Back to Journals » Infection and Drug Resistance » Volume 16
Antimicrobial Peptides and Cell-Penetrating Peptides: Non-Antibiotic Membrane-Targeting Strategies Against Bacterial Infections
Received 7 November 2022
Accepted for publication 2 February 2023
Published 28 February 2023 Volume 2023:16 Pages 1203—1219
DOI https://doi.org/10.2147/IDR.S396566
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Xucheng Huang,1,2 Guoli Li1,2
1Department of Clinical Laboratory, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Key Laboratory of Precision Medicine in Diagnosis and Monitoring Research of Zhejiang Province, Hangzhou, People’s Republic of China
Correspondence: Guoli Li, Department of Clinical Laboratory, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, 3 East Qingchun Road, Hangzhou, Zhejiang, 310016, People’s Republic of China, Tel +86-571-86002260, Fax +86-571-86044817, Email [email protected]
Abstract: The prevalence of antimicrobial resistance (AMR) has been rising quickly in recent years. AMR has emerged as a significant obstacle to the treatment of infectious diseases, and many attempts have been made over the past decades to find the best antimicrobials to overcome it. Therefore, it is crucial to find new medicines to combat the global rise of AMR. Antimicrobial peptides (AMPs) and cell-penetrating peptides (CPPs), which target membranes, are promising antibiotic substitutes. AMPs and CPPs are short amino acid sequences with antibacterial activity as well as possible therapeutic benefits. In this review, we provide a thorough and systematic introduction to the advancement of research on AMPs and CPPs, including information on their classification, mechanism of action, current state of application, limitations and optimization.
Keywords: antimicrobial peptides, cell‐penetrating peptides, membrane-targeting peptides, cellular uptake process, bacterial infection
Introduction
Due to the increase in resistant pathogens worldwide, antimicrobial resistance (AMR) is becoming a threat to global health. In 2019, the World Health Organization (WHO) identified 32 antibiotics in clinical development that address the WHO list of priority pathogens, of which only six were classified as innovative (https://www.who.int). AMR has raised concerns because it threatens the effectiveness of treatment and prevention of infectious diseases globally.1 The discovery of new antibiotics, particularly those with new mechanisms, is currently a very important public health issue.
The integrity of the bacterial membrane is crucial for bacterial survival, and numerous medications must cross the membrane to reach intracellular targets. This membrane barrier is also responsible for establishing concentration and electrical gradients between the bacteria and its environment.2,3 Antibiotics that impair this permeability barrier are urgently needed.4 Many peptides can damage or penetrate the peripheral membrane to disrupt cell functions. Antimicrobial peptides (AMPs) and cell‐penetrating peptides (CPPs) are short, cationic peptides with antimicrobial activity. AMPs can be synthetic or natural, with 10–60 amino acid residues that contribute to the cell killing of bacteria, viruses and fungi. The majority of AMPs are cationic, consisting of short segments of positively charged amino acids, while also being amphiphilic. CPPs are positively charged with short compounds consisting of 5–30 natural or artificial amino acid residues, which can penetrate a wide range of biological membranes and have been used as tools to deliver various types of conjugated cargo.5 AMPs exert their antimicrobial activity by damaging bacterial membranes, and CPPs can penetrate the microbial envelope barrier and enhance the transport of antimicrobials. AMPs and CPPs are considered new weapons to fight against infections caused by AMR bacteria, and AMPs and CPPs have shown considerable abilities in the treatment of infections by AMR pathogens.3,6,7 In this review, we described the characteristics of AMPs and CPPs and summarized recent insights into the mechanisms and designs of AMPs and CPPs.
Antimicrobial Peptides
AMPs are synthetic or natural peptides with 10–60 amino acid residues and possess antibacterial activity. AMPs are present in all forms of life, from multicellular organisms to bacteria, and they were initially discovered in the late 1930s. For hundreds of millions of years, natural AMPs have demonstrated a pivotal role in the innate immune system as a host defence mechanism by destroying invading pathogens such as bacteria, fungi, parasites, and viruses.8,9 Moreover, many AMPs are involved in the regulation of cell proliferation, epithelialization, angiogenesis, wound healing, the inflammatory response and adaptive immunity.10 Currently, AMPs are viewed as a possible substitute for traditional antimicrobials due to their broad-spectrum antimicrobial activities against a variety of bacteria, fungi, protozoans, and viruses.11 A total of 3425 AMPs have been reported in the antimicrobial peptide database (APD) updated on June 30, 2022.12 The search for novel antiviral compounds to combat severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has recently boosted interest in AMPs.13 Increasing evidence shows that AMPs can inhibit SARS-CoV-2, paving the way for their use as therapeutic drugs.14 Taken together, AMPs have attracted increasing attention in recent years as an attractive class of antimicrobials for the treatment of microbial infections, especially those caused by AMR pathogens.
Structural and Functional Properties of AMPs
Although AMP sequences and sources are quite diverse, they can be divided into four categories based on their secondary structures, including α-helical peptides (α), β-sheet peptides (β), linear extension structure (non-αβ), and peptides with both α-helix and β-sheet structures (αβ)15 (Figure 1).
 |
Figure 1 Different structures of AMPs. (A) Clavanin A, fowlicidin-1 and plantaricin A have typical α-helical conformations. (B) Gomesin, hepcidin-20 and thanatin have β-sheet conformations. (C) Termicin, phormicin and plectasin have both alpha-helix and beta-sheet conformations. (D) Drosocin, indolicidin and tritrpticin are AMPs with linear extension structures. All the structures were taken from the DBAASP v3: database.112 |
Three crucial characteristics of the antibacterial activity of AMPs include their cationic properties, amphiphilicity, and hydrophobicity.113 Almost all AMPs have positive charges ranging from +2 to +9. The cationic properties of AMPs contribute to their antibacterial activity by electrostatic interactions between the cationic AMPs and anionic bacterial membranes. The amphipathic and hydrophobicity of AMPs contribute to their ability to interact with hydrophilic or hydrophobic components.15,16 It was observed that the right combination of hydrophobicity, charge density, and peptide length could influence the antimicrobial activity of AMPs. Furthermore, the position of charged amino acids and the size of hydrophobic regions can affect the secondary structure of AMPs and consequently affect their antibacterial activities. Amphiphilicity and hydrophobicity are frequently employed to describe how peptides interact with and permeate bacterial membranes, respectively.
The Mechanisms of AMPs
The bacterial membrane is essential because it is vital for homeostasis and metabolic energy transduction and houses approximately one-third of cell proteins, which regulate several crucial roles.17 Bacterial membranes represent a highly selective permeability barrier between intra- and extracellular media, which maintains cell integrity and facilitates normal cellular functions.18 The primary mechanism of AMPs is their direct activity on the bacterial membrane, which results in membrane permeabilization and structural disruption.19 Membrane interactions are mediated by electrostatic forces between positively charged AMPs and negatively charged microbial surfaces.
In gram-negative bacteria, the lipopolysaccharide (LPS)-rich outer membrane is the first-line defence and is highly impermeable. It is the main intrinsic antibiotic resistance factor and a major reason why gram-negative bacteria are much more resilient to antibiotic attacks than gram-positive bacteria.2 It has been found that some AMPs, most prominently polymyxins, can target the outer membrane.20 Some AMPs can cross the membrane by charge-exchange mechanisms, in which cationic peptides compete with Ca2+ and Mg2+ bound to LPS, possibly promoted by binding to outer membrane proteins.21 Gram-positive bacterial cell-envelope structures differ significantly from those of gram-negative bacteria. In comparison to gram-negative bacteria, gram-positive bacteria lack the outer membrane, and they typically have thicker cell walls with multiple peptidoglycan layers. This structure is essential for bacterial survival by protecting bacteria against mechanical and osmotic stress. The cell wall synthesis machinery is the most common target of clinical antibiotics as well as AMPs.22 Previous studies suggested that interactions of AMPs with the membrane severely disturb the synthesis of peptidoglycan precursor lipid II.23 AMPs also act via various mechanisms in different membrane environments. Membrane fluidity has recently been found to play a vital role in the mechanism of antibiotics and AMPs.24 Many membrane-active compounds, including several AMPs, affect the distribution of membrane domains.25 In addition, some AMPs can bind to and inhibit DNA/RNA or protein synthesis processes and lead to inactivation of essential intracellular enzymes.26
Once AMPs cross the outer membrane and the cell wall, their interaction with the cytoplasmic membrane and internal targets may follow similar mechanisms.27 Biologically, the main process involved in AMP-host interactions can be summarized in five steps: (1) initial contact with the target membrane via either biochemical or biophysical affinity; (2) structural adjustment in the target cell membrane; (3) accumulation up to a threshold AMP concentration; (4) disturbing the target cell membrane by permeabilization or depolarization; and (5) accessing the ultimate targets.10 In summary, the interaction between AMPs and bacterial membranes leads to a breakdown of membrane potential, an alteration in membrane permeability, and metabolite leakage, ultimately causing bacterial cell death.
After initial binding and conformational transitions, AMPs exert their antimicrobial activity via further structural changes, such as conformational changes into a helix or barrel structure. Membrane permeabilization is a key step in allowing certain AMPs to translocate into the bacterial cytoplasm.28 Several models have been proposed to describe various AMP-mediated membrane interactions, which vary depending on both the physical properties of the AMPs and the cell membrane composition; the following are three typical models: carpet model, barrel-stave model, and toroidal pore model29(Figure 2). (1) The carpet model involves parallel accumulation of peptides via electrostatic attractions to the anionic cell surface in a carpet-like fashion. AMPs become associated with the acidic lipid-rich regions of the membrane, thus “carpeting” the surface. Peptides such as ovisprin and cecropins employ the carpeting mechanism to start the initial peptide binding process.30 (2) The barrel-stave model involves the accumulation of monomer peptides on the cell surface followed by conformational changes and aggregation to form barrel-shaped multimers within the bacterial membrane. The barrel-stave model is often associated with hydrophobic peptides bearing 20 amino acids or more, such as alamethicin. (3) The toroidal model is an intermediate templating between the carpet and barrel-stave models. The toroidal pore model involves the formation of “wormhole-like” pores in the membrane, in which phospholipid head groups of membrane lipids remain associated with the hydrophilic portion of the peptides continuously from the outer to inner leaflets of the membrane. This type of mechanism has been proposed for peptides such as PGLa and magainins.31,32 These models are not necessarily mutually exclusive, and certain AMPs may adopt features of more than one model.
 |
Figure 2 Three models of action for extracellular AMP activity include the carpet model, barrel stave model and toroidal pore model.114 Notes: Reproduced from Huan Y, Kong Q, Mou H and Yi H (2020) Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 11:582779. Creative Commons Attribution License (CC BY).114 |
The Therapeutic Potential of AMPs
The cell membrane is a potential target for clinical therapy, and serious damage to the cell membrane can initiate cell autophagy, apoptosis and necrosis.
Since AMPs have multiple modes of action and many AMPs act on evolutionarily conserved components of the cell membrane, bacterial membrane disruptions often occur rapidly. Thus, it is unlikely that a mechanism would evolve that can save bacteria from AMPs; multiple mutations would be necessary in bacterial membrane structures, which would require a long time.33 It has been postulated that the rapid microbicidal action of AMPs precludes the evolution of strong resistance. In brief, even though antimicrobial resistance is a major challenge, the mechanism by which AMPs kill pathogens makes it relatively more difficult to develop resistance.29 Overall, the broad-spectrum activity, minimal resistance generation and rapid bactericidal action of AMPs make AMPs promising candidates for antimicrobial drugs.34
Currently, the enormous number of AMPs entering clinical trials reflects their medicinal potential.35 Some AMPs are approved today for clinical use as anti-infectives, such as polymyxins and daptomycin. Clinical application examples of AMPs in antibacterial are shown in Table 1. Polymyxins are typically applied for ocular infection treatment, selective decontamination of the digestive tract, and systemic treatment of infections caused by drug-resistant gram-negative pathogens. Daptomycin is a cyclic AMP recently applied in clinical practice for the treatment of complex infections of the skin and skin structure caused by gram-positive bacteria, particularly Staphylococcus aureus.36–38 In addition, antibiotic resistance may be avoided or reduced with the use of combinational antimicrobial peptides and antibiotic treatment.39 For example, combination therapy with the antimicrobial peptide DP7 (Sequence: VQWRIRVAVIRK) eradicated vancomycin and azithromycin resistance in Staphylococcus aureus, Pseudomonas aeruginosa, and Escherichia coli.40 Moreover, bioinspired AMPs are suitable building blocks for antimicrobial coatings due to their versatile design, scalability, and environmentally friendly properties.41
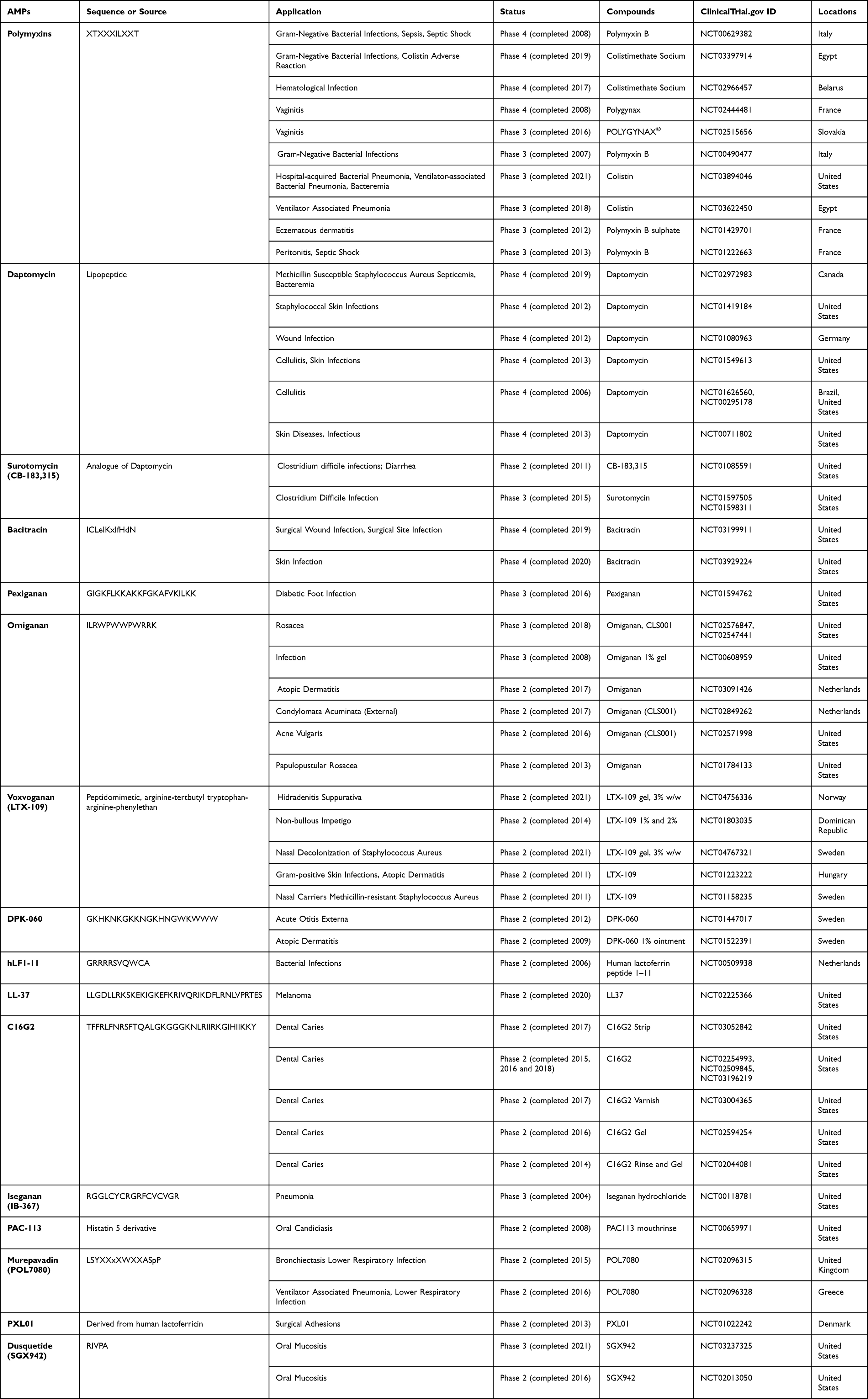 |
Table 1 Selection of AMPs in Clinical Phase of Development |
Additionally, considering that AMPs can regulate proinflammatory responses, enhance cell proliferation, and accelerate wound healing by modulating cell migration, angiogenesis, chemotaxis, and cytokine release, AMPs have the potential for a variety of clinical uses, including antioxidant, immunomodulatory, and anti-inflammatory activities.42 Due to advantages, such as their broad antimicrobial activity, the lack of quick resistance evolution, low accumulation in tissues and different sources available in nature, AMPs exhibit many benefits over commonly used antibiotics.43 Thus, AMPs represent a good starting point for the development of a new generation of antimicrobial drugs.
The Limitations and Designs of AMPs
When considering AMPs for clinical use, it is important to consider their toxicity to cells. Natural AMPs often have very long sequences and are not structurally optimized. They can easily be degraded due to the presence of certain delicate amino acids. Additionally, they may contain sequences that are easily cleavable to enzymatic digestion. AMPs might also damage the mammalian membrane and cause haemolytic side effects.29 In summary, the main limitations of AMPs that hamper their clinical utilization are their systemic toxicity, low in vivo stability, low bioavailability in the physiological concentration of salts, and rapid degradation.34 Antimicrobial peptides frequently fail to reach the market because they do not exhibit enhanced activity over already existing antibiotics for a particular indication. Although AMPs are less likely to cause bacterial resistance than traditional antibiotics, pathogens still have a chance of becoming resistant to AMPs. It was reported that pathogens can survive under prolonged AMP exposure and evolve resistance.44 Furthermore, rising production costs and technical problems also limit the manufacture of AMPs. Despite all these limitations, AMPs still have relevant advantages, such as the abundance of organisms able to produce new AMPs. Therefore, designing AMPs and membrane-active synthetic polymers as antibiotic alternatives has attracted increasing attention. The discovery of noncytotoxic AMPs requires a challenging compound optimization process towards appropriate physicochemical properties.45 Synthetic mimics of AMPs represent a promising class of novel antibiotics. Synthetic mimics of AMPs are designed in the laboratory to retain an antimicrobial pharmacophore while allowing flexibility in the chemical structure to adjust for desirable properties such as improved activity, reduced cytotoxicity, and proteolysis. Several principles must be considered in AMP design, including chain length, amphiphilicity, secondary structure, net charge, truncation, incorporation of unnatural amino acids or arginine enrichment.29 Biomimetic or de novo designed AMPs have become popular over the past two decades. De novo-generated peptide sequences have been adopted by numerous research teams to increase their activity, stability and reduce toxicity.46 They mimic the structure, function and mode of action of native AMPs while being resistant to enzymatic degradation and exhibiting better pharmacological properties. Several of these substances have shown promise in clinical trials, such as the defensin-mimetic brilacidin.47 Chemical modifications have improved the stability of peptides, including the addition of D-amino acids or unnatural amino acids, rational amino acid substitution, lipidation, halogenation, cyclization/stapling, acetylation, and peptidomimetics.48 Peptidomimetics were prepared by cyclization of linear peptides and coupling of stable unnatural amino acids, which has recently come to light. Against natural linear AMPs such as LL-37, these cyclizing peptidomimetics have shown improvement in potency.49 Chu et al described a strategy to design AMPs with enhanced salt resistance and antiendotoxin activities by linking two helical AMPs with Ala-Gly-Pro hinges, which provides a new approach for designing AMPs with antimicrobial and antiendotoxin activities for potential therapeutic applications.50 Faya et al found that a novel formulation of AMPs could enhance their activity and penetration, thereby improving the treatment of bacterial infections.51 In another recent study, it was shown that when the magainin derivative 9P2-2 is conjugated to ampicillin, the conjugate has a higher antimicrobial activity than either of the two components alone or when delivered together. The conjugate has been shown to be effective against laboratory E. coli and clinical A. Baumannii strains but was noncytotoxic against human HEK 293 cells.52 Insights could be gained to develop shorter amphiphilic peptides with greater antimicrobial activity and less cytotoxicity to mammalian host cells. To conclude, optimization of the chemical structure to create more effective synthetic peptides represents a promising strategy for the development of AMPs as a new class of drugs to prevent and treat systemic and topical infections.53 Although AMPs are a promising family of antibacterial agents, further investigation is still needed.
Cell-Penetrating Peptides
CPPs, also known as protein transduction domains, are positively charged with short compounds consisting of 5–30 natural or artificial amino acid residues, which can pass through the cell membrane via energy-dependent or energy-independent mechanisms with no interactions with specific receptors.54,55 CPPs have a high permeability rate, cross the membrane of different cell types, present low cytotoxicity and do not activate the immune response of the host.56 It has been extensively reported that CPPs are capable of transporting a wide variety of bioactive cargoes into cells, including proteins, peptides, plasmids, DNAs, siRNAs, and small drugs.57 Thus, they are considered to be peptidic delivery factors.58 Since the discovery of the first CPP, transcription activator of the human immunodeficiency virus type 1 (TAT) peptide, hundreds of CPPs have been discovered thus far with varied lengths and physicochemical properties.59,60 The utilization of CPPs as innovative carriers for intracellular cargo delivery has drawn greater interest in recent decades. The extraordinary versatility of CPPs has opened new venues for the use of CPPs in both research and therapeutic applications.
Classification of CPPs
The variety of CPPs makes it challenging to categorize them, as CPP subclasses frequently overlap. CPPs can be grouped based on their source, physicochemical characteristics, cargoes, and other factors. (1) CPPs can be classified into three classes based on the origin of peptides: protein-derived CPPs, chimeric CPPs and synthetic or artificial CPPs. Protein-derived CPPs are usually short peptide sequences responsible for translocation. Chimeric CPPs are derived from a combination of hydrophilic and hydrophobic peptide fragments from different sources. Synthetic CPPs are sequences usually designed based on model amphipathic peptides and peptides of the polyarginine family. (2) According to their physicochemical properties, CPPs can be mainly classified into cationic, amphipathic, and hydrophobic peptides. The cationic class comprises peptides with highly positive net charges at physiological pH that primarily originate from the basic short strands of arginine and lysine residues.61,62 Amphipathic CPPs generally exhibit a common structural motif, an α-helical structure, in which hydrophilic and hydrophobic amino acids are grouped in separate faces of the helix.63 Hydrophobic CPPs have a high content of hydrophobic amino acid residues, resulting in a low net charge. (3) According to the CPP-cargo conjugations, CPPs can also be divided into covalent bonded CPPs and noncovalent bonded CPPs.64,65 Covalent bonding of CPPs means that covalent bonds, such as amide, disulfide and thioester linkages, are required for attachment between the cargo and CPPs, and each type of cargo needs unique covalent conjugation.66 Noncovalent CPP-cargo complexes rely on electrostatic and/or hydrophobic interactions between large, negatively charged cargoes and positively charged CPPs. A noncovalent manner of bonding can protect the bioactive conjugates from protease or nuclease degradation, thereby increasing the serum half-life of cargoes and making it appropriate for a variety of cargo delivery applications.67
Cellular Uptake Mechanisms of CPPs
Although cellular internalization of CPPs has been extensively studied, the precise pathways involved in this intricate process remain unclear. The internalization of CPPs begins with the interaction with the membrane or directly with the phospholipid bilayer, followed by membrane permeation and finally the release of CPP into the cytosol.68 It was reported that CPP uptake could be influenced by a variety of factors, including cell type, membrane structure, linkage method, cargo size and type, concentration, incubation time, temperature, dose and physiochemical properties of the CPPs. The entry routes are broadly divided into two groups: energy-independent direct penetration into the plasma membrane and energy-dependent endocytosis69 (Figure 3). The main difference between direct penetration and endocytosis is in the membrane permeation and release steps. Whether one pathway is predominant mostly depends on the size and physicochemical nature of the cargo molecule, and some CPPs have been shown to use different routes simultaneously.70–72
 |
Figure 3 Different cellular uptake mechanisms of CPPs. (A) Direct translocation models including (a) the barrel-stave model; (b) the toroidal model; (c) inverted micelle formation; and (d) the carpet model.82 (B) Pinocytosis models including (a) macropinocytosis; (b) clathrin-mediated endocytosis; (c) caveolin-mediated endocytosis; and (d) clathrin/caveolin-independent endocytosis. The short green curves represent CPPs. Notes: Adapted from Szabó I, Yousef M, Soltész D, Bató C, Mező G, Bánóczi Z. Redesigning of cell-penetrating peptides to improve their efficacy as a drug delivery system. Pharmaceutics. 2022;14(5):907. Copyright © 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/.82 |
Energy-Independent Direct Penetration
The process of direct penetration is energy independent and can occur even at low temperatures or in the presence of inhibitors of endocytosis. It involves multiple entry routes that are initially based on the interaction of positively charged CPPs with negatively charged membrane components such as heparan sulfate and the phospholipid bilayer. This interaction is followed by peptide entrance via various mechanisms, dependent on the peptide sequence, dose, and lipid structure of the cell membrane, which include the pore formation model, inverted micelle formation model and carpet-like74 model (Figure 3A). (1) The pore formation mechanism contains two submodels: the barrel stave model and the toroidal pore model. In the barrel-stave model, helical CPPs form a barrel through hydrophobic residues, which are near the lipid chains, and hydrophilic residues create the central pore. In the toroidal model, peptides penetrate into the lipid bilayer of the membrane and cause bending of the lipid monolayer into the interior, forming a hydrophilic gap in the plasma membrane.74 The insertion of CPPs induces continuous bending of the lipid monolayers towards the pore. In this model, both CPP and lipids form a pore,75 and CPPs such as MPG and Pep-1 can enter cells via the pore formation mechanism.76 (2) The “inverted micelle” is another mechanism of direct penetration, as observed in the penetration of peptides.77 After the primary binding of positively charged residues of CPPs to the negatively charged phospholipids of the membrane, CPPs traverse the cell membrane towards the cytoplasm, forming pocket-like micelles.78 Then, these micelles cross and invert the cell membrane for the release of CPPs and their cargo into the cytoplasm. The HIV-1 TAT peptide and octaarginine are effectively internalized through inverted micelle formation.67 (3) In the carpet-like model, CPPs cover the surface of the membrane in a carpet-like manner through linkages between charged domains of CPPs and the cell membrane. Consequently, the hydrophobic part of the peptide is flipped by the hydrophobic core of the membrane, contributing to the disruption of the membrane, which finally allows the translocation of cargo.79
Energy-Dependent Endocytosis
Endocytosis is a natural and energy-dependent process occurring in all cells, in which the plasma membrane folds inwards to carry materials from the outside to the inside of cells. Bypassing endocytosis allows CPP-based delivery systems greater defence against the degradation of protein-based drugs than other drug delivery systems.80 Endocytosis of CPPs consists of two steps: endocytic entry followed by endosomal escape.81 Depending on the cargo type, endocytic entry involves several different pathways, including macropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and clathrin/caveolin-independent endocytosis83–85 (Figure 3B). Macropinocytosis results in the formation of vesicles called macropinosomes, which are formed during the inwards folding of the plasma membrane.67 Generally, energy-dependent macropinocytosis is a primary endocytotic pathway responsible for CPP-mediated intracellular delivery of large molecules and nanoparticles and their subsequent enhanced release from endosomes into the cell cytoplasm.75 Clathrin and caveolin are proteins that are present in the intracellular part of the cell membrane during endocytosis. Clathrin and caveolin are required for the invagination of the membrane and the formation of vesicles that are coated with them. Clathrin-coated vesicles are a few hundred nanometres in diameter, while caveolin-coated vesicles are less than one hundred nanometres in diameter. Clathrin-mediated endocytosis is a selective route for translocation of material into cells through binding of material to specific receptors on the surface of the cell.83 Arginine-rich CPPs, as highly positively charged molecules, typically undergo clathrin-mediated endocytosis for internalization. Amphipathic Pro-rich or TAT-dominated CPPs were often found to be internalized through caveolin-mediated endocytosis.84 Further research is required to determine the mechanism by which CPPs enter the cell by endocytosis independent of clathrin and caveolin. While the exact mechanisms of each of these processes differ concerning vesicle structure and the machinery utilized, evidence suggests that peptides remain trapped in endosomes during endocytosis. Therefore, endosomal escape must occur after CPP and CPP-cargo complex ingestion by endocytosis to prevent the cargo from being degraded in lysosomes. Some different modification strategies have been proposed to facilitate this process.64,78 However, the precise mechanism of endosomal escape remains elusive.
Preclinical and Clinical Use of CPPs
CPPs are effective tools for the delivery of therapeutic molecules and are generally considered to be safer and less cytotoxic than other currently available delivery systems. The delivery efficiency of CPPs may depend on some parameters, such as the size of the cargo-CPP complex, the nature of the CPP, and the type of peptide sequence.80 Because of their low toxicity in cells, CPPs can deliver poorly permeable or impermeable generic drugs into cells and tissues or through the skin, conjunctiva of the eyes, and the blood‒brain barrier. In recent years, CPPs have been applied for both in vitro and in vivo delivery of various therapeutic molecules, including peptides, therapeutic proteins, DNA, siRNA, drugs, imaging agents, and nanoparticles, for a wide variety of biomedical applications with direct antimicrobial, antifungal and antiparasitic functions.73 CPPs are desirable components for vaccine delivery because they are inexpensive, easy to manufacture, and often nontoxic. Among them, TAT has been most intensively studied. VP22 and polyarginine were able to significantly improve vaccine penetration and efficacy.85 Some CPPs have been applied for the delivery of peptide antidiabetics, including insulin and exendin-4, for the treatment of diabetes and Alzheimer’s disease.86
Few CPP-linked medications have been approved for topical and systemic administration in the clinic, although various CPP-based treatments have entered preclinical and clinical studies and have proven to be an effective delivery route for therapeutic compounds. To date, no CPP or CPP-conjugated drug has been approved by the US Food and Drug Administration (FDA) due to various negative characteristics, such as problems with toxicity, endosomal entrapment, immunogenicity, and in vivo stability issues.87 Although there are still several challenges to be solved before CPPs may be widely exploited in the development of novel therapeutics, CPP techniques have greatly improved the development of CPP-conjugated peptide therapeutics in human therapies. Great promise for clinical use has been demonstrated by considerable preclinical advancements made over the past few decades in the evaluation of different CPP-derived peptide therapies. Data obtained from several preclinical and clinical trials have demonstrated the ability of CPPs to transport therapeutic molecules of various types across cell and tissue barriers, thereby allowing them to reach their targets. Thus, the outcome of these investigations opened new perspectives for CPP application in the development of unprecedented human therapies. Growing knowledge of molecular mechanisms may encourage the use of CPPs as delivery vehicles for a range of treatments.
Limitations of CPPs
Clinical trials have demonstrated some obstacles to be addressed, including toxicity, immunological reactions, stability, and tissue specificity.88 Kidney and liver toxicity, as the two main elimination pathways, must be carefully evaluated and weighed against the therapeutic value. Moreover, the immunogenicity of CPPs, especially following chronic administration, needs to be carefully studied before clinical application.89 Stability issues of CPPs include lack of oral bioavailability, short half-life in blood and short duration of action. The lack of oral bioavailability can be overcome by using innovative administration routes, including formulations for intranasal delivery, inhalation and injection. In addition, before entering the tissue, medicines should have the ability to avoid being metabolized by blood proteases.90 Inactivation of CPPs by proteases leads to their short half-life in blood, thus, CPPs must be stabilized via the incorporation of nonproteinogenic amino acids or through cyclization to avoid inactivation by proteases. Additionally, some CPPs must be uptaken into intracellular endosomes and then released to reach their targets. Thus, endosomal escape efficiency is important for CPPs. The addition of auxiliary compounds or charged polymers might improve the escape efficiency but destroy the stability of CPPs and therefore lead to a short duration of action. To avoid this endosome-associated instability, new CPPs with nonendosomal uptake mechanisms should be developed.66 It has been shown that the optimal concentration of CPP and drug complexes is closely related to the ability of CPP to promote endosomal escape and higher cargo delivery rates.91
CPPs have a general lack of target specificity, which limits their clinical applications.92 Nonselective membrane penetration of CPPs might cause a wide biodistribution and drug exposure to healthy tissue.93 For example, cationic or hydrophobic CPPs can transduce into a variety of cells and tissues in vivo, which might lead to a higher chance of off-target, nonspecific effects, thereby increasing the likelihood of adverse side effects and thus limiting their utility.89 Hence, for therapeutic applications, these types of CPPs should be administered directly to the target cell. Higher specificity can be obtained by using a cell-specific CPP to transport the therapeutic agent or using a nonspecific CPP to transport cargo with targeted activity.66,92 It is proposed that a substantially lower dose of therapeutic CPP-drug complex will be needed to minimize toxicity and side effects associated with higher doses. This necessitates the development of efficient techniques for delivering the cargo precisely to the intended organ or tissue.
Optimization of CPPs
Improvements to CPPs are essential for their therapeutic usage. Improvements include reducing the degradation of CPPs by enzymes circulating in the plasma, improving the endosomal escape efficiency, and enhancing cell/tissue specificity. Typically, CPPs must be functionalized or chemically modified to create effective delivery vectors for targeting specific cells or tissues. Chemical and structural modifications of CPPs have led to more elaborate CPP designs that not only address internalization efficiency but also solve problems such as endosomal escape, circulation time, specificity and selectivity (for cells, tissues, diseases), protease stability and cytotoxicity. Properly developed CPPs and their conjugates with therapeutics offer a very promising way to deliver lower concentrations of toxic drugs to targeted tissues.
Chemical and Structural Modifications to Enhance Therapeutic Delivery and Stability
Numerous research teams concentrate on creating new CPP sequences or improving those that already exist. Their work consists of finding the shortest sequence necessary for cell entrance with the best delivery efficiency (cellular uptake and endosomal escape) and the best stability.87 Furthermore, it is possible to enhance cellular uptake by changing peptides into cyclic peptides94 or dendrimers95 or transforming the side chains.96,97 A study showed that peptides with α-helical regions can more effectively enter cells.98 The replacement of L-amino acids with their nonnatural D-stereoisomer has been described as an effective strategy to increase stability; when lysine residues are replaced with ornithine residues, the peptide becomes more resistant to cellular degradation.99 The side chain of arginine containing the guanidinium group is essential for the facilitation of cellular uptake because the hydrophobic counterion complex around the guanidinium-rich backbone can “coat” the highly cationic structure with lipophilic moieties and act as an activator.100 The addition of trifluoromethylquinoline moieties or replacement of certain residues with histidines is a common strategy to make endosmotic CPPs.101–103 Studies also found nanoparticles conjugation can enhance the antimicrobial activity of CPPs.104,105
Targeting Strategies to Improve the Specificity of CPPs
When using CPPs as a delivery mechanism, it is important to keep in mind that drug delivery often must be highly specific. Many efforts have been made to improve the specificity of CPPs. For instance, targetable CPPs can be created by combination with cell-specific targeting ligands such as small molecules, peptides, and proteins.100 The surface modification of liposomes, micelles, polymeric and inorganic nanoparticles with CPPs allows for obtaining efficient vectors. Several strategies have been introduced to constrain those peptides to be inactivated and build “off-on” switches for CPP activity based on sensitivity to external triggers (eg, UV light, ultrasound and temperature) and endogenous triggers (eg, enzymatic reaction and pH).106,107 Another important advance to improve CPP specificity could be represented by activatable CPPs (ACPPs), which are stimuli-responsive CPPs with specific sequences that promote localization to the proper cellular organelles.108
Conclusion
As widely announced by the WHO, there is an alarming rise globally in resistance towards conventional antimicrobials, posing a potential serious risk to public health.109 The exhaustion of the traditional antibiotic pipeline prompted research into alternate antimicrobial strategies. A broad range of bioactive molecules, including drugs, peptides, and proteins, cannot efficiently cross cell membranes. Because of their weak membrane-crossing capabilities, many promising candidate medications were abandoned before further development could be completed. Under the current global endeavour of fighting against antimicrobial resistance, cell membrane-targeting therapy has recently raised great interest.110 Attachment to membrane-targeting peptides that act as delivery vectors is an attractive approach to improving cellular uptake. AMPs and CPPs are membrane-targeting peptides that share similar physicochemical properties. AMPs are characterized by disruption or destabilization of cell membranes, pore formation, and enhancement of the immune response, and CPPs are involved in cell pore penetration and delivery of different cargos. As previously mentioned, AMPs are a promising class of molecules that might address the increase in bacterial resistance to conventional antibiotics. CPPs offer exciting potential to transport various types of therapeutic drugs across the cell membrane, which could help to develop effective drugs for the treatment of different types of viral and bacterial infections. Therefore, AMPs and CPPs, with antibacterial activities, have shown great potential to treat microbial infections due to their capacities to enhance drug bioavailability and improve therapeutic efficiency.111
Considering that AMPs and CPPs have been proposed as potential alternatives to antibiotics, deep insight and understanding into their possible mechanism of resistance by bacteria will be of great importance for the proper design and modification of AMPs and CPPs for use against extensive drug-resistant pathogens. In summary, it is essential to promote the long-term exploration of highly targeted membrane peptides. We believe that further research into this cutting-edge field will result in a significant breakthrough in treating drug-resistant infections.
Funding
This study was supported by grants from the Zhejiang Provincial Medical and Health Science and Technology plan (2023KY119).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Laxminarayan R, Matsoso P, Pant S, et al. Access to effective antimicrobials: a worldwide challenge. Lancet. 2016;387(10014):168–175.
2. Epand RM, Walker C, Epand RF, Magarvey NA. Molecular mechanisms of membrane targeting antibiotics. Biochim Biophys Acta Biomembr. 2016;1858(5):980–987.
3. Lima PG, Oliveira JTA, Amaral JL, Freitas CDT, Souza PFN. Synthetic antimicrobial peptides: characteristics, design, and potential as alternative molecules to overcome microbial resistance. Life Sci. 2021;278:119647.
4. Ruiz N, Kahne D, Silhavy TJ. Advances in understanding bacterial outer-membrane biogenesis. Nat Rev Microbiol. 2006;4(1):57–66.
5. Hadjicharalambous A, Bournakas N, Newman H, Skynner MJ, Beswick P. Antimicrobial and cell-penetrating peptides: understanding penetration for the design of novel conjugate antibiotics. Antibiotics. 2022;11(11):1636.
6. Claro B, González-Freire E, Calvelo M, et al. Membrane targeting antimicrobial cyclic peptide nanotubes – an experimental and computational study. Colloids Surf B Biointerfaces. 2020;196:111349.
7. Pham TN, Loupias P, Dassonville‐Klimpt A, Sonnet P. Drug delivery systems designed to overcome antimicrobial resistance. Med Res Rev. 2019;39(6):2343–2396.
8. Hamoen LW, Wenzel M. Editorial: antimicrobial peptides - interaction with membrane lipids and proteins. Front Cell Dev Biol. 2017;5:4.
9. Bechinger B, Gorr SU. Antimicrobial peptides: mechanisms of action and resistance. J Dent Res. 2016;96(3):254–260.
10. Moravej H, Moravej Z, Yazdanparast M, et al. Antimicrobial peptides: features, action, and their resistance mechanisms in bacteria. Microb Drug Resist. 2018;24(6):747–767.
11. da Cunha NB, Cobacho NB, Viana JFC, et al. The next generation of antimicrobial peptides (AMPs) as molecular therapeutic tools for the treatment of diseases with social and economic impacts. Drug Discov Today. 2017;22(2):234–248.
12. Wang G, Li X, Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44(D1):D1087–D1093.
13. Kurpe SR, Grishin SY, Surin AK, et al. Antimicrobial and amyloidogenic activity of peptides. can antimicrobial peptides be used against SARS-CoV-2? Int J Mol Sci. 2020;21(24):9552.
14. Mabrouk DM. Antimicrobial peptides: features, applications and the potential use against covid-19. Mol Biol Rep. 2022;49(10):10039–10050.
15. Hancock RE. Peptide antibiotics. Lancet. 1997;349(9049):418–422.
16. Bahar A, Ren D. Antimicrobial peptides. Pharmaceuticals. 2013;6(12):1543–1575.
17. Qiu W-X, Zhang M-K, Liu L-H, et al. A self-delivery membrane system for enhanced anti-tumor therapy. Biomaterials. 2018;161:81–94.
18. Dias C, Rauter AP. Membrane-targeting antibiotics: recent developments outside the peptide space. Future Med Chem. 2019;11(3):211–228.
19. Pushpanathan M, Gunasekaran P, Rajendhran J. Antimicrobial peptides: versatile biological properties. Int J Pept. 2013;2013:675391.
20. Vaara M. Agents that increase the permeability of the outer membrane. Microbiol Rev. 1992;56(3):395–411.
21. Anunthawan T, de la Fuente-Núñez C, Hancock RE, Klaynongsruang S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim Biophys Acta. 2015;1848(6):1352–1358.
22. Schneider T, Sahl H-G. An oldie but a goodie – cell wall biosynthesis as antibiotic target pathway. Int J Med Microbiol. 2010;300(2–3):161–169.
23. Müller A, Wenzel M, Strahl H, et al. Daptomycin inhibits cell envelope synthesis by interfering with fluid membrane microdomains. Proc Natl Acad Sci. 2016;113(45):E7077–E7086.
24. Omardien S, Drijfhout JW, Vaz FM, et al. Bactericidal activity of amphipathic cationic antimicrobial peptides involves altering the membrane fluidity when interacting with the phospholipid bilayer. Biochim Biophys Acta Biomembr. 2018;1860(11):2404–2415.
25. Schäfer A-B, Wenzel M. A how-to guide for mode of action analysis of antimicrobial peptides. Front Cell Infect Microbiol. 2020;10:540898.
26. Scocchi M, Mardirossian M, Runti G, Benincasa M. Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr Top Med Chem. 2016;16(1):76–88.
27. Malanovic N, Lohner K. Gram-positive bacterial cell envelopes: the impact on the activity of antimicrobial peptides. Biochim Biophys Acta. 2016;1858(5):936–946.
28. Pfalzgraff A, Brandenburg K, Weindl G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front Pharmacol. 2018;9:281.
29. Ciumac D, Gong H, Hu X, Lu JR. Membrane targeting cationic antimicrobial peptides. J Colloid Interface Sci. 2019;537:163–185.
30. Teixeira V, Feio MJ, Bastos M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog Lipid Res. 2012;51(2):149–177.
31. Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55(1):27–55.
32. Baxter AA, Lay FT, Poon IKH, Kvansakul M, Hulett MD. Tumor cell membrane-targeting cationic antimicrobial peptides: novel insights into mechanisms of action and therapeutic prospects. Cell Mol Life Sci. 2017;74(20):3809–3825.
33. Marr AK, Gooderham WJ, Hancock RE. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol. 2006;6(5):468–472.
34. Mahlapuu M, Håkansson J, Ringstad L, Björn C. Antimicrobial peptides: an emerging category of therapeutic agents. Front Cell Infect Microbiol. 2016;6:194.
35. Browne K, Chakraborty S, Chen R, et al. A new era of antibiotics: the clinical potential of antimicrobial peptides. Int J Mol Sci. 2020;21(19):7047.
36. Mahlapuu M, Björn C, Ekblom J. Antimicrobial peptides as therapeutic agents: opportunities and challenges. Crit Rev Biotechnol. 2020;40(7):978–992.
37. Zavascki AP, Goldani LZ, Li J, Nation RL. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother. 2007;60(6):1206–1215.
38. Sierra JM, Fusté E, Rabanal F, Vinuesa T, Viñas M. An overview of antimicrobial peptides and the latest advances in their development. Expert Opin Biol Ther. 2017;17(6):663–676.
39. Doern CD. When does 2 plus 2 equal 5? A review of antimicrobial synergy testing. J Clin Microbiol. 2014;52(12):4124–4128.
40. Wu X, Li Z, Li X, et al. Synergistic effects of antimicrobial peptide DP7 combined with antibiotics against multidrug-resistant bacteria. Drug Des Devel Ther. 2017;11:939–946.
41. Freitas ED, Bataglioli RA, Oshodi J, Beppu MM. Antimicrobial peptides and their potential application in antiviral coating agents. Colloids Surf B Biointerfaces. 2022;217:112693.
42. Moretta A, Scieuzo C, Petrone AM, et al. Antimicrobial peptides: a new hope in biomedical and pharmaceutical fields. Front Cell Infect Microbiol. 2021;11. doi:10.3389/fcimb.2021.668632
43. Mba IE, Nweze EI. Antimicrobial peptides therapy: an emerging alternative for treating drug-resistant bacteria. Yale J Biol Med. 2022;95(4):445–463.
44. Boparai JK, Sharma PK. Mini review on antimicrobial peptides, sources, mechanism and recent applications. Protein Pept Lett. 2020;27(1):4–16.
45. Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov. 2011;11(1):37–51.
46. Annunziato G, Costantino G. Antimicrobial peptides (AMPs): a patent review (2015–2020). Expert Opin Ther Pat. 2020;30(12):931–947.
47. Kuppusamy R, Willcox M, Black DS, Kumar N. Short cationic peptidomimetic antimicrobials. Antibiotics. 2019;8(2):44.
48. Drayton M, Kizhakkedathu JN, Straus SK. Towards robust delivery of antimicrobial peptides to combat bacterial resistance. Molecules. 2020;25(13):3048.
49. Rounds T, Straus SK. Lipidation of antimicrobial peptides as a design strategy for future alternatives to antibiotics. Int J Mol Sci. 2020;21(24):9692.
50. Chu H-L, Chih Y-H, Peng K-L, et al. Antimicrobial peptides with enhanced salt resistance and antiendotoxin properties. Int J Mol Sci. 2020;21(18):6810.
51. Faya M, Hazzah HA, Omolo CA, et al. Novel formulation of antimicrobial peptides enhances antimicrobial activity against methicillin-resistant Staphylococcus aureus (MRSA). Amino Acids. 2020;52(10):1439–1457.
52. Yamauchi R, Kawano K, Yamaoka Y, et al. Development of antimicrobial peptide-antibiotic conjugates to improve the outer membrane permeability of antibiotics against gram-negative bacteria. ACS Infect Dis. 2022;8(11):2339–2347.
53. Cardoso MH, Orozco RQ, Rezende SB, et al. Computer-aided design of antimicrobial peptides: are we generating effective drug candidates? Front Microbiol. 2019;10:3097.
54. Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med. 2015;21(9):560–570.
55. Ye J, Liu E, Yu Z, et al. CPP-assisted intracellular drug delivery, what is next? Int J Mol Sci. 2016;17(11):1892.
56. Ruseska I, Zimmer A. Internalization mechanisms of cell-penetrating peptides. Beilstein J Nanotechnol. 2020;11:101–123.
57. Del Rio G, Trejo Perez Mario A, Brizuela Carlos A. Antimicrobial peptides with cell-penetrating activity as prophylactic and treatment drugs. Biosci Rep. 2022;42(9):BSR20221789.
58. Grdisa M. The delivery of biologically active (therapeutic) peptides and proteins into cells. Curr Med Chem. 2011;18(9):1373–1379.
59. Milletti F. Cell-penetrating peptides: classes, origin, and current landscape. Drug Discov Today. 2012;17(15–16):850–860.
60. Gautam A, Chaudhary K, Kumar R, et al. In silico approaches for designing highly effective cell penetrating peptides. J Transl Med. 2013;11:74.
61. De Coupade C, Fittipaldi A, Chagnas V, et al. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J. 2005;390(Pt 2):407–418.
62. Kim H, Kitamatsu M, Ohtsuki T. Enhanced intracellular peptide delivery by multivalent cell-penetrating peptide with bioreducible linkage. Bioorg Med Chem Lett. 2018;28(3):378–381.
63. Deshayes S, Konate K, Aldrian G, Heitz F, Divita G. Interactions of amphipathic CPPs with model membranes. Methods Mol Biol. 2011;683:41–56.
64. Vasconcelos L, Pärn K, Langel U. Therapeutic potential of cell-penetrating peptides. Ther Deliv. 2013;4(5):573–591.
65. Gros E, Deshayes S, Morris MC, et al. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim Biophys Acta. 2006;1758(3):384–393.
66. Derakhshankhah H, Jafari S. Cell penetrating peptides: a concise review with emphasis on biomedical applications. Biomed Pharmacother. 2018;108:1090–1096.
67. Böhmová E, Machová D, Pechar M, et al. Cell-penetrating peptides: a useful tool for the delivery of various cargoes into cells. Physiol Res. 2018;67(Suppl 2):S267–S279.
68. Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES. Cell-penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des. 2012;80(5):639–646.
69. Zorko M, Langel Ü. Studies of cell-penetrating peptides by biophysical methods. Q Rev Biophys. 2022;1–55:e3.
70. Maiolo JR, Ferrer M, Ottinger EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta. 2005;1712(2):161–172.
71. Ramsey JD, Flynn NH. Cell-penetrating peptides transport therapeutics into cells. Pharmacol Ther. 2015;154:78–86.
72. Gestin M, Dowaidar M, Langel Ü. Uptake mechanism of cell-penetrating peptides. In: Peptides and Peptide-Based Biomaterials and their Biomedical Applications. Springer;2017:255–264.
73. Kardani K, Milani A, Shabani S, Bolhassani A. Cell penetrating peptides: the potent multi-cargo intracellular carriers. Expert Opin Drug Deliv. 2019;16(11):1227–1258.
74. Ruczynski J, Wierzbicki PM, Kogut-Wierzbicka M, Mucha P, Siedlecka-Kroplewska K, Rekowski P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem Cytobiol. 2014;52(4):257–269.
75. Wadia JS, Stan RV, Dowdy SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med. 2004;10(3):310–315.
76. Thorén PE, Persson D, Isakson P, Goksör M, Onfelt A, Nordén B. Uptake of analogs of penetratin, Tat(48–60) and oligoarginine in live cells. Biochem Biophys Res Commun. 2003;307(1):100–107.
77. Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8(2):84–87.
78. Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38(4):406–424.
79. Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones AT. Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans. 2007;35(Pt 4):784–787.
80. Nam SH, Park J, Koo H. Recent advances in selective and targeted drug/gene delivery systems using cell-penetrating peptides. Arch Pharm Res. 2023;46:18–34.
81. Bechara C, Sagan S. Cell‐penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587(12):1693–1702.
82. Szabó I, Yousef M, Soltész D, Bató C, Mező G, Bánóczi Z. Redesigning of cell-penetrating peptides to improve their efficacy as a drug delivery system. Pharmaceutics. 2022;14(5):907.
83. Ter-Avetisyan G, Tünnemann G, Nowak D, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284(6):3370–3378.
84. Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896.
85. Yang J, Luo Y, Shibu MA, Toth I, Skwarczynskia M. Cell-penetrating peptides: efficient vectors for vaccine delivery. Curr Drug Deliv. 2019;16(5):430–443.
86. Maeng J, Lee K. Systemic and brain delivery of antidiabetic peptides through nasal administration using cell-penetrating peptides. Front Pharmacol. 2022;13:1068495.
87. Habault J, Poyet J-L. Recent advances in cell penetrating peptide-based anticancer therapies. Molecules. 2019;24(5):927.
88. Bolhassani A, Jafarzade BS, Mardani G. In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides. 2017;87:50–63.
89. Zahid M, Robbins P. Cell-type specific penetrating peptides: therapeutic promises and challenges. Molecules. 2015;20(7):13055–13070.
90. Feni L, Neundorf I. The current role of cell-penetrating peptides in cancer therapy. In: Peptides and Peptide-Based Biomaterials and Their Biomedical Applications. Springer;2017:279–295.
91. Lundberg P, El-Andaloussi S, Sütlü T, Johansson H, Langel U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007;21(11):2664–2671.
92. Reissmann S. Cell penetration: scope and limitations by the application of cell-penetrating peptides. J Pept Sci. 2014;20(10):760–784.
93. Skotland T, Iversen T, Torgersen M, Sandvig K. Cell-penetrating peptides: possibilities and challenges for drug delivery in vitro and in vivo. Molecules. 2015;20(7):13313–13323.
94. Park SE, Sajid MI, Parang K, Tiwari RK. Cyclic cell-penetrating peptides as efficient intracellular drug delivery tools. Mol Pharm. 2019;16(9):3727–3743.
95. Kozhikhova KV, Andreev SM, Shilovskiy IP, et al. A novel peptide dendrimer LTP efficiently facilitates transfection of mammalian cells. Org Biomol Chem. 2018;16(43):8181–8190.
96. Tesei G, Vazdar M, Jensen MR, et al. Self-association of a highly charged arginine-rich cell-penetrating peptide. Proc Natl Acad Sci U S A. 2017;114(43):11428–11433.
97. Demizu Y, Oba M, Okitsu K, et al. A preorganized β-amino acid bearing a guanidinium side chain and its use in cell-penetrating peptides. Org Biomol Chem. 2015;13(20):5617–5620.
98. Copolovici DM, Langel K, Eriste E, Langel Ü. Cell-penetrating peptides: design, synthesis, and applications. ACS nano. 2014;8(3):1972–1994.
99. Ezzat K, Andaloussi SE, Zaghloul EM, et al. PepFect 14, a novel cell-penetrating peptide for oligonucleotide delivery in solution and as solid formulation. Nucleic Acids Res. 2011;39(12):5284–5298.
100. Kang Z, Ding G, Meng Z, Meng Q. The rational design of cell-penetrating peptides for application in delivery systems. Peptides. 2019;121:170149.
101. Andaloussi SE, Lehto T, Mäger I, et al. Design of a peptide-based vector, PepFect6, for efficient delivery of siRNA in cell culture and systemically in vivo. Nucleic Acids Res. 2011;39(9):3972–3987.
102. El-Sayed A, Futaki S, Harashima H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: ways to overcome endosomal entrapment. AAPS J. 2009;11(1):13–22.
103. Endoh T, Ohtsuki T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv Drug Deliv Rev. 2009;61(9):704–709.
104. Zhang W, Taheri-Ledari R, Hajizadeh Z, et al. Enhanced activity of vancomycin by encapsulation in hybrid magnetic nanoparticles conjugated to a cell-penetrating peptide. Nanoscale. 2020;12(6):3855–3870.
105. Taheri-Ledari R, Ahghari MR, Ansari F, et al. Synergies in antimicrobial treatment by a levofloxacin-loaded halloysite and gold nanoparticles with a conjugation to a cell-penetrating peptide. Nanoscale Adv. 2022;4(20):4418–4433.
106. Huang Y, Jiang Y, Wang H, et al. Curb challenges of the “Trojan Horse” approach: smart strategies in achieving effective yet safe cell-penetrating peptide-based drug delivery. Adv Drug Deliv Rev. 2013;65(10):1299–1315.
107. Tang B, Zaro JL, Shen Y, et al. Acid-sensitive hybrid polymeric micelles containing a reversibly activatable cell-penetrating peptide for tumor-specific cytoplasm targeting. J Control Release. 2018;279:147–156.
108. Pescina S, Ostacolo C, Gomez-Monterrey IM, et al. Cell penetrating peptides in ocular drug delivery: state of the art. J Control Release. 2018;284:84–102.
109. Luong HX, Thanh TT, Tran TH. Antimicrobial peptides - advances in development of therapeutic applications. Life Sci. 2020;260:118407.
110. Bhattacharjya S, Straus SK. Design, engineering and discovery of novel α-helical and β-boomerang antimicrobial peptides against drug resistant bacteria. Int J Mol Sci. 2020;21(16):5773.
111. Splith K, Neundorf I. Antimicrobial peptides with cell-penetrating peptide properties and vice versa. Eur Biophys J. 2011;40(4):387–397.
112. Pirtskhalava M, Amstrong AA, Grigolava M, et al. DBAASP v3: database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021;49(D1):D288–D297.
113. Riahifard N, Mozaffari S, Aldakhil T, et al. Design, synthesis, and evaluation of amphiphilic cyclic and linear peptides composed of hydrophobic and positively-charged amino acids as antibacterial agents. Molecules. 2018;23:10.
114. Huan Y, Kong Q, Mou H, Yi H. Antimicrobial peptides: classification, design, application and research progress in multiple fields. Front Microbiol. 2020;11:582779.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Antimicrobial Peptides: Mechanisms, Applications, and Therapeutic Potential
Alzain M, Daghistani H, Shamrani T, Almoghrabi Y, Daghistani Y, Alharbi OS, Sait AM, Mufrrih M, Alhazmi W, Alqarni MA, Saleh BH, Zubair MA, Juma NA, Niyazi HA, Niyazi HA, Halabi WS, Altalhi R, Kazmi I, Altayb HN, Ibrahem K, Alfadil A
Infection and Drug Resistance 2025, 18:4385-4426
Published Date: 27 August 2025
Hypervirulent and Drug-Resistant Klebsiella pneumoniae: Clinical Challenges and Alternative Treatment Strategies
Bani Abdel-Rahman S, Altarawneh H, Alfreahat HAH, Alhazmi KA, Alharbi OS, Gazzaz M, Alharthi TM, Ismaeel LA, Halabi WS, Sharif AT, Redwan B, Alkuwaity KK, Alhazmi W, Saleh BH, Alqarni MA, Alsaedi A, Alhussainy NH, Niyazi HA, Niyazi HA, AbdulMajed H, Juma NA, Ibrahem K
International Journal of General Medicine 2026, 19:548733
Published Date: 20 May 2026