")
Back to Journals » OncoTargets and Therapy » Volume 11
Antimetastatic effect of epigenetic drugs, hydralazine and valproic acid, in Ras-transformed NIH 3T3 cells
Authors Pérez-Cárdenas E , Taja-Chayeb L , Trejo-Becerril C, Chanona-Vilchis J, Chávez-Blanco A , Domínguez-Gómez G, Langley E , García-Carrancá A , Dueñas-González A
Received 14 September 2018
Accepted for publication 9 November 2018
Published 7 December 2018 Volume 2018:11 Pages 8823—8833
DOI https://doi.org/10.2147/OTT.S187306
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Enrique Pérez-Cárdenas,1 Lucía Taja-Chayeb,1 Catalina Trejo-Becerril,1 José Chanona-Vilchis,2 Alma Chávez-Blanco,1 Guadalupe Domínguez-Gómez,1 Elizabeth Langley,1 Alejandro García-Carrancá,3,4 Alfonso Dueñas-González3,4
1Division of Basic Research, Instituto Nacional de Cancerología, Mexico City, Mexico; 2Department of Pathology, Instituto Nacional de Cancerología, Mexico City, Mexico; 3Unit of Biomedical Research on Cancer, Biomedical Research Institute, Universidad Nacional Autonoma de Mexico, Mexico City, Mexico; 4Unit of Biomedical Research on Cancer, Instituto Nacional de Cancerologia, Mexico City, Mexico
Introduction: Metastasis involves the accumulation of genetic and epigenetic alterations leading to activation of prometastatic genes and inactivation of antimetastatic genes. Among epigenetic alterations, DNA hypermethylation and histone hypoacetylation are the focus of intense translational research because their pharmacological inhibition has been shown to produce antineoplastic activity in a variety of experimental models.
Aims: This study aimed to evaluate the antimetastatic effect of the DNA-methylation inhibitor, hydralazine, and the histone deacetylase inhibitor, valproic acid.
Methods: NIH 3T3-Ras murine cells were treated with hydralazine and valproic acid to evaluate their effects upon cell proliferation, cell motility, chemotaxis, gelatinase activity, and gene expression. Lung metastases were developed by intravenous injection of NIH 3T3-Ras cells in BALB/c nu/nu mice and then treated with the drug combination.
Results: Treatment induced a growth-inhibitory effect on NIH 3T3-Ras cells, showed a trend toward increased gelatinase activity of MMP2 and MMP9, and inhibited chemotaxis and cell motility. The combination led to a strong antimetastatic effect in lungs of nude mice.
Conclusion: Hydralazine and valproic acid, two repositioned drugs as epigenetic agents, exhibit antimetastatic effects in vitro and in vivo and hold potential for cancer treatment.
Keywords: hydralazine, valproic acid, epigenetics, metastasis, ras
Corrigendum for this paper has been published.
Introduction
Metastases are the result of a complex series of cellular events in cancer cells and surrounding stromata, as well as in distant organs.1 During these processes, cells must exhibit the ability to survive the harsh conditions imposed by physiological and anatomical barriers, and hence must acquire advantageous genetic and epigenetic traits to do so.2–5 It is thought that epigenetic changes may aid tumor cells to turn metastasis-related genes off and on in a timely manner, as gene expression is not only dependent on the presence of the appropriate transcription factors that interact with enhancer or promoter elements in a cell-type- or tissue-specific manner but also dependent on the availability of binding sites regulated by higher-ordered chromatin structures, specifically DNA methylation and histone deacetylation, among others.6 For instance, demethylation and reexpression of E-cadherin are thought to be essential for the correct incorporation of metastatic cells into their new and normal cellular environment.7 Other metastasis-related genes, such as NM23 and RECK, are rarely mutated in cancer and their silencing may occur through epigenetic mechanisms, which could be reverted by epigenetic drugs, DNA methyltransferase inhibitors (DNMTIs), and histone deacetylase inhibitors (HDACIs).8–12
Hydralazine and valproic acid are repositioned DNMTIs and HDACIs, respectively.13 This combination of drugs has shown clinical activity in myelodysplastic syndrome,14 cutaneous T-cell lymphoma,15 and in combination with chemotherapy or radiation against several solid tumors.16–18 Furthermore, it is known that cancer progression and metastases may present downregulation of HLA class I antigens by DNA hypermethylation and histone deacetylation, which can be reverted by DNMTIs and HDACIs. In addition, altered NK-cell activity may favor cancer metastasis.19,20 Interestingly, we and others have shown that hydralazine–valproic acid treatment upregulates HLA class I antigen expression and antigen-specific cytotoxic T-cell-lymphocyte response in carcinoma cells, as well as upregulating NKG2D ligands and enhancing NK-cell cytotoxicity in several carcinoma cell lines.21–23 In a proof-of-principle study on breast cancer, this drug combination was able to upregulate a number of tumor-suppressor genes, including the anti-metastatic gene NM23.24 It is well known that the presence of RAS mutations associates with the presence of metastases,25 so to further evaluate the potential role of this combination as antimetastatic therapy, we used the widely known Hras-transfected NIH 3T3 system, since these cells readily form lethal tumor nodules in the lungs of nude mice after tail vein injection.26
Methods
Reagents and cell lines
Valproic acid and hydralazine hydrochloride were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Polyclonal or monoclonal antibodies against pan-Ras, Nm23-H1, E-cadherin, RECK, and actin were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA), the polyclonal antibody against Smad4 was obtained from Cell Signaling Technologies (Danvers, MA, USA), and gelatin type A was obtained from Sigma-Aldrich. Embryonic mouse NIH 3T3 fibroblasts were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) containing 10% FBS (Thermo Fisher Scientific), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma-Aldrich), and incubated at 37°C in 5% CO2.
Generation of stable NIH 3T3-Ras cell line
NIH 3T3 cells were plated at a density of 5×105 cells per dish (10 cm diameter) in DMEM containing 10% FBS. Semiconfluent cells were electroporated with linearized pEJ-neo (Hrasval12). Two days after transfection, 800 μg/mL G418 antibiotic (Thermo Fisher Scientific) was added to the medium. Stable NIH 3T3 cells transfected with this plasmid were designated NIH 3T3-Ras.27
Cell-viability assay
Cells were seeded into 24-well Falcon plates (Becton Dickinson, Franklin Lakes, NJ, USA) at appropriate densities in 1 mL of complete medium. At 24 hours later, they were treated with hydralazine (10 μM) and valproic acid (1 mM) for 5 days. Thereafter, the medium was aspirated and the cells were fixed in 0.5 mL 10% buffered formaldehyde for 24 hours. Cells were washed and stained with 0.75% crystal violet in phosphate solution. Cells were then washed and the dye eluted with 33% acetic acid solution. Cell viability was assessed by dye absorbance measured at 570 nm on an automated ELISA reader. All assays were reproduced sixfold in triplicate. The cytotoxic effect of each treatment was expressed as a percentage of cell viability relative to untreated control cells (percentage of control).
Cell-migration assay
The migratory ability of cells was evaluated using a wound-healing assay. Cell lines were grown in 24-well plates and treated with 10 μM hydralazine and 1 mM valproic acid for 5 days. At day 6, the cell monolayer was mechanically wounded in the center of each well with a sterile 200 mL pipette tip. Cells were then washed three times to remove detached cells and debris. Wound healing was monitored at different time points with inverted bright-field microscopy under a 4× objective. Only the cell-free area was selected, then measured, photographed, and quantified using ImageJ 1.43 software, in agreement with the protocol described by Valster et al,28 and calculated as percentage wound healing using the equation (1 − [wound area at Tx hours/wound area at T0)], where Tx is the respective time point and T0 the time immediately after wounding.
Chemotaxis assay
NIH 3T3-Ras cells were assayed for chemotaxis using a modified Boyden-chamber method. Briefly, cells were treated with 10 μM hydralazine and 1 mM valproic acid for 5 days. After this time, cells were detached and suspended in serum-free DMEM. Thereafter, 15×103 viable cells, as assessed by trypan blue exclusion, were placed in the upper compartment of a microchemotaxis chamber (Neuro Probe, Gaithersburg, MD, USA), onto gelatin-coated (type A; Sigma-Aldrich) cellulose nitrate membranes with 8 μm pore diameter (Neuro Probe). DMEM containing no, 2.5%, 5%, 7.5%, 10%, and 15% FBS was placed in the lower compartment of the chambers. After 24 hours of incubation, cells that had moved to the lower surface of the filters were fixed, stained with azure dye, and counted. Cellular motility observed with DMEM supplemented with 10% FBS was taken as 100% migration.
Gelatinase activity
Gelatinase activity was evaluated by zymography. Cells were cultured in 75 cm2 culture flasks in 10 mL medium and treated with hydralazine and valproic acid at 10 μM and 1 mM, respectively. After 5 days, cells were cultured in unsupplemented medium (serum- and phenol red-free). At 24 hours later, conditioned medium was removed, centrifuged at 1,500 g for 10 minutes, and concentrated by ultrafiltration (Amicon). Proteins with molecular weight >10 kDa were retained. Protein content was measured by Bradford assay using BSA (Sigma-Aldrich) as a standard, and gelatinase activity was determined by zymography. Gelatin zymography was performed as previously described.29 Briefly, 5 mg of each concentrated conditioned medium were loaded onto nonreducing 8% SDS-PAGE copolymerized with 1% gelatin. After electrophoresis, gels were rinsed in 2.5% Triton X-100, incubated in TNC buffer (Tris–HCl 50 mM, NaCl 150 mM, and CaCl2 20 mM, pH 7.4) at 37°C for 14–16 hours, and stained with Coomassie blue R250 (Sigma-Aldrich). MMPs were detected as transparent bands (digested area) on the blue background of the stained gel. Levels of proteolytic activity were quantified by densitometry using ImageJ 1.45.
Real-time PCR array
Total RNA was extracted from NIH 3T3-Ras cells treated for 5 days in the presence or absence of hydralazine and valproic acid (10 μM and 1 mM, respectively), using Trizol. Analysis of metastatic gene expression was performed with an RT2 Profiler PCR array kit to examine mRNA levels of 84 genes associated with tumor metastasis, including five housekeeping genes, one genomic DNA control, three reverse-transcription controls, and three positive PCR controls, in 96-well plates, following the manufacturer’s protocol (Qiagen, Venlo, the Netherlands). Briefly, cDNAs were synthesized from 1 μg total RNA using the RT2 First Strand cDNA reagent kit according to the manufacturer’s protocol. The reaction mixtures (20 μL) were incubated at 42°C for 15 minutes, followed by incubation at 95°C for 5 minutes, and then cooled on ice. PCR arrays were performed independently using a 7,500 real-time PCR system (Thermo Fisher Scientific). Values were obtained for the threshold cycle (Ct) for each gene and normalized using the average of four housekeeping genes on the same array. Resulting values are reported as fold change. Only genes showing double or greater change were considered for the analysis.
Western blots
Whole-cell extracts were prepared in lysis buffer containing 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5% Nonidet P40, 1 mM EDTA, and a protease inhibitor cocktail (Sigma-Aldrich). For immunoblot analysis, 20 μg protein from cell lysates were separated on 10% SDS-PAGE. Subsequently, proteins were electrotransferred to a polyvinylidene difluoride filter (Amersham Hybond) and probed with antibodies against pan-Rasval12 (mouse monoclonal antibody; Sigma-Aldrich), Nm23-H1 (rabbit polyclonal IgG antibody; Santa Cruz Biotechnology), E-cadherin (mouse monoclonal IgG antibody; Santa Cruz Biotechnology Inc.), Smad4 and RECK (rabbit polyclonal IgG antibody; Cell Signaling Technology), and actin (goat polyclonal IgG antibody; Santa Cruz Biotechnology). Detection using species-specific IgG-HRP secondary antibodies (Santa Cruz Biotechnology Inc.) was performed with the Immobilon Western chemiluminescent HRP substrate (Merck KGaA, Darstadt, Germany).
Lung-metastasis assay
Animal studies were approved by the Ethics Committee of the National Cancer Institute of Mexico and the Institutional Committee of Animal Care and Use and conducted in accordance with federal laws in adherence with the Mexican Official Norm (NOM-062-ZOO-1999) policy. Six-week-old male BALB/c nu/nu mice (Harlan Laboratories) received 106 NIH 3T3 cells or NIH-Ras cells injected in 0.1 mL PBS through the tail vein. Five mice were inoculated with NIH 3T3 cells, nine mice with NIH-Ras cells, and six mice with NIH-Ras cells. Two days later, the animals in the last group were treated intraperitoneally with hydralazine at 5 mg/kg body weight and valproic acid 300 mg/kg body weight every day for 1 month. After 4 weeks, mice were killed by cervical dislocation under anesthesia to assess the presence of metastases in the internal organs. Macroscopic examinations were carried out for all internal organs, including lungs, and microscopic examination of lung metastases was performed on multiple sections of formalin-fixed, paraffin-embedded lung tissue stained with H&E. Metastatic lesion counts in the lungs of each mouse were carried out by microscopic examination.
Statistical analysis
Unless specified, results are expressed as mean ± SD of data collected from at least three independent experiments carried out in triplicate. Statistical significance was determined using SigmaStat 2.03 or InerStat-A 1.3 software by ANOVA, and P<0.05 was considered significant.
Results
Hydralazine and valproic acid decrease expression of Ras oncogene and viability of NIH 3T3-Ras cells
NIH 3T3 cells stably transfected with mutated (Val12) human c-H-RAS gene and cultured under G418 selection, were treated for 5 days with 10 μM hydralazine and 1 mM valproic acid, and then tested for Ras Val12 protein expression. The presence of the transfected Hras oncogene in the NIH 3T3-Ras cell line was confirmed by Western blot, and was found absent in untransfected wild-type cells (Figure 1A). To determine whether NIH 3T3-Ras cells remained viable after 5 days of treatment with hydralazine and valproic acid at 10 μM and 1 mM, respectively, we assayed cell viability. The results showed that after treatment, around 25.01%±1.8% of NIH 3T3-Ras cells remained viable compared to 39.7%±12.1% of wild-type cells (P<0.05; Figure 1B). Morphologically, as expected after transformation, untreated NIH 3T3-Ras cells showed transformed foci, whereas those treated with the combination showed a reduction in transformed foci and some cells showed rounded morphology (Figure 1C).
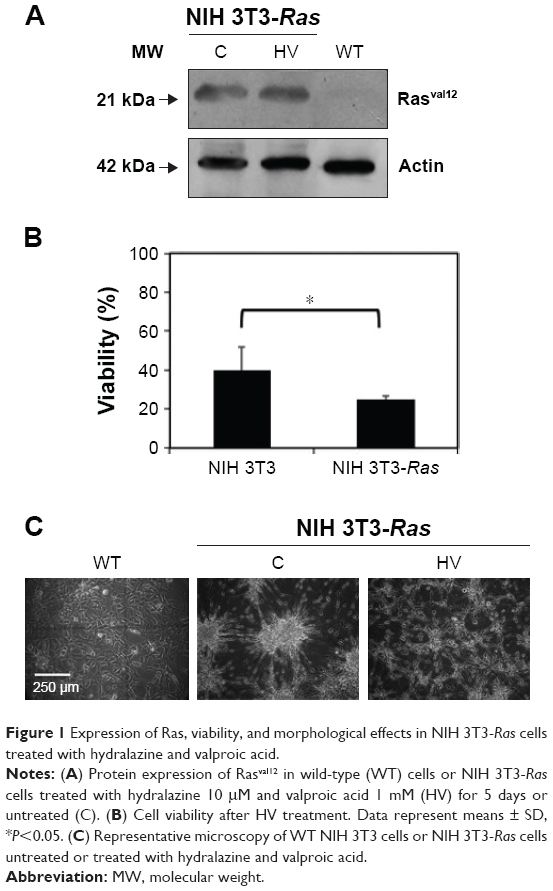 | Figure 1 Expression of Ras, viability, and morphological effects in NIH 3T3-Ras cells treated with hydralazine and valproic acid. |
Hydralazine and valproic acid decrease cell motility and chemotaxis
The effects of hydralazine and valproic acid on cell motility were evaluated using the wound-healing assay and cell chemotaxis after 5-day treatments. The results showed a 35% decrease in wound healing compared with untreated cells, though this effect was seen only at 24 hours after wounding and not earlier (P<0.05; Figure 2A and B). Chemotaxis of NIH 3T3-Ras cells evaluated in the Neuro Probe chamber also showed that treatment with hydralazine and valproic acid resulted in an almost complete lack of migration to the lower compartment of the chamber (Figure 2C and D).
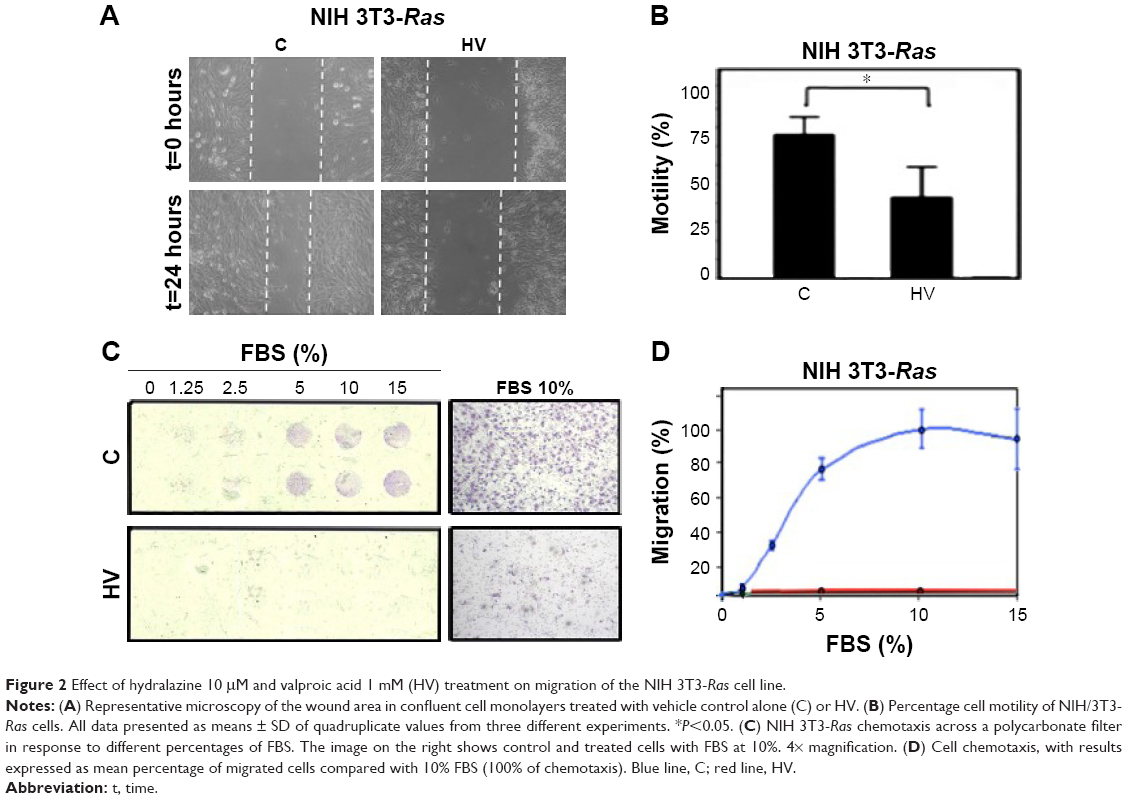 | Figure 2 Effect of hydralazine 10 μM and valproic acid 1 mM (HV) treatment on migration of the NIH 3T3-Ras cell line. |
Hydralazine and valproic acid do not change gelatinase activity
Evaluation of gelatinase activity of NIH 3T3-Ras cells treated with hydralazine and valproic acid showed a trend toward increased MMP9 and MMP2 activity, though this change was not statistically significant (P=0.603 and P=0.194, respectively; Figure 3). Inhibition with EDTA indicated the metalloproteinase nature of these bands (data not shown). In addition, we verified equal amounts of protein in each sample by staining the proteins after denaturing PAGE (data not shown).
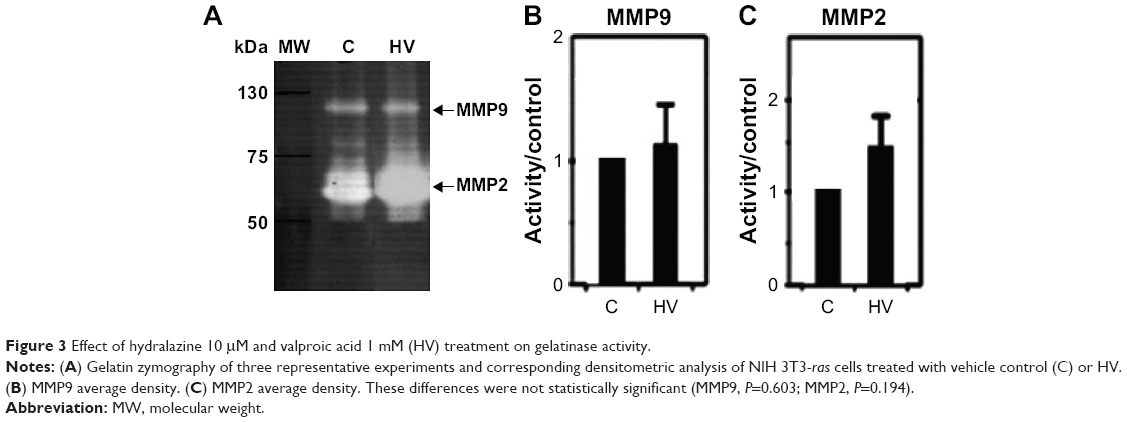 | Figure 3 Effect of hydralazine 10 μM and valproic acid 1 mM (HV) treatment on gelatinase activity. |
Hydralazine and valproic acid decrease the metastatic ability of NIH 3T3-Ras
To evaluate the overall effect of these epigenetic drugs upon metastatic ability, NIH 3T3-Ras cells were injected into tail veins of male nu/nu mice. Two days after injection, epigenetic drugs were administered intraperitoneally daily for one month. The results indicated a strong antimetastatic effect of these epigenetic drugs compared to untreated controls (Figure 4A and B; Table 1). All untreated animals developed numerous lung metastases, with a mean of 47.3 lung nodules, whereas the average for hydralazine and valproic acid treatment was 1.5 (P<0.001 compared to control). Figure 4C shows representative microscopy of histological sections. No macroscopic metastases were observed in other organs.
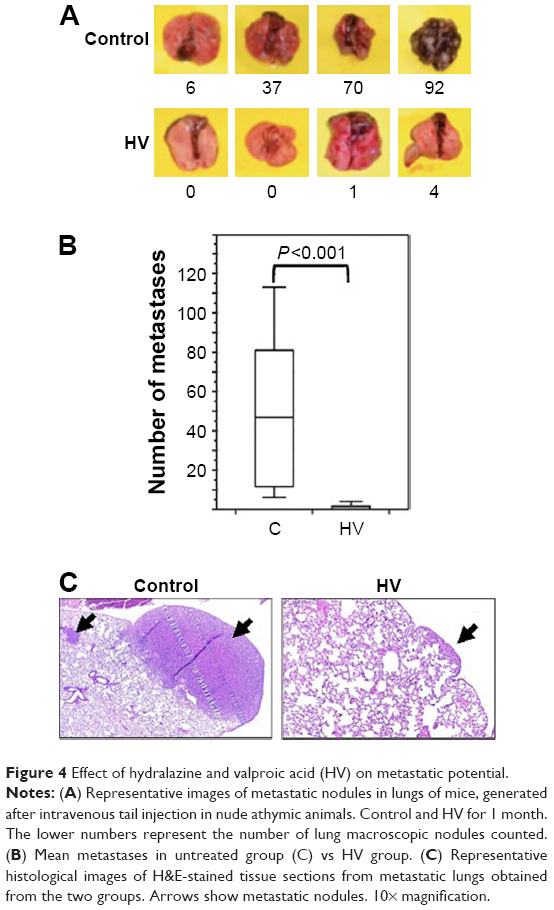 | Figure 4 Effect of hydralazine and valproic acid (HV) on metastatic potential. |
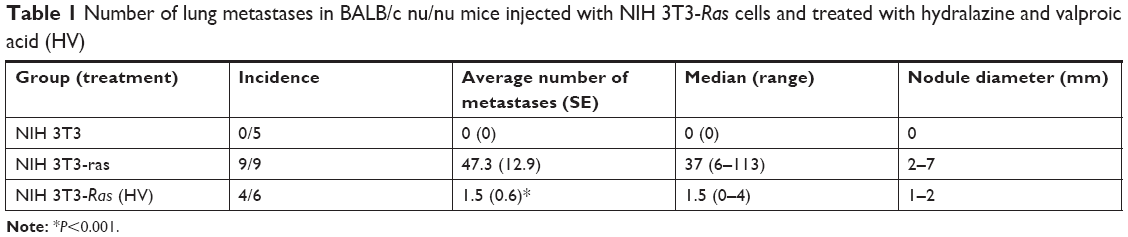 | Table 1 Number of lung metastases in BALB/c nu/nu mice injected with NIH 3T3-Ras cells and treated with hydralazine and valproic acid (HV) |
Hydralazine and valproate changes in gene expression
To analyze transcriptional changes in genes involved in metastasis contained in a mouse-tumor-metastasis RT profiler, we compared differential gene-expression profiles of 84 genes between NIH 3T3-Ras cells treated or not with valproic acid and hydralazine. These genes encode several classes of protein factors, including cell adhesion, extracellular matrix components, cell cycle, cell growth, proliferation, apoptosis, transcription factors, and regulators, and other genes that participate in metastasis. The results showed that there were 15 downregulated and 33 upregulated genes with greater than twofold change. The mean ΔCt for the downregulated genes was 28.43 vs 32.34 (95% CI −7.1998 to −0.6202 P=0.0229), whereas the corresponding mean ΔCt for the upregulated genes was 28.18 vs 26.18 (95% CI 1.32–2.66, P=0.0263; Table 2). To get a better picture of gene-expression changes, Figure 5 shows the genes that underwent greater than fivefold change in expression, either up or down. Among the downregulated genes were CCL7, HTATIP, CXCL12, CDH11, and MYCL1, whereas those upregulated were MMP3, SMAD4, CXRC2, EPHB2, and FXYD5. The well-known metastasis-suppressor gene NM23-H1 was upregulated, but only 1.72-fold (not statistically significant). Validation of protein expression by Western blot confirmed increased expression of both NM23-H1 and SMAD4 (Figure 6). Interestingly, E-cadherin, which was upregulated in the microarray by 3.41-fold, was actually slightly decreased in protein expression, though this difference was not statistically significant. The RECK gene was not included in the mouse-tumor-metastasis RT profiler; nevertheless, it was analyzed by Western blot because it reverses Ras activation in fibroblasts, and as expected there was important upregulation.
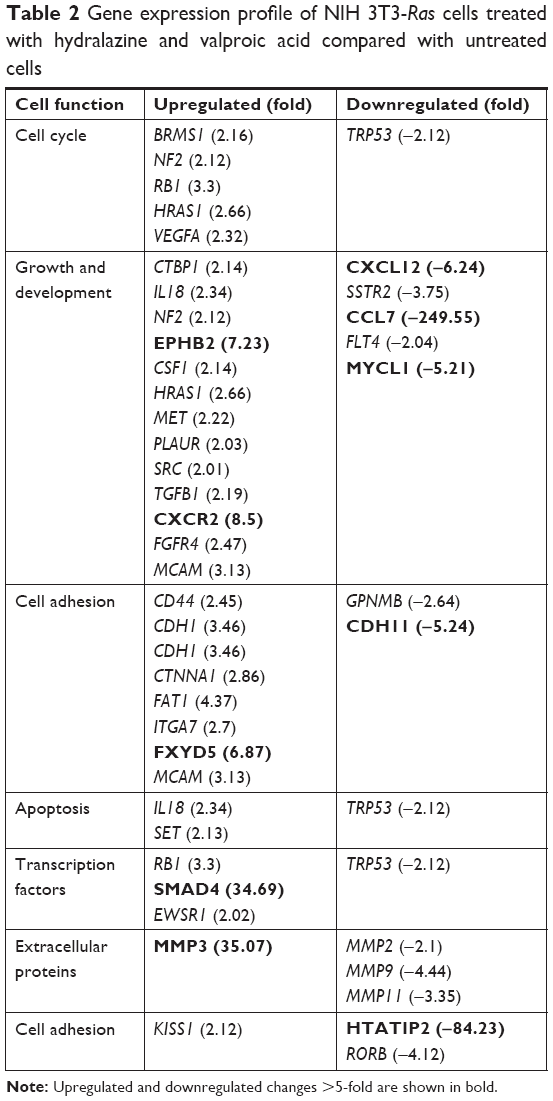 | Table 2 Gene expression profile of NIH 3T3-Ras cells treated with hydralazine and valproic acid compared with untreated cells |
 | Figure 5 Genes that changed their expression fivefold (up/downregulated) upon treatment with hydralazine and valproic acid compared with untreated cells. |
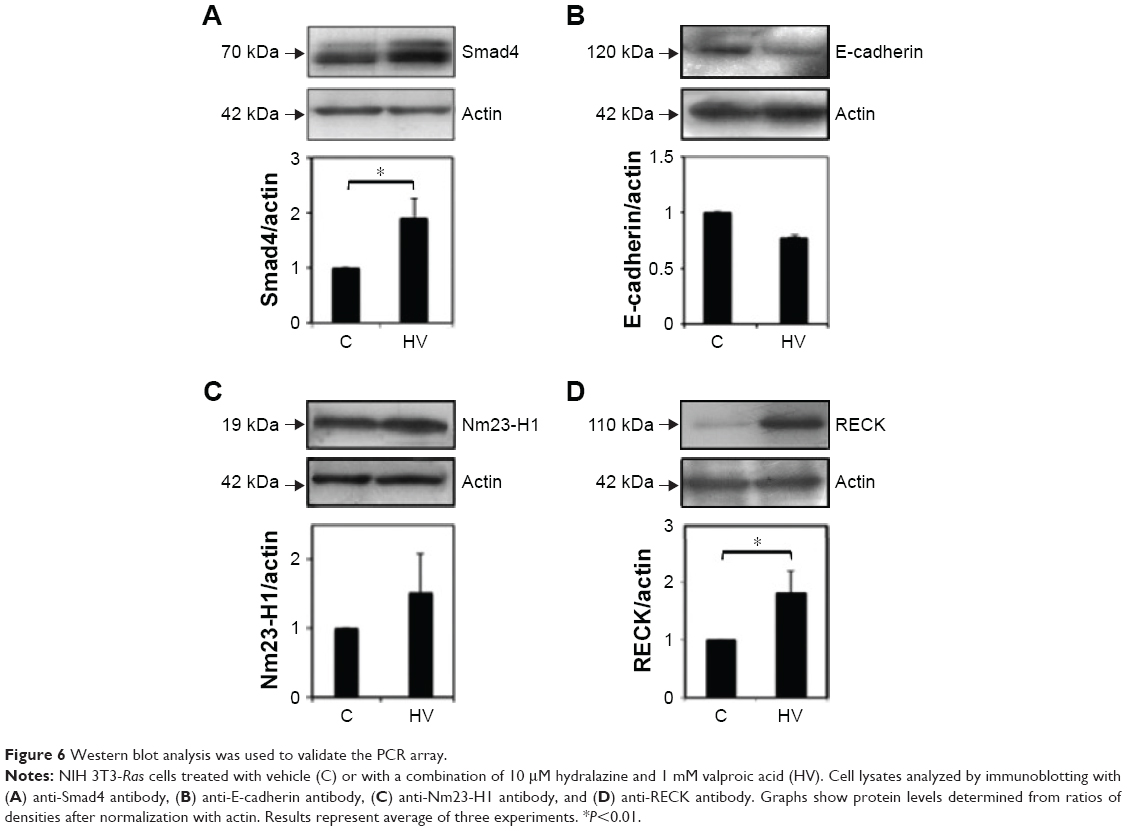 | Figure 6 Western blot analysis was used to validate the PCR array. |
Discussion
The results of this study show that the combination of hydralazine and valproic acid has distinct in vitro effects upon several phenotypic traits known to be involved in the metastatic process, such as cell proliferation, invasion, and migration and gelatinase activity. These agents, while only showing a tendency to increase metalloproteinase activity, do reduce cell motility and cell migration. Whether this reduction in cell motility and migration results from hydralazine–valproic acid-induced gene-expression changes in metastasis-related genes or a direct effect of these drugs on viability remains to be analyzed. However, the overall result is a striking effect showing reduction of lung metastases in vivo.
A significant number of metastasis-related genes are known to be dysregulated via epigenetic alterations, specifically CpG-island hypermethylation and disruption of normal patterns of histone acetylation/deacetylation.30,31 As such, DNA demethylating drugs and HDACIs, may have a role as chemo- or radiosensitization agents by inducing synergistic gene reactivation.32–37 However, there is a paucity of studies with these agents regarding their antimetastatic effects. An early study with 5-Aza-2′-deoxycytidine (5-Aza-dC) showed reexpression of NM23-H1 with a reduction in motility of breast cancer cells.9 Another study showed that 5-Aza-dC treatment of MDA-MB435 cells reduced the rate of lung metastases, which was attributed to demethylation and reexpression of SYK, a metastasis-suppressor gene.38 Additionally, some class 1, 6, and 8 HDACs have been found to be critical for invasion in breast cancer cells,39 and the HDACIs apicidin, vorinostat, entinostat, and romidepsin reduce basal and CXCL12-mediated cell migration in several human cancer cell lines by impairing CXCL12-dependent signaling cascades through STAT3 and cSRC, suggesting that they delay or prevent metastasis.40 Likewise, Zhang et al showed that valproic acid inhibits MDA-MB231 cell motility by survivin downregulation.41 The ability of several HDACIs to modulate the expression of invasive gene expression has also been demonstrated in bladder cancer cell lines.42 Studies combining agents that have targeted DNA methylation and histone deacetylation in metastasis models are limited. In breast cancer cells, 5-Aza-dC plus depsipeptide induces greater reactivation of both maspin and gelsolin compared to each agent alone.43 Furthermore, the same combination shows synergy in expression of E-cadherin and TIMP3.44
The in vivo antimetastatic effect of the combination of hydralazine and valproic acid has not been previously evaluated. A study showed that they synergize in demethylation-induced reexpression of E-cadherin in the human biliary tract carcinoma cell line QBC939, which was associated with a sharp decrease in invasiveness, and that was only slightly decreased with either drug alone.45 While hydralazine has not been tested alone, valproic acid has been shown to reduce clonogenicity and invasion in Matrigel assays to different extents in two bladder cell lines46 and to reduce growth significantly of T24 bladder cancer xenografts.47
Despite the fact that most studies demonstrate that epigenetic drugs decrease one or more of the metastatic cascade processes, it must be acknowledged that epigenetic drugs are pleiotropic agents, and might activate prometastatic genes while inhibiting antimetastatic genes. Therefore, the combination, at least in vitro, could promote the metastatic process. In this regard, 5-Aza-dC increases the expression of the metalloproteinases MMP1, -2, -3, -7, -9, and -14, increases cell motility and in vitro invasion, and upregulates the prometastatic genes UPA, VEGF, TGF, SNCG, and BAX in pancreatic and MCF7 breast cancer cells.48,49 Similarly, HDAC inhibition by trichostatin A, sodium butyrate, and scriptaid, increases Matrigel invasion through the overexpression of uPA.50 Therefore, it seems that while these opposite effects may result solely from the experimental system used, the majority of studies point to the antimetastatic effects of epigenetic drugs, suggesting that the overall effect upon global gene expression may favor an “antimetastatic profile”.
Our results on gene-expression point toward an overall antimetastatic gene profile. Among the most downregulated genes is CCL7, which in cross talk with CCR3 and promotes invasion and metastases in colon and oral cancer,51,52 while EGFR, CCR3, and CCR7 predict breast cancer metastasis.53 Downregulation of HTATIP, also known as TIP30, is required to inhibit the prometastatic effects of sorafenib by metformin in liver carcinoma.54 CXCL12 derived from tumor fibroblasts is known to promote tumor-cell invasion in breast cancer.55,56 CDH11, an adhesion molecule, is involved in pancreatic cell migration,57 and its overexpression, combined with Cdh6 and CD44, is associated with lymph-node metastases and poor prognosis in oral carcinoma.58 Finally, MYCL1 has not been reported to play a role in metastasis, but is recurrently amplified in Merkel-cell carcinoma59 and its genetic deletion in lung cancer models leads to strong tumor suppression.60
With regard to the upregulated genes observed, SMAD4 is a well-known antimetastatic gene in colorectal cancer,61,62 as is EPHB2, since its loss is associated with invasion, migration, and metastasis in colorectal cancer.63,64 Moreover, overexpression of EphA4 plus low expression of EPHB2 correlates with liver metastases in colorectal cancer.65 Nevertheless, CXRC2 is well demonstrated to have a role in tumor progression and metastasis,66 while FXYD5, also known as dysadherin, facilitates cell motility and metastatic potential in pancreatic cancer.67 Both genes were found to be upregulated, as was MMP3, which is associated with metastatic potential in a number of tumors.68 Interestingly, MMP9 and MMP2 were found to be downregulated (−4.4 and −2.1, respectively); however, their gelatinase activity was increased, although not statistically significantly. These data are intriguing, as at least one study has reported that MMP9 expression decreases the metastatic potential of chondrosarcoma.72 In summary, five downregulated genes are reported as pro-metastatic and one oncogenic, whereas only two of six upregulated genes are known metastasis suppressors. Interestingly, as oncogenic RAS inhibits RECK expression70 via promoter methylation71 and histone deacetylation,69 and RECK negatively regulates the activities of matrix metalloproteinases, we analyzed the expression of the RECK protein. Our results confirmed that hydralazine and valproic acid upregulated its expression, suggesting that this effect may also contribute to the almost complete inhibition of metastasis in vivo.
Conclusion
While our results strongly suggest that this combination of the epigenetic agents hydralazine and valproic acid could be of value for the treatment of metastases, the meaning of our results is limited by the use of the model of Hras-transfected murine cells. Therefore, additional research on human tumor models for metastasis are needed. Of note, epigenetic agents are already in clinical trials for solid tumors, and thus a detailed analysis of patient outcome regarding the course of metastatic disease could also provide information on whether or not epigenetic drugs can be clinically tested as antimetastatic agents.
Acknowledgment
This work is submitted in partial fulfillment of the requirements for the PhD degree (in Biomedical Sciences) of Zeferino Enrique Pérez-Cárdenas at the Universidad Nacional Autónoma de México.
Disclosure
The authors report no conflicts of interest in this work.
References
Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5(5):402–418. | ||
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. | ||
Yan J, Huang Q. Genomics screens for metastasis genes. Cancer Metastasis Rev. 2012;31(3–4):419–428. | ||
Lujambio A, Esteller M. How epigenetics explain human metastasis. Cell Cycle. 2009;8(3):377–382. | ||
Minn AJ, Bevilacqua E, Yun J, Rosner MR. Identification of novel metastasis suppressor signaling pathways for breast cancer. Cell Cycle. 2012;11(13):2452–2457. | ||
Wang Y, Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Exp Cell Res. 2013;319(2):160–169. | ||
Graff JR, Gabrielson E, Fujii H, Baylin SB, Herman JG. Methylation patterns of the E-cadherin 5′ CpG island are unstable and reflect the dynamic, heterogeneous loss of E-cadherin expression during metastatic progression. J Biol Chem. 2000;275(4):2727–2732. | ||
Cho CY, Wang JH, Chang HC, Chang CK, Hung WC. Epigenetic inactivation of the metastasis suppressor RECK enhances invasion of human colon cancer cells. J Cell Physiol. 2007;213(1):65–69. | ||
Prabhu VV, Siddikuzzaman, Grace VM, Guruvayoorappan C. Targeting tumor metastasis by regulating Nm23 gene expression. Asian Pac J Cancer Prev. 2012;13(8):3539–3548. | ||
Hartsough MT, Clare SE, Mair M, et al. Elevation of breast carcinoma Nm23-H1 metastasis suppressor gene expression and reduced motility by DNA methylation inhibition. Cancer Res. 2001;61(5):2320–2327. | ||
Li GF, Qian TL, Li GS, et al. Sodium valproate inhibits MDA-MB-231 breast cancer cell migration by upregulating NM23H1 expression. Genet Mol Res. 2012;11(1):77–86. | ||
Jeon HW, Lee YM. Inhibition of histone deacetylase attenuates hypoxia-induced migration and invasion of cancer cells via the restoration of RECK expression. Mol Cancer Ther. 2010;9(5):1361–1370. | ||
Dueñas-Gonzalez A, Coronel J, Cetina L, González-Fierro A, Chavez-Blanco A, Taja-Chayeb L. Hydralazine-valproate: a repositioned drug combination for the epigenetic therapy of cancer. Expert Opin Drug Metab Toxicol. 2014;10(10):1433–1444. | ||
Candelaria M, Herrera A, Labardini J, et al. Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial. Ann Hematol. 2011;90(4):379–387. | ||
Espinoza-Zamora JR, Labardini-Méndez J, Sosa-Espinoza A, et al. Efficacy of hydralazine and valproate in cutaneous T-cell lymphoma, a phase II study. Expert Opin Investig Drugs. 2017;26(4):481–487. | ||
Candelaria M, Gallardo-Rincón D, Arce C, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18(9):1529–1538. | ||
Coronel J, Cetina L, Pacheco I, et al. A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results. Med Oncol. 2011;28(Suppl 1):S540–S546. | ||
Candelaria M, Cetina L, Pérez-Cárdenas E, et al. Epigenetic therapy and cisplatin chemoradiation in FIGO stage IIIB cervical cancer. Eur J Gynaecol Oncol. 2010;31(4):386–391. | ||
Kitamura H, Torigoe T, Asanuma H, Honma I, Sato N, Tsukamoto T. Down-regulation of HLA class I antigens in prostate cancer tissues and up-regulation by histone deacetylase inhibition. J Urol. 2007;178(2):692–696. | ||
Sers C, Kuner R, Falk CS, et al. Down-regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer. 2009;125(7):1626–1639. | ||
Mora-García ML, Duenas-González A, Hernández-Montes J, et al. Up-regulation of HLA class-I antigen expression and antigen-specific CTL response in cervical cancer cells by the demethylating agent hydralazine and the histone deacetylase inhibitor valproic acid. J Transl Med. 2006;4:55. | ||
Chávez-Blanco A, De la Cruz-Hernández E, Domínguez GI, et al. Upregulation of NKG2D ligands and enhanced natural killer cell cytotoxicity by hydralazine and valproate. Int J Oncol. 2011;39(6):1491–1499. | ||
Yamanegi K, Yamane J, Kobayashi K, et al. Valproic acid cooperates with hydralazine to augment the susceptibility of human osteosarcoma cells to Fas- and NK cell-mediated cell death. Int J Oncol. 2012;41(1):83–91. | ||
Arce C, Pérez-Plasencia C, González-Fierro A, et al. A proof-of-principle study of epigenetic therapy added to neoadjuvant doxorubicin cyclophosphamide for locally advanced breast cancer. PLoS One. 2006;1:e98. | ||
Spella M, Marazioti A, Arendt KAM, Stathopoulos GT. RAS oncogenes direct metastasis. Mol Cell Oncol. 2017;4(5):e1345711. | ||
Bradley MO, Kraynak AR, Storer RD, Gibbs JB. Experimental metastasis in nude mice of NIH 3T3 cells containing various ras genes. Proc Natl Acad Sci U S A. 1986;83(14):5277–5281. | ||
Perez-Cardenas E. Participación de los genes ras y nm23 en la capacidad metastásica de células transformadas. Ras and NM23 genes participation in the metastatic abiity of transformed cells [master’s thesis]. Mexico City: National Autonomous University of Mexico; 2001. | ||
Valster A, Tran NL, Nakada M, Berens ME, Chan AY, Symons M. Cell migration and invasion assays. Methods. 2005;37(2):208–215. | ||
Argüello-Ramírez J, Pérez-Cárdenas E, Delgado-Chávez R, Solorza-Luna G, Villa-Treviño S, Arenas-Huertero F. Matrix metalloproteinases-2, -3, and -9 secreted by explants of benign and malignant lesions of uterine cérvix. Int J Gynecol Cancer. 2004;14(2):333–340. | ||
Cock-Rada A, Weitzman JB. The methylation landscape of tumour metastasis. Biol Cell. 2013;105(2):73–90. | ||
Sahin M, Sahin E, Gümüşlü S, Erdoğan A, Gültekin M. DNA methylation or histone modification status in metastasis and angiogenesis-related genes: a new hypothesis on usage of DNMT inhibitors and S-adenosylmethionine for genome stability. Cancer Metastasis Rev. 2010;29(4):655–676. | ||
Schneider-Stock R, Ocker M. Epigenetic therapy in cancer: molecular background and clinical development of histone deacetylase and DNA methyltransferase inhibitors. IDrugs. 2007;10(8):557–561. | ||
Hellebrekers DM, Griffioen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta. 2007;1775(1):76–91. | ||
Zhu WG, Otterson GA. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents. 2003;3(3):187–199. | ||
Zhu WG, Lakshmanan RR, Beal MD, Otterson GA. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2001;61(4):1327–1333. | ||
Ferguson LR, Tatham AL, Lin Z, Denny WA. Epigenetic regulation of gene expression as an anticancer drug target. Curr Cancer Drug Targets. 2011;11(2):199–212. | ||
Sigalotti L, Fratta E, Coral S, et al. Epigenetic drugs as pleiotropic agents in cancer treatment: biomolecular aspects and clinical applications. J Cell Physiol. 2007;212(2):330–344. | ||
Xia TS, Shi JP, Ding Q, et al. Reactivation of Syk gene by AZA suppresses metastasis but not proliferation of breast cancer cells. Med Oncol. 2012;29(2):448–453. | ||
Park SY, Jun JA, Jeong KJ, et al. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep. 2011;25(6):1677–1681. | ||
Ierano C, Basseville A, To KK, et al. Histone deacetylase inhibitors induce CXCR4 mRNA but antagonize CXCR4 migration. Cancer Biol Ther. 2013;14(2):175–183. | ||
Zhang L, Wang G, Wang L, et al. VPA inhibits breast cancer cell migration by specifically targeting HDAC2 and down-regulating Survivin. Mol Cell Biochem. 2012;361(1–2):39–45. | ||
Gould JJ, Kenney PA, Rieger-Christ KM, et al. Identification of tumor and invasion suppressor gene modulators in bladder cancer by different classes of histone deacetylase inhibitors using reverse phase protein arrays. J Urol. 2010;183(6):2395–2402. | ||
Primeau M, Gagnon J, Momparler RL. Synergistic antineoplastic action of DNA methylation inhibitor 5-AZA-2′-deoxycytidine and histone deacetylase inhibitor depsipeptide on human breast carcinoma cells. Int J Cancer. 2003;103(2):177–184. | ||
Gagnon J, Shaker S, Primeau M, Hurtubise A, Momparler RL. Interaction of 5-aza-2′-deoxycytidine and depsipeptide on antineoplastic activity and activation of 14-3-3sigma, E-cadherin and tissue inhibitor of metalloproteinase 3 expression in human breast carcinoma cells. Anticancer Drugs. 2003;14(3):193–202. | ||
Li H, Chen S, Shu Y, et al. Effects of hydralazine and valproate on the expression of E-cadherin gene and the invasiveness of QBC939 cells. Front Med China. 2009;3(2):153–157. | ||
Byun SS, Kim FJ, Khandrika L, et al. Differential effects of valproic acid on growth, proliferation and metastasis in HTB5 and HTB9 bladder cancer cell lines. Cancer Lett. 2009;281(2):196–202. | ||
Chen CL, Sung J, Cohen M, et al. Valproic acid inhibits invasiveness in bladder cancer but not in prostate cancer cells. J Pharmacol Exp Ther. 2006;319(2):533–542. | ||
Ateeq B, Unterberger A, Szyf M, Rabbani SA. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia. 2008;10(3):266–278. | ||
Sato N, Maehara N, Su GH, Goggins M. Effects of 5-aza-2′-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J Natl Cancer Inst. 2003;95(4):327–330. | ||
Pulukuri SM, Gorantla B, Rao JS. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J Biol Chem. 2007;282(49):35594–35603. | ||
Lee YS, Kim SY, Song SJ, et al. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget. 2016;7(24):36842–36853. | ||
Jung DW, Che ZM, Kim J, et al. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int J Cancer. 2010;127(2):332–344. | ||
Liu Y, Ji R, Li J, et al. Correlation effect of EGFR and CXCR4 and CCR7 chemokine receptors in predicting breast cancer metastasis and prognosis. J Exp Clin Cancer Res. 2010;29:1–16. | ||
Guo Z, Cao M, You A, et al. Metformin inhibits the prometastatic effect of sorafenib in hepatocellular carcinoma by upregulating the expression of TIP30. Cancer Sci. 2016;107(4):507–513. | ||
Ahirwar DK, Nasser MW, Ouseph MM, et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene. 2018;37(32):4428–4442. | ||
Meng W, Xue S, Chen Y. The role of CXCL12 in tumor microenvironment. Gene. 2018;641:105–110. | ||
Birtolo C, Pham H, Morvaridi S, et al. Cadherin-11 is a cell surface marker up-regulated in activated pancreatic stellate cells and is involved in pancreatic cancer cell migration. Am J Pathol. 2017;187(1):146–155. | ||
Ma C, Zhao JZ, Lin RT, et al. Combined overexpression of cadherin 6, cadherin 11 and cluster of differentiation 44 is associated with lymph node metastasis and poor prognosis in oral squamous cell carcinoma. Oncol Lett. 2018;15(6):9498–9506. | ||
Paulson KG, Lemos BD, Feng B, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol. 2009;129(6):1547–1555. | ||
Kim DW, Wu N, Kim YC, et al. Genetic requirement for Mycl and efficacy of RNA Pol I inhibition in mouse models of small cell lung cancer. Genes Dev. 2016;30(11):1289–1299. | ||
Voorneveld PW, Kodach LL, Jacobs RJ, et al. Loss of SMAD4 alters BMP signaling to promote colorectal cancer cell metastasis via activation of Rho and ROCK. Gastroenterology. 2014;147(1):196–208. | ||
Zhang B, Halder SK, Kashikar ND, et al. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138(3):969–980.e1–e3. | ||
Guo DL, Zhang J, Yuen ST, et al. Reduced expression of EphB2 that parallels invasion and metastasis in colorectal tumours. Carcinogenesis. 2006;27(3):454–464. | ||
Senior PV, Zhang BX, Chan ST. Loss of cell-surface receptor EphB2 is important for the growth, migration, and invasiveness of a colon cancer cell line. Int J Colorectal Dis. 2010;25(6):687–694. | ||
Oshima T, Akaike M, Yoshihara K, et al. Overexpression of EphA4 gene and reduced expression of EphB2 gene correlates with liver metastasis in colorectal cancer. Int J Oncol. 2008;33(3):573–577. | ||
Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics. 2017;7(6):1543–1588. | ||
Shimamura T, Yasuda J, Ino Y, et al. Dysadherin expression facilitates cell motility and metastatic potential of human pancreatic cancer cells. Cancer Res. 2004;64(19):6989–6995. | ||
Overall CM, Dean RA. Degradomics: systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006;25(1):69–75. | ||
Malcherczyk D, Heyse TJ, El-Zayat BF, et al. Expression of MMP-9 decreases metastatic potential of chondrosarcoma: an immunohistochemical study. BMC Musculoskelet Disord. 2018;19(1):9. | ||
Yeh HH, Tseng YF, Hsu YC, et al. Ras induces experimental lung metastasis through up-regulation of RbAp46 to suppress RECK promoter activity. BMC Cancer. 2015;15:172. | ||
Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer Res. 2006;66(17):8413–8420. | ||
Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras-induced down-regulation of the metastasis suppressor RECK. Cell Signal. 2004;16(6):675–679. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.