strains were identified as SCCmec type I, II, III, IV or V by performing PCR using nine pairs of primers and the primers for the mecA gene as previously described.17 The SCCmec types were determined on the basis of the band pattern obtained.
Spa typing was performed according to the website https://spa.ridom.de/. The isolates were assigned to the specific spa types according to the guidelines described by the publicly available Center of Genomic Epidemiology (https://cge.cbs.dtu.dk/services/spaTyper-1.0/).23
MLST was performed based on seven housekeeping genes (arcC, aroE, glpF, gmK, pta, tpiA and yqiL) obtained from the MLST database as previously described.24 Isolates were assigned sequence types (STs) according to the MLST database (http://saureus.mlst.net).
Whole genome sequencing (WGS)
WGS was performed for three selected isolates, QY152, L2316 and L2317. QY152 was selected for its relatedness to ST398, which is not common in China, but common in Europe. Both L2316 and L2317 were selected for their TGC resistance (the only two TGC-resistant isolates) as well as their relatedness to ST9. The ST398 isolate (SO385, GenBank: AM990992) was used as a reference strain for comparative genomics.
The resulting draft genomes were described. DNA libraries were constructed with 350 bp paired-end fragments. The isolate reads were produced by Illumina Hiseq2000 platform. Reads were assembled using SPAdes. The genome annotation was performed using PROKKA, E-value setting as 1e−5 25 and Rapid Annotations Subsystems Technology. Bacteriophages detection was performed using PHAge Search Tool.26 Antimicrobial resistance genes were identified using the Comprehensive Antibiotic Resistance Database (https://card.mcmaster.ca/) and the virulence factors were screened using the Virulence Factors Database (http://www.mgc.ac.cn/VFs/). This Whole Genome Shotgun project of the three selected isolates has been deposited at GenBank under the following accession numbers: RDQU000000, RDQT000000 and RDQS000000, respectively.
Statistical analysis
Statistical analysis for resistance and virulence genes as well as for the resistance rates to the tested antibiotics among our MRSA was performed using chi-squared test (SPSS version 20). P-values <0.01 were considered statistically significant.
Results
MRSA identification
Among 187 identified S. aureus samples, 103 (55.1%) were confirmed as MRSA depending on mecA gene and resistance to FOX. The majority of these MRSA (58/103, 56.31%) were recovered from SSTI, followed by sputum (25/103, 24.3%), BSI (15/103, 14.6%) and others (5/103, 4.9%), as shown in Table 1.
 | Table 1 Prevalence and resistance pattern of MRSA isolates Note: a,bEach subscript letter denotes a subset of group categories whose column proportions do not differ significantly from each other at the 0.01 level. Abbreviations: BSI, blood stream infection; CHL, chloramphenicol; CIP, ciprofloxacin; CLI, clindamycin; ERY, erythromycin; FOX, cefoxitin; GEN, gentamicin; MDR, mutidrug resistance; MRSA, methicillin-resistant Staphylococcus aureus; NIT, nitrofurantoin; RFD, rifampicin; SSTI, skin and soft tissue infection; SXT, trimethoprim– sulfamethoxazole; TGC, tigecycline; VAN, vancomycin. |
Antimicrobial susceptibility testing
All MRSA isolates were found to be resistant to penicillin and FOX, while all were susceptible to VAN and NIT. Some of the tested antimicrobials revealed high resistance pattern, including ERY (66/103, 64.1%), CLI (50/103, 48.5%), GEN (38/103, 36.9%) and CIP (35/103, 33.98%), while others presented relatively low resistance rates, including RFD (19/103, 18.5%), CHL (12/103, 11.65%), SXT (11/103, 10.7%), Q/D (9/103, 8.74%) and TGC (2/103, 1.94%). All the isolates were found to be resistant to at least two antibiotics, and 68/103 (66.2%) were multidrug resistant (MDR), exhibiting resistance to three or more antibiotic classes, where 44.12% (30/68) of MDR isolates were recovered from SSTI, 35.3% (24/68) from sputum, 13.2% (9/68) from BSI and 7.4% (5/68) from other sites. The resistance rates of GEN, ERY, CIP, RFD and Q/D were found to be higher in sputum than in SSTI or BSI; however, statistically differences were only found in GEN and CIP (P<0.01, Table 1; Figure 1).
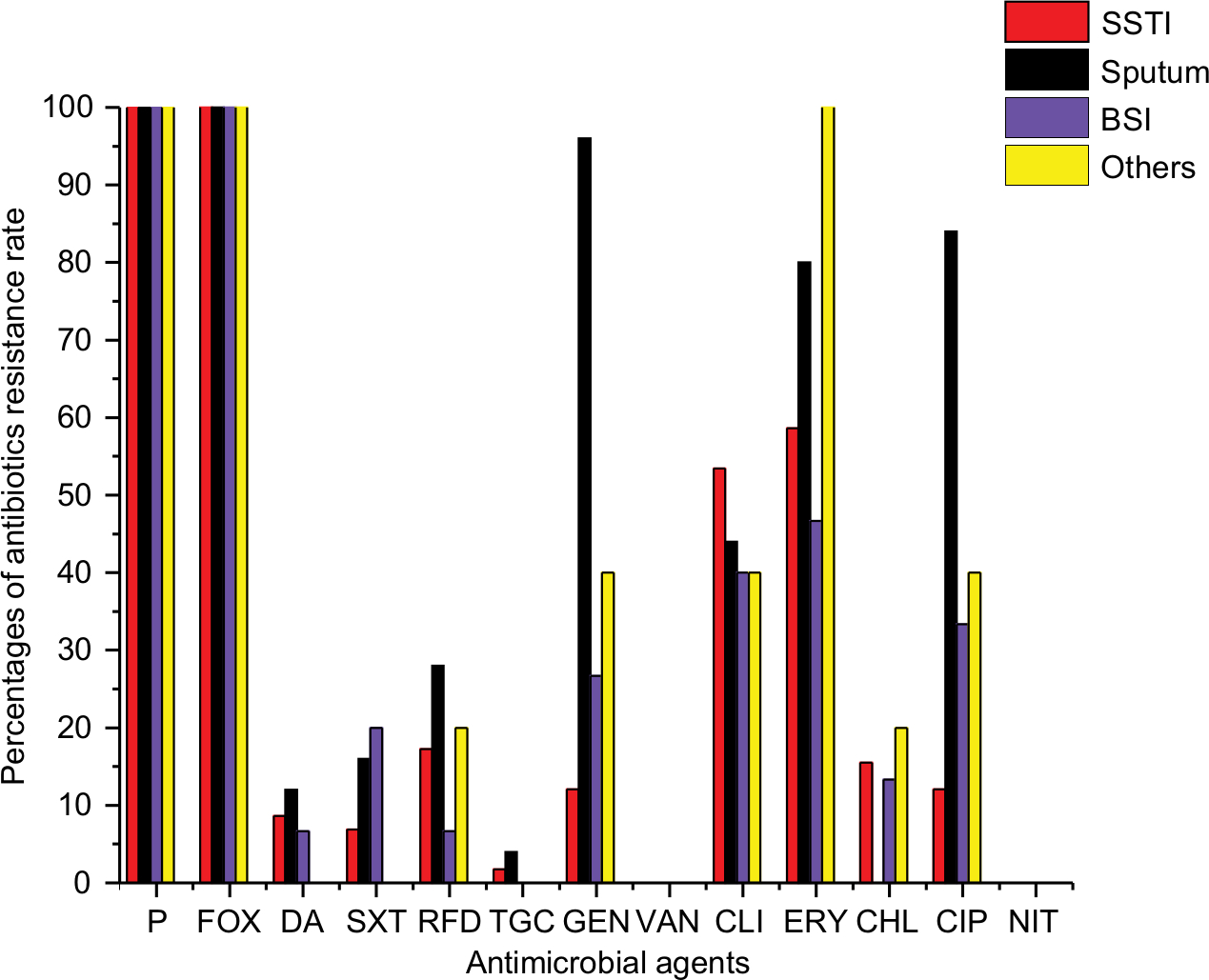 | Figure 1 Percentages of antibiotic resistance rates of MRSA isolated from various clinical sources. Abbreviations: BSI, blood stream infection; CHL, chloramphenicol; CIP, ciprofloxacin; CLI, clindamycin; ERY, erythromycin; FOX, cefoxitin; GEN, gentamicin; MRSA, methicillin-resistant Staphylococcus aureus; NIT, nitrofurantoin; P, penicillin; RFD, rifampicin; SSTI, skin and soft tissue infection; SXT, trimethoprim–sulfamethoxazole; TGC, tigecycline; VAN, vancomycin. |
PCR amplification and sequencing of antibiotic resistance genes
Screening for fluoroquinolone resistance-encoding genes revealed the presence of gyrA (37/103, 35.92%), gyrB (1/103, 1.94%), grlA (21/103, 20.4%) and grlB (11/103, 10.68%), as shown in Table S1. Macrolide-encoding resistance genes detected were as follows: ermA (22/103, 21.4%), ermB (19/103, 18.44%) and ermC (22/103, 21.4%). Aac(6′)-aph(2″) gene which is correlated with GEN resistance was detected in 25/103 (24.3%) of MRSA. Regarding CHL, fexA (2/103, 1.94%) was detected, while no cfr was found, indicating the low antimicrobial resistance rate of MRSA to CHL. The rpoB gene was detected in 22/103 (21.4%) isolates, lnuA gene in 19/103 (18.44%) isolates, while dfrG was detected in 11/103 (10.7%) of MRSA isolates (Table 2).
 | Table 2 Resistance pattern and virulence genes of MRSA isolates from different clinical sources Note: a,b,cEach subscript letter denotes a subset of group categories whose column proportions do not differ significantly from each other at the 0.01 level. Abbreviations: BSI, blood stream infection; MRSA, methicillin-resistant Staphylococcus aureus; SSTI, skin and soft tissue infection. |
Virulence genes profiling
Multiplex PCR revealed that 97/103 (94.2%) MRSA isolates were harboring virulence genes, including four enterotoxin genes, sea (12/103, 11.7%), seb (22/103, 21.4%), sec (1/103, 0.97%) and sed (1/103, 0.97%), and six exotoxin genes, whereas for hemolysin toxins (hla, hlb, hlg and hld), results revealed that hla gene was the highest detected gene that was harbored by 89/103 isolates (86.41%), followed by hld (55/103, 53.4%), hlb (18/103, 17.48%) and hlg (11/103, 10.68%). The majority of hla-positive strains were recovered from SSTI (44/89, 49.4%) followed by sputum (25/89, 28.1%), BSI (15/89, 16.85%) and others (5/89, 5.62%). Concerning tsst-1 gene, it was harbored by 4/103 isolates (3.9%). PCR assay for detection of pvl gene showed that 28/103 (27.2%) MRSA were pvl positive and the majority of them were isolated from SSTI (25/28, 89.3%; Table 2).
Molecular typing of MRSA
Results of SCCmec typing indicated that five types of SCCmec elements were detected. Type III was the most predominant (39/103, 37.86%) followed by type IV (17/103, 16.5%), wherein the majority of isolates belonged to type IVa (15/17 88.2%) then type IVb (2/17, 11.8%). The prevalence of SCCmec types II and V was very low (2/103, 1.94% and 1/103, 0.97%, respectively), while no isolates belonging to type I were found. Out of 103 MRSA isolates, 44 (42.7%) were classified as non-typable MRSA. The majority of the isolates harboring hla gene (37/89, 41.57%) were found to be belonging to SCCmec type III, followed by type IV (12/89, 13.48%), type II (2/89, 2.24%) and type V (1/89, 1.12%), while 36/89 (40.44%) of them were found to be associated with non-typable MRSA. Tsst-1 gene was associated with SCCmec type II (2/4, 50%). Regarding PVL-positive isolates, 16/28 (57.1%) isolates belonged to the predominant SCCmec type III.
Spa typing revealed a diverse range of spa types, wherein 25 types were detected among MRSA. t437 (22/103, 21.4%) was the most predominant type, followed by t37 (18/103, 17.5%). The remaining spa types were distributed homogeneously among MRSA isolates. Also, 17/103 (16.5%) of MRSA isolates were not identified in the spa database (Table S2).
According to the MLST typing, MRSA isolates were assigned to 23 different STs; also, 18 novel STs were detected and deposited in the database of PubMLST (https://pubmlst.org/saureus/), as shown in Table S2. ST239 was the most prevalent among our isolates (24/103, 23.3%), followed by ST338 (18/103, 17.5%). ST239 was mainly distributed in H1, while ST338 was circulating in H3. It is noteworthy that 65 different clones have been detected where ST239-SCCmecA III-t37 clone was found to be the most predominant genotype circulating in H1 and H2 (9/103, 8.7%), followed by ST338-SCCmecA III-t437 (6/103, 5.83%), which was found to be circulating mainly in H3.
Isolates harboring hla gene were related to different clones, while the majority were associated with ST338 (13/89, 14.61%) and t437 (16/89, 17.97%). Of the 28/103 isolates harboring pvl gene, 7/28 (25%) isolates belonged to ST338-SCCmecA III-t437 clone. The tsst-1 gene (4/103) was associated with ST1, ST72, ST641 and ST863 (Figure 2).
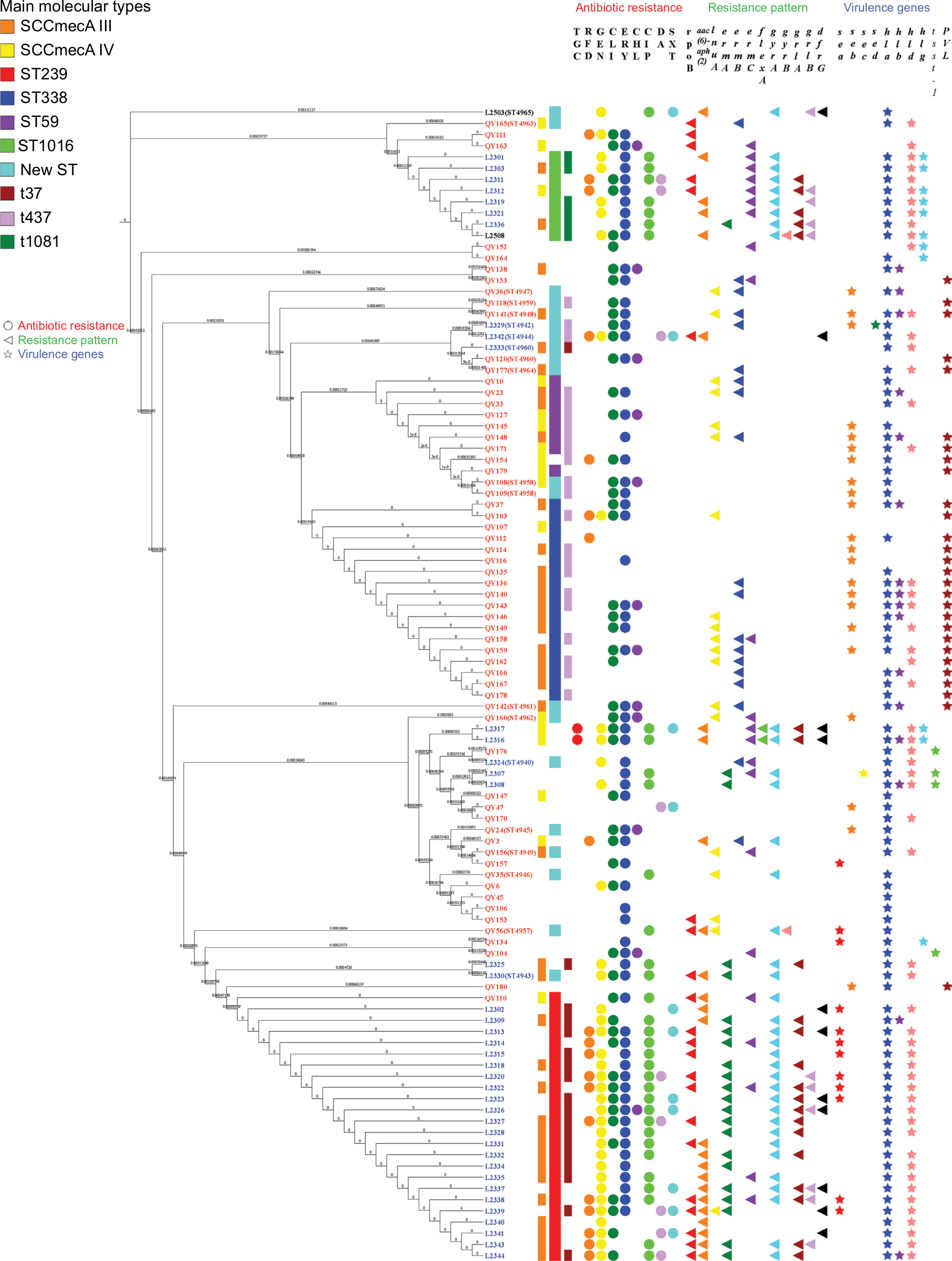 | Figure 2 Phylogenetic tree of 103 MRSA isolates. Notes: A maximum likelihood phylogeny was used of every MRSA isolate. Strain names are color-coded based on the hospital: blue is hospital 1, black is hospital 2 and red is hospital 3. The main molecular types are shown in various colors as shown in the legend. The inner bar indicates the main SCCmecA and the outer bar indicates the main spa types. Main STs are indicated by the middle bar. Abbreviations: MRSA, methicillin-resistant Staphylococcus aureus; SCCmec, staphylococcal chromosomal cassette mec; ST, sequence type. |
Whole genome sequencing
WGS was performed for three selected isolates (QY152, L2316 and L2317). QY152 was found to be harboring six resistance genes and two common virulence factors: hemolysin and adhesin. The mepA and mepR genes were detected among the TGC-resistant isolates (Table 3).
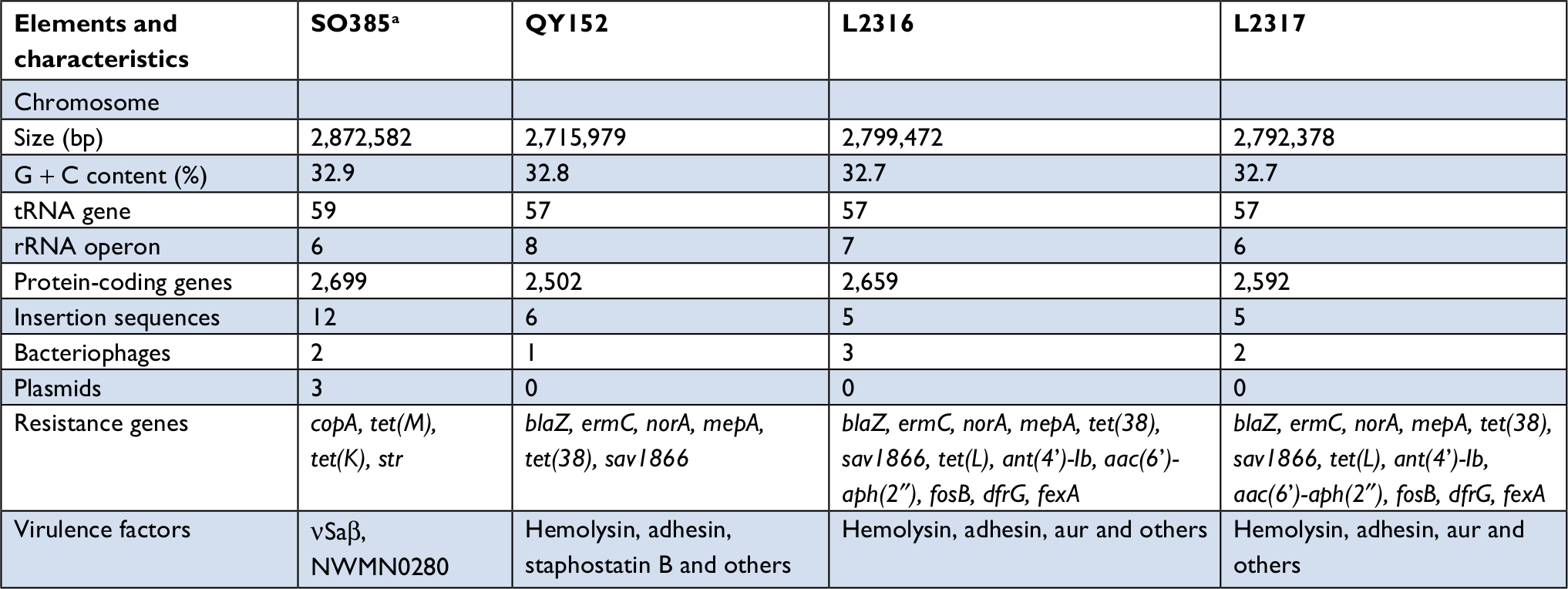 | Table 3 General characteristics of the genomes of QY152, L2316 and L2317 in comparison with SO385 Note: aSO385 livestock-associated MRSA (ST398 isolate) from the Netherlands. GenBank: AM990992. Abbreviation: MRSA, methicillin-resistant Staphylococcus aureus. |
Discussion
As a clinically important drug-resistance pathogen, MRSA can cause a variety of infections, including SSTI, respiratory tract infection, BSI and others. Herein, we investigated the prevalence, antibiotic resistance, virulence factors and the molecular characteristics of MRSA isolated from different clinical sources as shown in Table 1. The prevalence of MRSA in the present study was high. However, the prevalence among SSTI was much lower than that reported from a burn center in southeastern China in 2014–2015,16 but was consistent with that reported in a study from Taiwan in 2008.27 The prevalence of our MRSA isolates in BSI was lower than those reported from China in 2016 and Latin America in 2011–2014.28,29 The detection rate of MRSA among our sputum samples was much higher than that reported from Dongguan and Shenzhen, two cities of Guangdong Province,12,30 but much lower than that reported in a multicenter study in China from 2007 to 2009.31
Among our isolates, high drug resistance was observed. More than half of the isolates were MDR, which mostly came from sputum and SSTI and were found related to ST239 (23/24) and ST1016 (7/8). The high MDR problem of ST239-SCCmecA III-t37/t30 clone is still extremely serious. The MDR rate in our study was a little lower than that reported in a previous study in Beijing Children’s Hospital, China.32
We believe that different clinical samples show differences in developing drug resistance, which is attributed to the different selection processes. However, in contrast to our hypothesis, the antimicrobial resistance rate among our MRSA isolates did not show a statistical difference, except for CIP and GEN, where acc(6′)-aph(2″), gyrA, gyrB, grlA and grlB were found to be more prevalent among sputum samples (P<0.01). We hypothesized that the reason was that fluoroquinolones were often used in pneumonia, which may increase the resistance rate.33,34
According to the US Food and Drug Administration black box, TGC was a last-line agent for MRSA infections until September 2010.36 In the present study, TGC resistance was very low (2/103, 1.94%), similar to that reported in a study from Zhejiang Province. The rate of TGC resistance in China is still low, but the number of MRSA isolates resistant to TGC is actually growing.35 Here, WGS revealed the occurrence of the efflux protein coding gene mepA among the whole genome analyzed sequences, which was found to be related to decreasing the susceptibility to TGC. MepA is rarely reported in China, while it is more prevalent in African countries.37,38
Regarding the virulence genes, our study showed relatively similar findings for sea, seb, sec, sed, tsst-1, pvl and hla with those reported of a burn center in southeastern China.16
Isolates recovered from SSTI were associated with >15 types of MLST and spa types, which indicated high diversity and complexity in their genetic background. Our results were consistent with those reported in a multicenter study of SSTI in China39 where the predominant spa type was t437. In the present study, ST338-t437 was the predominant clone, which is inconsistent with other studies which revealed that ST59-t437 clone was predominant.28 However, it has been reported that both ST338-t437 and ST59-t437 belonged to the same clonal complex (CC59), where ST59 was the predominant ST, while single-locus variants belonging to ST338 were obtained.40 Therefore, we could probably suppose that ST239-t30 clone which was predominant in 2013 had been gradually replaced by ST338-t437 or ST59-t437.
Our results were similar to those of a study conducted in Wenzhou, China from 2012 to 2013 in which all the isolates belonging to CC59 were susceptible to NIT, Q/D and SXT; however, 81.8% of them were resistant to ERY and CLI.41
Noteworthy, two of our isolates recovered from SSTI at the same hospital were found to be belonging to ST398, which has been known as a livestock-associated MRSA (LA-MRSA) clone.42 In previous studies, it was reported that most of the LA-MRSA isolates in Asia belonged to ST9, while ST398-related isolates were mainly reported in European and North American countries.43,44 Generally, due to the lack of several common virulence factors, the ST398 MRSA in Europe did not cause the frequent invasive diseases in humans.45 However, a recent report indicated that the Chinese LA-MRSA ST398 isolates are far different from those of other continents, because they likely evolved from human-adapted MSSA.46 Recently, a report about the emergence of ST398 in a pig farm in Guangdong, China was published,13 which indicated the transmission of this clone between different continents. Therefore, we supposed that the two isolates belonging to ST398 may be transmitted via livestock, which is in accordance with other reports that showed the possibility of transmission of ST398 from the animal reservoir to humans.28,47,48 Also, since the source of infection was SSTI, we can conclude that the possible way of transmission of MRSA was via skin contact with the livestock, which requires certain precautions.
Among the sputum clinical samples, 6/25 (24%) of MRSA belonged to t1081 type, which is supposed to be more prevalent in long-term care facilities (LTCFs).49 Several reports indicated that t1081 had spread from the Netherlands to Hong Kong and Taiwan.50–54 The present study was done in Guangdong, which is a province located near Taiwan and Hong Kong; therefore, presumably, t1081 might not only be introduced from LTCF residents to hospitalized patients, but also from Hong Kong and Taiwan to Mainland China.
Conclusion
In conclusion, our study elucidated and gave insight on the high prevalence of MRSA in southern China, especially from SSTI and sputum clinical samples. Notably, GEN and CIP should be used carefully in respiratory tract infections with MRSA and rational use of antibiotics should be undertaken to slow the epidemic spread of MDR in China. Our study also showed that ST239-SCCmecA III-t37 clone was the most prevalent among our isolates, indicating that ST239-t30 clone, which was previously predominant worldwide, had been replaced by a new clone, providing confirmation that clonal replacement is persistent.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (grant numbers 81471988 and 81722030).
Disclosure
The authors report no conflicts of interest in this work.
References
Goudarzi M, Seyedjavadi SS, Nasiri MJ, Goudarzi H, Sajadi Nia R, Dabiri H. Molecular characteristics of methicillin-resistant Staphylococcus aureus (MRSA) strains isolated from patients with bacteremia based on MLST, SCCmec, spa, and agr locus types analysis. Microb Pathog. 2017;104:328–335. | ||
Jiang B, Yin S, You B, et al. Antimicrobial resistance and virulence genes profiling of methicillin-resistant Staphylococcus aureus isolates in a burn center: a 5-year study. Microb Pathog. 2018;114:176–179. | ||
Papadopoulos P, Papadopoulos T, Angelidis AS, et al. Prevalence of Staphylococcus aureus and of methicillin-resistant S. aureus (MRSA) along the production chain of dairy products in north-western Greece. Food Microbiol. 2018;69:43–50. | ||
Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA. ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov. 2015;14(8):529–542. | ||
Savoldi A, Azzini AM, Baur D, Tacconelli E. Is there still a role for vancomycin in skin and soft-tissue infections? Curr Opin Infect Dis. 2018;31(2):120–130. | ||
El-Baz R, Rizk DE, Barwa R, Hassan R. Virulence characteristics and molecular relatedness of methicillin resistant Staphylococcus aureus harboring different staphylococcal cassette chromosome mec. Microb Pathog. 2017;113:385–395. | ||
Khosravi AD, Jenabi A, Montazeri EA. Distribution of genes encoding resistance to aminoglycoside modifying enzymes in methicillin-resistant (MRSA) strains. Kaohsiung J Med Sci. 2017;33(12):587–593. | ||
Lee AS, de Lencastre H, Garau J, et al. Methicillin-resistant Staphylococcus aureus. Nat Rev Dis Primers. 2018;4:18033. | ||
Peng Y, Ou Q, Lin D, et al. Metro system in Guangzhou as a hazardous reservoir of methicillin-resistant staphylococci: findings from a point-prevalence molecular epidemiologic study. Sci Rep. 2015;5(1):16087. | ||
Lin J, Xu P, Peng Y, et al. Prevalence and characteristics of Staphylococcus aureus and methicillin-resistant Staphylococcus aureus nasal colonization among a community-based diabetes population in Foshan, China. J Diabetes Investig. 2017;8(3):383–391. | ||
Xie X, Dai X, Ni L, et al. Molecular epidemiology and virulence characteristics of Staphylococcus aureus nasal colonization in medical laboratory staff: comparison between microbiological and non-microbiological laboratories. BMC Infect Dis. 2018;18(1):122. | ||
Hu L, Li Y, Lu Y, et al. Clinical characteristics, virulence factors and molecular typing of methicillin-resistant Staphylococcus aureus infections in Shenzhen City, China. Epidemiol Infect. 2016;144(14):3037–3045. | ||
Li W, Liu JH, Zhang XF, et al. Emergence of methicillin-resistant Staphylococcus aureus ST398 in pigs in China. Int J Antimicrob Agents. 2018;51(2):275–276. | ||
Liu B, Sun H, Pan Y, et al. Prevalence, resistance pattern, and molecular characterization of Staphylococcus aureus isolates from healthy animals and sick populations in Henan Province, China. Gut Pathog. 2018;10(1):31. | ||
Gu FF, Hou Q, Yang HH, et al. Characterization of Staphylococcus aureus isolated from non-native patients with skin and soft tissue infections in Shanghai. PLoS One. 2015;10(4):e0123557. | ||
Chen K, Lin S, Li P, et al. Characterization of Staphylococcus aureus isolated from patients with burns in a regional burn center, southeastern China. BMC Infect Dis. 2018;18(1):51. | ||
Zhang K, McClure JA, Elsayed S, Louie T, Conly JM. Novel multiplex PCR assay for characterization and concomitant subtyping of staphylococcal cassette chromosome mec types I to V in methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 2005;43(10):5026–5033. | ||
Kehrenberg C, Schwarz S. Distribution of florfenicol resistance genes fexA and cfr among chloramphenicol-resistant Staphylococcus isolates. Antimicrob Agents Chemother. 2006;50(4):1156–1163. | ||
Lim JA, Kwon AR, Kim SK, Chong Y, Lee K, Choi EC. Prevalence of resistance to macrolide, lincosamide and streptogramin antibiotics in Gram-positive cocci isolated in a Korean hospital. J Antimicrob Chemother. 2002;49(3):489–495. | ||
Schmitz FJ, Fluit AC, Gondolf M, et al. The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 European hospitals. J Antimicrob Chemother. 1999;43(2):253–259. | ||
Argudín MA, Tenhagen BA, Fetsch A, et al. Virulence and resistance determinants of German Staphylococcus aureus ST398 isolates from nonhuman sources. Appl Environ Microbiol. 2011;77(9):3052–3060. | ||
Lozano C, Aspiroz C, Sáenz Y, et al. Genetic environment and location of the lnu(A) and lnu(B) genes in methicillin-resistant Staphylococcus aureus and other staphylococci of animal and human origin. J Antimicrob Chemother. 2012;67(12):2804–2808. | ||
Wu D, Wang Z, Wang H, et al. Predominance of ST5-II-t311 clone among healthcare-associated methicillin-resistant Staphylococcus aureus isolates recovered from Zhejiang, China. Int J Infect Dis. 2018;71:107–112. | ||
Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J Clin Microbiol. 2000;38(3):1008–1015. | ||
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. | ||
Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39(Web Server issue):W347–W352. | ||
Changchien CH, Chen SW, Chen YY, Chu C. Antibiotic susceptibility and genomic variations in Staphylococcus aureus associated with skin and soft tissue infection (SSTI) disease groups. BMC Infect Dis. 2016;16(1):276. | ||
Li S, Sun S, Yang C, et al. The changing pattern of population structure of Staphylococcus aureus from bacteremia in China from 2013 to 2016: ST239-030-MRSA replaced by ST59-t437. Front Microbiol. 2018;9:332. | ||
Arias CA, Reyes J, Carvajal LP, et al. A prospective cohort multicenter study of molecular epidemiology and phylogenomics of Staphylococcus aureus bacteremia in nine Latin American countries. Antimicrob Agents Chemother. 2017;61(10). | ||
He X, Xie M, Li S, et al. Antimicrobial resistance in bacterial pathogens among hospitalized children with community acquired lower respiratory tract infections in Dongguan, China (2011–2016). BMC Infect Dis. 2017;17(1):614. | ||
Li DZ, Chen YS, Yang JP, et al. Preliminary molecular epidemiology of the Staphylococcus aureus in lower respiratory tract infections: a multicenter study in China. Chin Med J (Engl). 2011;124(5):687–692. | ||
Yang X, Qian S, Yao K, et al. Multiresistant ST59-SCCmec IV-t437 clone with strong biofilm-forming capacity was identified predominantly in MRSA isolated from Chinese children. BMC Infect Dis. 2017;17(1):733. | ||
Musher DM, Thorner AR. Community-acquired pneumonia. N Engl J Med. 2014;371(17):1619–1628. | ||
American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med 2005;171:388–416. | ||
Wu D, Wang Z, Wang H, et al. Predominance of ST5-II-t311 clone among healthcare-associated methicillin-resistant Staphylococcus aureus isolates recovered from Zhejiang, China. Int J Infect Dis. 2018;71:107–112. | ||
Molina KC, Huang V. Resistance to non-glycopeptide agents in serious Staphylococcus aureus infections. Curr Infect Dis Rep. 2016;18(12):47. | ||
Conceição T, Coelho C, de Lencastre H, Aires-de-Sousa M. High prevalence of biocide resistance determinants in Staphylococcus aureus isolates from three African countries. Antimicrob Agents Chemother. 2016;60(1):678–681. | ||
McAleese F, Petersen P, Ruzin A, et al. A novel MATE family efflux pump contributes to the reduced susceptibility of laboratory-derived Staphylococcus aureus mutants to tigecycline. Antimicrob Agents Chemother. 2005;49(5):1865–1871. | ||
Liu Y, Xu Z, Yang Z, Sun J, Ma L. Characterization of community-associated Staphylococcus aureus from skin and soft-tissue infections: a multicenter study in China. Emerg Microbes Infect. 2016;5(12):e127. | ||
Li J, Wang L, Ip M, et al. Molecular and clinical characteristics of clonal complex 59 methicillin-resistant Staphylococcus aureus infections in mainland China. PLoS One. 2013;8(8):e70602. | ||
Yu F, Liu Y, Lv J, et al. Antimicrobial susceptibility, virulence determinant carriage and molecular characteristics of Staphylococcus aureus isolates associated with skin and soft tissue infections. Braz J Infect Dis. 2015;19(6):614–622. | ||
Fitzgerald JR. Livestock-associated Staphylococcus aureus: origin, evolution and public health threat. Trends Microbiol. 2012;20(4):192–198. | ||
Pantosti A. Methicillin-resistant Staphylococcus aureus associated with animals and its relevance to human health. Front Microbiol. 2012; 3:127. | ||
Chuang YY, Huang YC. Livestock-associated methicillin-resistant Staphylococcus aureus in Asia: an emerging issue? Int J Antimicrob Agents. 2015;45(4):334–340. | ||
Schijffelen MJ, Boel CH, van Strijp JA, Fluit AC. Whole genome analysis of a livestock-associated methicillin-resistant Staphylococcus aureus ST398 isolate from a case of human endocarditis. BMC Genomics. 2010;11(1):376. | ||
He L, Zheng HX, Wang Y, et al. Detection and analysis of methicillin-resistant human-adapted sequence type 398 allows insight into community-associated methicillin-resistant Staphylococcus aureus evolution. Genome Med. 2018;10(1):5. | ||
Wang X, Li X, Liu W, Huang W, Fu Q, Li M. Molecular characteristic and virulence gene profiles of community-associated methicillin-resistant Staphylococcus aureus isolates from pediatric patients in Shanghai, China. Front Microbiol. 2016;7:1818. | ||
Gu FF, Chen Y, Dong DP, et al. Molecular epidemiology of Staphylococcus aureus among patients with skin and soft tissue infections in two Chinese hospitals. Chin Med J (Engl). 2016;129(19):2319–2324. | ||
Gruteke P, Ho PL, Haenen A, Lo WU, Lin CH, de Neeling AJ. MRSA spa t1081, a highly transmissible strain endemic to Hong Kong, China, in the Netherlands. Emerg Infect Dis. 2015;21(6):1074–1076. | ||
Ho CM, Lin CY, Ho MW, Lin HC, Peng CT, Lu JJ. Concomitant genotyping revealed diverse spreading between methicillin-resistant Staphylococcus aureus and methicillin-susceptible Staphylococcus aureus in central Taiwan. J Microbiol Immunol Infect. 2016;49(3):363–370. | ||
Lee TM, Yang MC, Yang TF, et al. Molecular characterization of community- and healthcare-associated methicillin-resistant Staphylococcus aureus isolates in southern Taiwan. Microb Drug Resist. 2015;21(6):610–621. | ||
Luk S, Ho AY, Ng TK, et al. Prevalence, prediction, and clonality of methicillin-resistant Staphylococcus aureus carriage at admission to medical units in Hong Kong, China. Infect Control Hosp Epidemiol. 2014;35(1):42–48. | ||
Cheng VC, Chan JF, Lau EH, et al. Studying the transmission dynamics of methicillin-resistant Staphylococcus aureus in Hong Kong using spa typing. J Hosp Infect. 2011;79(3):206–210. | ||
Cheng VC, Tai JW, Wong ZS, et al. Transmission of methicillin-resistant Staphylococcus aureus in the long term care facilities in Hong Kong. BMC Infect Dis. 2013;13(1):205. |
Supplementary materials
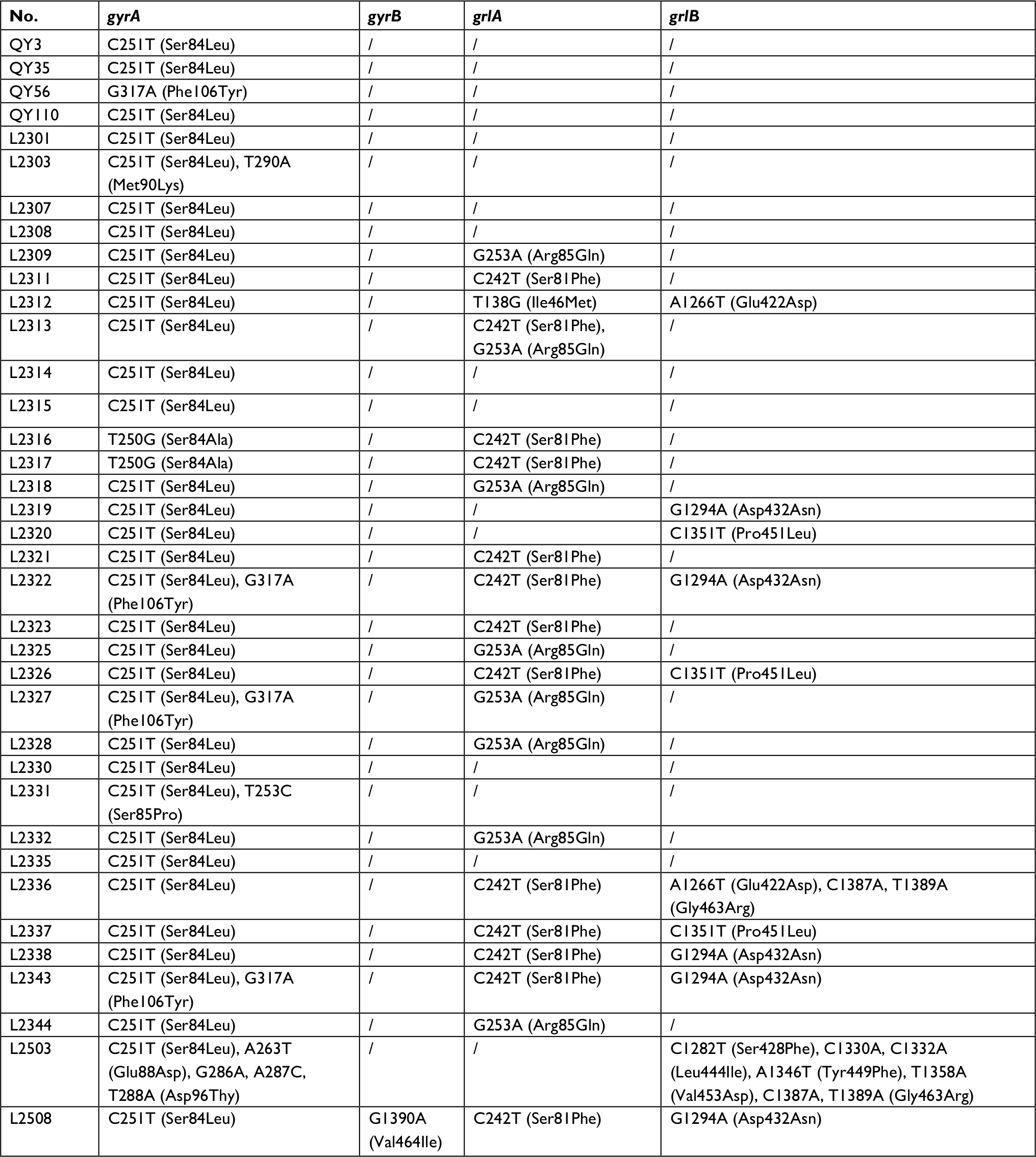 | Table S1 Mutation of gyrA, gyrB, grlA and grlB among MRSA isolates from different sources Note: Reference: GenBank: AASB02000026.1 (gyrA/gyrB), GenBank: AKYW15.1 (grlA/grlB). Abbreviation: MRSA, methicillin-resistant Staphylococcus aureus. |
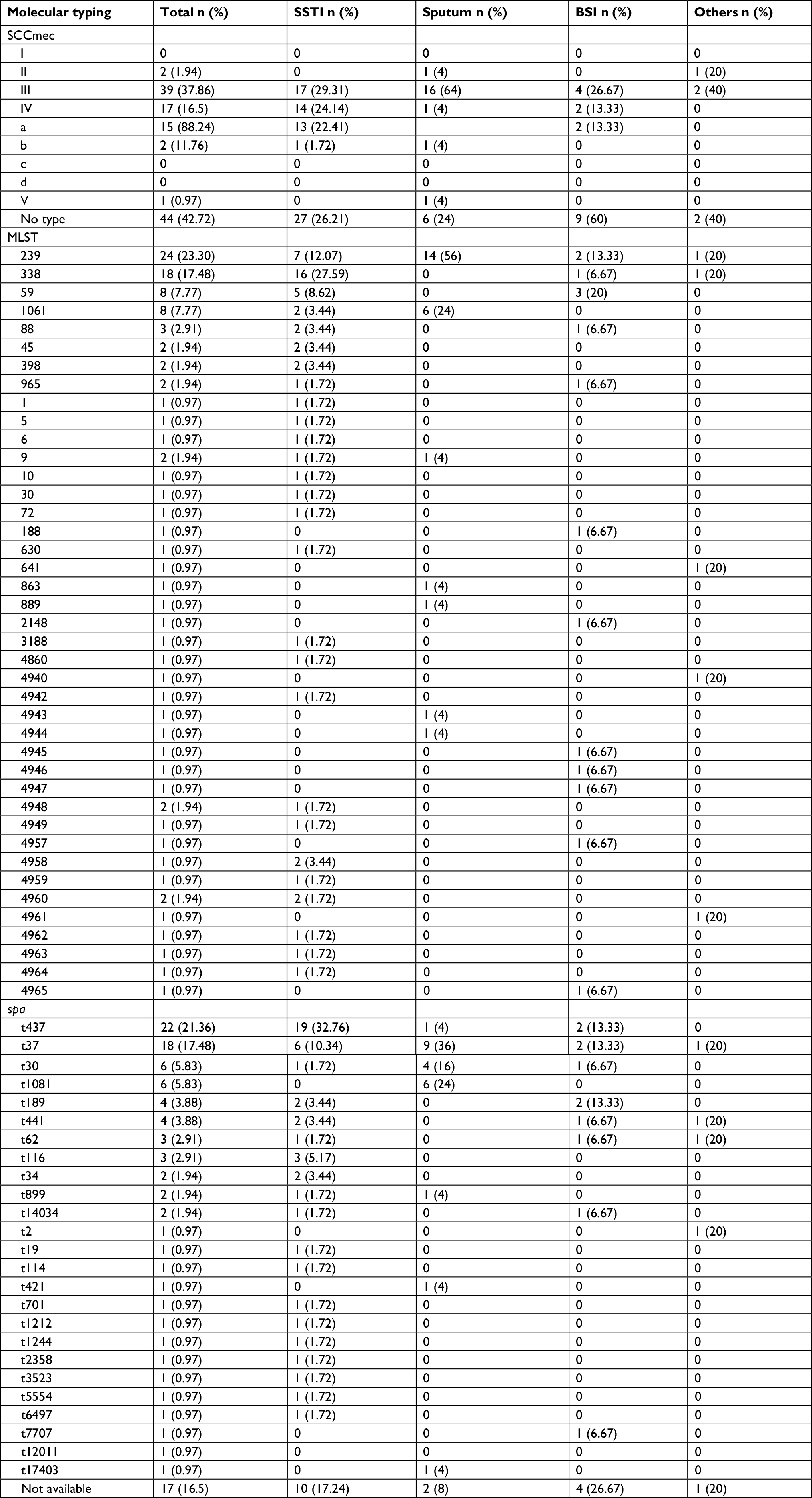 | Table S2 MLST, SCCmec types and spa types of MRSA isolates from different sources Abbreviations: BSI, blood stream infection; MLST, multilocus sequence typing; MRSA, methicillin-resistant Staphylococcus aureus; SCCmec, staphylococcal chromosomal cassette mec; SSTI, skin and soft tissue infection. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.