Back to Journals » Drug Design, Development and Therapy » Volume 8
Anticancer efficacy and absorption, distribution, metabolism, and toxicity studies of Aspergiolide A in early drug development
Authors Wang Y, Qi X, Li D, Zhu T, Mo X, Li J
Received 27 March 2014
Accepted for publication 21 May 2014
Published 17 October 2014 Volume 2014:8 Pages 1965—1977
DOI https://doi.org/10.2147/DDDT.S64989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Yuanyuan Wang, Xin Qi, Dehai Li, Tianjiao Zhu, Xiaomei Mo, Jing Li
Key Laboratory of Marine Drugs, Ministry of Education, School of Medicine and Pharmacy, Ocean University of China, Qingdao, People's Republic of China
Abstract: Since the first anthracycline was discovered, many other related compounds have been studied in order to overcome its defects and improve efficacy. In the present paper, we investigated the anticancer effects of a new anthracycline, aspergiolide A (ASP-A), from a marine-derived fungus in vitro and in vivo, and we evaluated the absorption, distribution, metabolism, and toxicity drug properties in early drug development. We found that ASP-A had activity against topoisomerase II that was comparable to adriamycin. ASP-A decreased the growth of various human cancer cells in vitro and induced apoptosis in BEL-7402 cells via a caspase-dependent pathway. The anticancer efficacy of ASP-A on the growth of hepatocellular carcinoma xenografts was further assessed in vivo. Results showed that, compared with the vehicle group, ASP-A exhibited significant anticancer activity with less loss of body weight. A pharmacokinetics and tissue distribution study revealed that ASP-A was rapidly cleared in a first order reaction kinetics manner, and was enriched in cancer tissue. The maximal tolerable dose (MTD) of ASP-A was more than 400 mg/kg, and ASP-A was not considered to be potentially genotoxic or cardiotoxic, as no significant increase of micronucleus rates or inhibition of the hERG channel was seen. Finally, an uptake and transport assay of ASP-A was performed in monolayers of Caco-2 cells, and ASP-A was shown to be absorbed through the active transport pathway. Altogether, these results indicate that ASP-A has anticancer activity targeting topoisomerase II, with a similar structure and mechanism to adriamycin, but with much lower toxicity. Nonetheless, further molecular structure optimization is necessary.
Keywords: aspergiolide A, anticancer, anthracyclines, ADMT, drug activity
Introduction
Because of particular living conditions, including salinity, nutrition, higher pressure, temperature variations, and competition with bacteria, viruses and other fungi, marine fungi may have developed specific secondary metabolic pathways compared with terrestrial fungi.1 In particular, the marine fungi associated with marine algae, sponge, invertebrates, and sediments appear to be a rich source for secondary metabolites, which have proven to be a promising source for the production of novel anticancer, antibiotic, and anti-inflammatory agents.2
In our continuous search for anticancer compounds from marine-derived microorganisms, a fungus, authenticated as Aspergillus glaucus, was obtained from the marine sediment around the mangrove roots collected in Fujian province of the People’s Republic of China. The isolation work of the active compounds on this fungus led to the finding of a novel anthraquinone derivative with a new skeleton, naphtho[1,2,3-de]chromene-2,7-dione skeleton, identified as aspergiolide A (ASP-A, Figure 1). The compound exhibited distinct cytotoxicities against cancer cell lines A-549, HL-60, BEL-7402, and P388.3 Anthracyclines rank among the most effective anticancer drugs ever developed,4 yet application of anthracyclines is usually limited by cumulative dose-related cardiotoxic effects; other side effects include nausea, alopecia or loss of hair, tissue necrosis, diarrhea, abdominal pain, thrombophlebitis, blood clots, fever, and chills. Since the discovery of the first anthracycline, daunorubicin, which was produced naturally by Streptomyces peucetius, many other related compounds have followed. The quest for high-efficiency and low-toxicity anthracycline analogues is ongoing.
 | Figure 1 The structure of ASP-A. |
A drug, besides active and special of its action, must be taken appropriately into the body (absorption), distributed to the right parts of the body, metabolized in a way that does not instantly remove its activity, and eliminated in a suitable manner.5 Recently, absorption, distribution, metabolism, elimination, and toxicity (ADMET) technologies are radically reshaping the pharmaceutical development process, and improvements in in vitro molecular and cellular assays enable compounds’ ADMET characteristics to be approximated well before the preclinical phase.6
Considering that a large amount of ASP-A has been prepared via optimization of medium composition, the purpose of this paper was to systematically assess efficacy, absorption, distribution, metabolism, and toxicity in the early drug development of ASP-A in order to find the beneficial and harmful effects of ASP-A, and to lay the foundation of further optimizing the drug candidates to introduce new formulations for development of novel anticancer anthracycline analogues.
Materials and methods
Cell culture and materials
HeLa, SGC-7901, MCF-7, MDA-MB-468, 786-O, SMMC-7721, U251, A431, SK-OV-3, BxPC3 cancer cell lines were obtained from Shanghai Cell Bank, Chinese Academy of Science. SGC-7901, MCF-7, MDA-MB-468, 786-O, SMMC-7721 cells were grown in Roswell Park Memorial Institute (RPMI)-1640 medium. A431, SK-OV-3 and Caco-2 cell lines were grown in Dulbecco’s Modified Eagle’s Medium. HeLa cells were maintained in Minimum Essential Medium. All culture media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were maintained at 37°C with 5% CO2 in a humidified incubator. ASP-A was isolated with a purity of 99.8%. Poly(ADP-ribose) polymerase (PARP), caspase-3, caspase-9, caspase-8, γ-H2AX, Bax, and Bcl-2 antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA). β-actin antibody was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA).
Cell proliferation assay
Cancer cells were plated into a 96-well microtiter plate containing 0.1 mL of growth media/well for 24 hours. Cells were incubated with various concentrations of ASP-A (0.625–20 μmol/mL) dissolved in dimethyl sulfoxide for 72 hours and evaluated for their cytotoxicities using the sulforhodamine B (SRB) test. The absorption value was detected by an enzyme-labeled instrument (Tecan Infinite F50, Tecan Group AG, Männedorf, Switzerland) at 540 nm. The 50% reduction of the cell viability compared to vehicle-treated cells was defined as the half-maximal inhibitory concentration (IC50) value.
Assay for DNA topoisomerase II inhibition in vitro
A modified and improved spectrofluorometric decatenation assay was used to determine the inhibition of topoisomerase IIα. Kinetoplast DNA (kDNA) consists of highly catenated networks of circular DNA. Topoisomerase IIα decatenates kDNA in an adenosine triphosphate (ATP)-dependent reaction to yield individual minicircles of DNA. Reactions contained 30 mM Tris-HCl, pH 7.6, 3 mM ATP, 15 mM 2-mercaptoethanol, 8 mM MgCl2, and 60 mM NaCl in a final volume of 20 μL. Reactions were incubated with 4 units of topoisomerase II in the presence or absence of the indicated ASP-A or adriamycin (ADM) for 30 minutes at 37°C. The reactions were terminated with 2 μL of 10% sodium dodecyl sulfate, followed by proteinase K treatment for 15 minutes at 37°C. Samples were resolved by electrophoresis on 1% agarose gel, stained with 0.5 mg/mL ethidium bromide, and visualized with ultraviolet light.
Western blot analysis
Following ASP-A treatment with different concentrations (2.5 μM, 5 μM, and 10 μM) for 12 hours, BEL-7402 cells were disrupted in lysate buffer on ice for 10 minutes. Lysates were clarified by centrifugation and 20 μL aliquots were separated by sodium dodecyl sulfate-polyacrylamide gelelectrophoresis on 10% gels before transfer to nitrocellulose membranes. Membranes were incubated with 5% skim milk in tris-buffered saline with 0.5% Tween for blocking nonspecific antigenic sites and immunoblotted with the indicated antibodies. The antibody–antigen complex was visualized and quantitated by chemiluminescence detection.
Cancer xenograft models
The H22 mouse hepatoma cells (2×105 cells/mouse) were inoculated subcutaneously into the right armpit of Kun Ming (KM) mice (eight animals/group). ASP-A (5 mg/kg, 15 mg/kg, 45 mg/kg) and ADM (2 mg/kg) dissolved in a mixed solution of HS-15 and N,N-Dimethylformamide (1:1, volume/volume) were injected directly into the intraperitoneal cavity. The mice were killed by dislocation of cervical vertebra after treatment. The transplanted tumor weights were measured and the inhibitory rates of cancer growth were calculated according to the following equation: rate of inhibition (%) = (mean cancer weight of control group – mean cancer weight of treated group)/mean cancer weight of control group × 100.
The BEL-7402 human hepatoma cells (5×106 cells/mice) were inoculated subcutaneously into the right armpit of nude mice (six animals/group), with a vernier caliper to measure the diameter of the transplanted cancers, until the cancers grew to about 100 mm3; then the mice were grouped randomly. ASP-A (7 mg/kg, 14 mg/kg, 28 mg/kg) and ADM (2 mg/kg) were injected directly into the intraperitoneal cavity. Cancer growth in nude mice was determined by measuring the length and width of the cancer every other day with a caliper, and cancer volume (V) was calculated on the basis of the following formula: V = (length × width2)/2. The individual relative tumor volume (RTV) was calculated as follows: RTV = Vt/V0, where Vt is the volume at each time, and V0 is the volume at the beginning of the treatment. The therapeutic effect of ASP-A was determined by the volume ratio of treatment to control (T/C). T/C (%) = (mean RTV of the treated group/mean RTV of the control group) × 100%.
The pharmacokinetics and tissue distribution study
For pharmacokinetics study
One hundred KM male mice were randomly assigned to the following two single dosages (15 mg/kg, 30 mg/kg) of ASP-A, with 50 mice for each dosage; these were further divided into ten subgroups with five mice for 5, 10, 30, 45, 60, 75, 90, 120, 180, and 240 minutes after ASP-A administration. At each time interval, whole blood samples were drawn from the retroorbital plexus of each mouse and placed into heparinized microfuge tubes and centrifuged at approximately 3,000 × g for 5 minutes. The resulting plasma layers were separated and stored in microcentrifuge tubes at −20°C until analysis was performed with the procedure described below. The use of animals was approved by the Institutional Animal Care and Use Committee of the Qingdao Experimental Animal and Animal Experiment Center, with confirmed adherence to the ethical guidelines for the care and use of animals.
For tissue distribution study
Fifteen male KM mice bearing H22 tumor were randomly assigned to three groups, each group consisting of five mice. After ASP-A was administered intraperitoneally (ip) at a dosage of 15 mg/kg, at 10 minutes, 30 minutes, and 60 minutes, respectively, the mice were immediately sacrificed, the hearts were perfused with saline, and the organs (heart, liver, spleen, lung, kidney, brain, stomach and tumor) were removed. Tissue samples were weighed rapidly and homogenized with physiologic saline in ice water, and then immediately centrifuged at 3,000 × g for 5 minutes. Afterwards, the tissue homogenate samples were drawn in tubes and frozen at −20°C until analysis was performed using the procedure described below.
Analysis of ASP-A in plasma and tissues
Concentrations of ASP-A in the plasma and tissue were determined by validated reverse-phase high-performance liquid chromatography (HPLC). The HPLC system consisted of a Waters 1525 4.6 mm × 250 mm Venmsil rotating packed bed column (Waters Corporation, Milford, MA, USA). The mobile phase was composed of acetonitrile–water (50:50, volume/volume) at a flow rate of 1 mL/minute, and the detection wavelength was 304 nm, at which ASP-A has the maximum absorption value. A volume of 20 μL supernatant of each sample was injected into the HPLC system for analysis of ASP-A. The ASP-A contents of the above samples were expressed as counts per gram of tissue or per milliliter of plasma. Concentrations of ASP-A were calculated from calibration curves. Pharmacokinetic parameters were analyzed by using 3P97 software (Chinese Pharmacological Society, Beijing, China).
Safety evaluation of ASP-A
Acute toxicity study
The acute toxicity study of ASP-A was carried out to investigate the toxic response and the target organ.7 Healthy male and female KM mice (6–8 weeks old) used for the acute toxicity study were bred and reared at the Animal House of School of Marine Medicine and Pharmacy, Ocean University of China. The animals were acclimatized to laboratory conditions for 1 week prior to treatment. The temperature in the animal room was maintained at 25°C±2°C with a relative humidity of 30%–70% and an illumination cycle set to 12 hours of light and 12 hours of dark. A starting dose of 40 mg/kg and two-fold increase doses of ASP-A were administered to mice by ip injection. After a single administration, mice were monitored for signs of possible toxicity every hour for the first 6 hours and every day for 14 days. Surviving animals were weighed daily and observed for any signs or symptoms of toxicity and mortality for up to 14 days.
Micronucleus test
ASP-A was administered ip at a dose of 100 mg/kg or 400 mg/kg for 30 hours, and all mice were sacrificed 6 hours after the second ip injection. The bone marrow fluid was then extruded. A drop of fetal calf serum was added on glass slides, and pushed after mixing with subsequently added equivalent amount of bone marrow. The slides were air-dried for 30 minutes, fixed in absolute ethanol for 15 minutes, stained in 10% Giemsa dye in phosphate buffer (pH 6.8) for 10 minutes, washed with distilled water and air-dried at room temperature, and then observed under an optical microscope. The frequencies of micronucleated polychromatic erythrocytes in 1,000 polychromatic erythrocytes per animal, and the ratio of polychromatic erythrocytes/normochromatic erythrocytes in 1,000 erythrocytes were determined under a BX51 light microscope (Olympus Corporation, Tokyo, Japan) with oil immersion, using 100× magnification. A negative control was also performed for each treatment.
hERG channels assay
Using the whole-cell patch clamp technique, we investigated the effects of ASP-A on hERG channels in HEK-293 cells. The HEK293 cells stably expressing hERG were maintained in Dulbecco’s Modified Eagle’s Medium containing 500 mg/mL G418 and 10% fetal bovine serum. Extracellular (bath) solution contained (mM) NaCl 137, KCl 4.0, MgCl2 1.0, CaCl2 1.8, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) 10.0, glucose 10, pH 7.4. The intracellular pipette solution contained (mM) KCl 130, MgCl2 1.0, HEPES 10, ethylene glycol-bis-(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid 5.0, Mg2-ATP, 5.0. pH 7.4. Cells were harvested from the flask and stored in extracellular solution containing 5% bovine serum albumin at 4°C. The holding potential was −80 mV, the cells were depolarized to 50 mV for 4 seconds to maximally activate hERG channels, followed by a test pulse to −50 mV for 5 seconds to measure the Ikr, the rapidly activating components of the delayed rectifier K+ current, encoded by the human HERG gene. The detectable concentrations of ASP-A were 1.25 μM, 2.5 μM, 5 μM, and 10 μM. Terfenadine acted as positive control (3 nM, 10 nM, 30 nM, 100 nM). Voltage protocols and data analyses were done using the pCLAMP 10.1 software (Molecular Devices, LLC, Sunnyvale, CA, USA). The inhibitory rates of the test compounds on the hERG current were calculated according to the following equation: (1 − current residual amplitude/control current amplitude) × 100.
Uptake and transport assay in Caco-2 cells
Hank’s Balanced Salt Solution (HBSS) containing 5 mM D-glucose and 5 mM 2-[N-morpholino]ethanesulfonic acid (for pH 6.0) was used as the uptake medium. HBSS containing 5 mM D-glucose and 5 mM HEPES (for pH 7.4) was used as the rinse medium. For cellular uptake studies, confluent monolayers of Caco-2 cells were grown in six-well plastic plates for 15 days postseeding to obtain an apical–basal polarity in human Caco-2 cells. Uptake was initiated by adding 1 mL ASP-A (25 μM, 50 μM, 100 μM, 200 μM, and 300 μM), after which the ASP-A solution was aspirated at the appropriate time to terminate the uptake and then the cells were rinsed twice with ice-cold rinse medium (pH 7.4). To determine the uptake amount of ASP-A, 1 mL of the mobile phase (see “Analysis of ASP-A in plasma and tissue”) was added. The cells were then detached and collected with a cell scraper, frozen with liquid nitrogen and thawed five times. The mixture was vortexed vigorously, centrifuged at 12,000× rpm for 15 minutes, and a 20 μL aliquot of the supernatant was injected into the HPLC. For transport studies, Caco-2 cells were seeded in polyester membrane chamber inserts (Transwell; Costar, Corning Corporation, Corning, NY, USA). Transport of ASP-A across the Caco-2 cell monolayers was studied 18–24 days postseeding. Transport was initiated by adding 1 mL of transport medium (HBSS pH 7.4) to the basolateral side of Caco cells and 0.5 mL ASP-A (50 μM, 100 μM, 200 μM, 400 μM) in transport medium to the apical side of Caco cells. At predetermined time points (10 minutes, 30 minutes, 60 minutes, 90 minutes, 120 minutes, 180 minutes), samples (0.5 mL) were taken from the basolateral side or apical side, and replaced by the same amount of transport medium. At the end of the transport experiment, all samples were analyzed by HPLC. Sample concentrations were calculated using the equation that described the best fit through the standard curve data. The apparent permeability coefficient (Papp cm/second) of ASP-A was calculated.

where dM/dt is the rate of appearance of substrate in the receiver phase (mmol/second), A is the area of the permeable support (cm2), and [So] is the initial concentration of substrate in the donor compartment (mmol/mL).
Statistical analysis
The statistical differences between two groups were evaluated by Student’s independent sample t-test. For all tests, statistical significance was defined as P<0.05. All descriptive parameters were expressed as mean ± standard deviation.
Results
ASP-A decreases the growth of human cancer cells in vitro
To investigate the cytotoxic effects of ASP-A against human cancer cells in vitro, an SRB assay was used after culture of cancer cells in the presence of ASP-A for 72 hours. The experimental results showed that ASP-A significantly inhibited the growth of 11 kinds of human cancer cell lines, with IC50 values in the range of 2.37–7.07 μM (Figure 2).
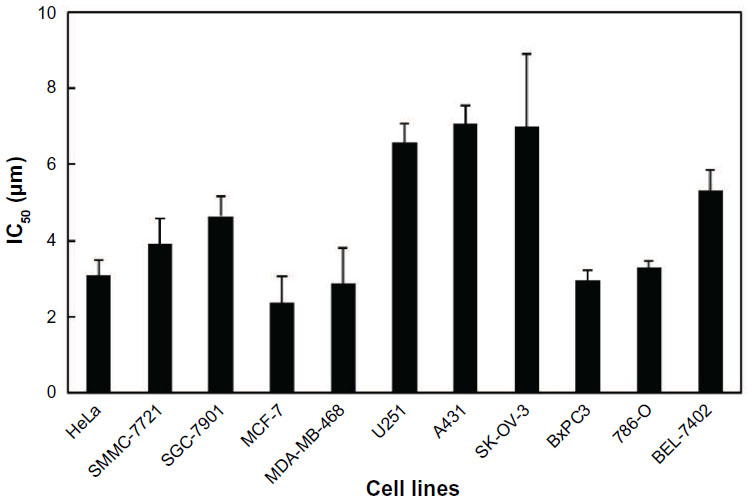 | Figure 2 Cytotoxicities of ASP-A on a panel of 11 human cancer cell lines. |
ASP-A induces apoptosis in BEL-7402 cells and inhibits DNA topoisomerase II activity
In order to determine whether the decrease in cancer cell growth evoked by ASP-A resulted from induction of apoptosis, we further investigated the possible involvement of caspases of the apoptosis pathway by detecting the active cleaved fragments of caspase-3, caspase-8, caspase-9, and PARP in hepatoma cells BEL-7402. According to the results of Western blot analysis (Figure 3A), procaspase-3, procaspase-8, procaspase-9, and PARP were cleaved after treatment with ASP-A for 12 hours, indicating that ASP-A induced caspase-dependent apoptosis of BEL-7402 cell line. Because the caspases, especially caspase-3, are known to act downstream of Bax/Bcl-2 control, we next examined the ratios of Bax (an apoptosis promoter) to Bcl-2 (an apoptosis inhibitor), as a rheostat to determine cell susceptibility to apoptosis. As shown in Figure 3B, decrease in Bcl-2 expression was accompanied by increased Bax expression after ASP-A treatment; thus, the ratios of Bax/Bcl-2 were upregulated compared with the control group (Figure 3C). Nuclear phosphorylation of the histone variant H2AX was recently shown to be a crucial component of apoptosis, in addition to having a well-studied role in DNA damage repair.8 We therefore also investigated whether γ-H2AX was activated by ASP-A. As shown in Figure 3D, γ-H2AX was induced after treatment with ASP-A.
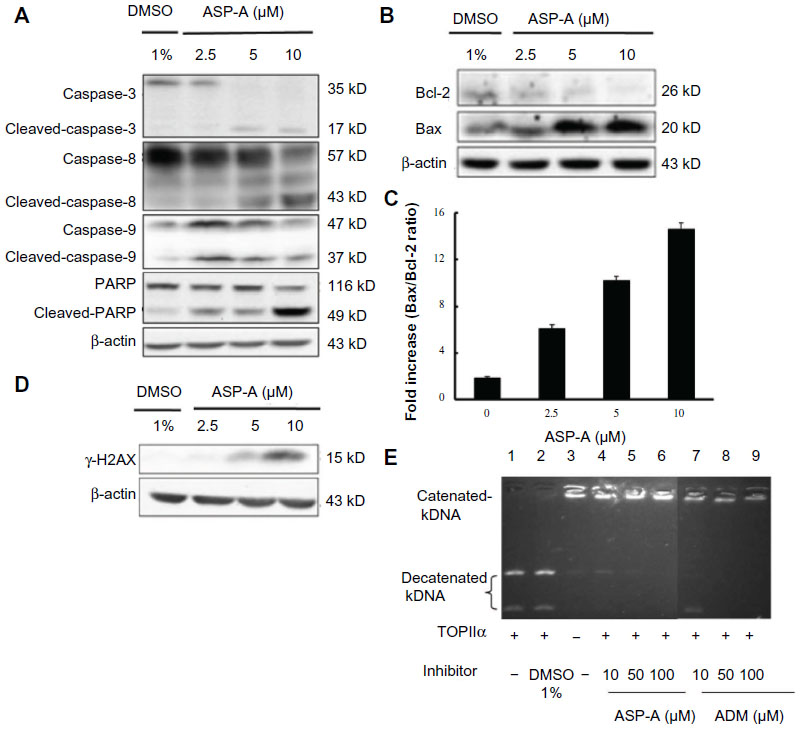 | Figure 3 ASP-A induces apoptosis in BEL-7402 cells and inhibits DNA topoisomerase II activity. |
Structure-activity relationship studies emphasize the critical role of topoisomerase II-mediated DNA cleavage on the apoptosis activity of anthracycline anticancer antibiotics.9 Therefore, a modified and improved spectrofluorometric decatenation assay was used to determine the inhibition of topoisomerase IIα in the presence of ASP-A. The result showed that ASP-A possessed activity against topoisomerase II that was comparable to that of ADM (Figure 3E).
The anticancer efficacy of ASP-A on the growth of hepatocellular carcinoma xenografts
To elucidate the anticancer efficacy of ASP-A in vivo, the cancer growth in H22 hepatoma-bearing mice following multiple doses of ASP-A treatment was investigated. In this experiment, ASP-A was administrated ip once a day after cancer tumor size reached 100 mm3 in volume. The mice were weighed twice per week and the cancer tumor weights were measured after the mice bearing H22 hepatoma were killed by dislocation of cervical vertebra. As shown in Figure 4A–C, ASP-A significantly inhibited the growth of H22 subcutaneous cancer (P<0.05) in a dose-dependent manner, with 55%, 62%, and 66% reduction at the doses of 5 mg/kg, 15 mg/kg, and 45 mg/kg respectively. ASP-A had little influence on the body weights of mice (P>0.05); however, it was noted that the inhibitory rate of the ADM (2 mg/kg) group was 51.9%, with much more loss of body weight.
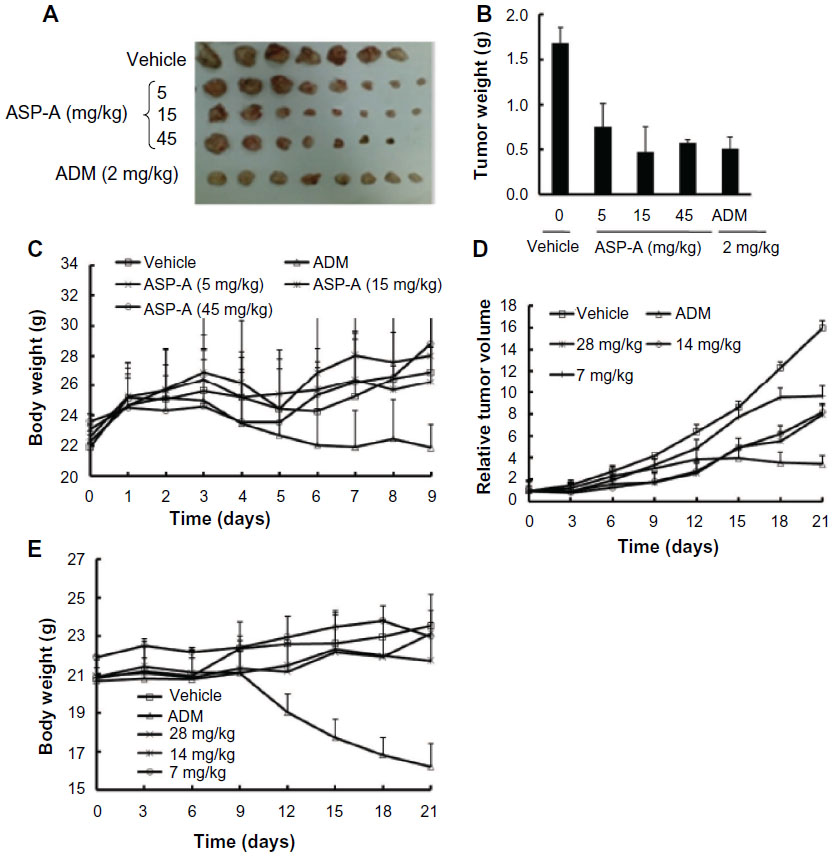 | Figure 4 The anticancer efficacy of ASP-A in vivo following ASP-A multiple doses of treatments. |
We further assessed the anticancer efficacy of ASP-A on the proliferation of human hepatocellular carcinoma xenografts. Compared with the vehicle group, ASP-A exhibited significant anticancer activity on BEL-7402 xenografts in nude mice (Figure 4D and E). The tumor volume significantly increased over time in vehicle-administered mice, while the increase was suppressed by the treatment with ASP-A. In the vehicle group, the cancer size on day 21 was 1,597±498 mm3, and at this time point the average tumor volumes on each of the groups after ASP-A treatment were 966±249 mm3, 817±84 mm3, and 803±168 mm3, and the values of T/C (%) were 59.9%, 51.2%, and 48.8%, respectively. The changes in body weights between the vehicle group and the ASP-A-treated groups were not remarkably different, but the body weights of the ADM group were significantly reduced, although the inhibitory rate of ADM reached 73.5%. These results suggest that ASP-A displays significant anticancer activity with lower toxicity compared with the positive control ADM.
Pharmacokinetics and tissue distribution in mice
The pharmacokinetic behavior of ASP-A was evaluated by determining the concentration of ASP-A versus time in mice plasma, after administration at a dose of 15 mg/kg and 30 mg/kg (Figure 5). Individual plasma concentration–time profiles were analyzed according to compartmental analysis. Median statistical summaries of pharmacokinetic parameters with two-compartment models are outlined in Table 1. The maximum plasma concentrations of ASP-A were 20.96 μg/mL and 39.50 μg/mL at doses of 15 mg/kg and 30 mg/kg, respectively. The mean terminal half-life of ASP-A was 40 minutes, indicating that ASP-A could be rapidly cleared in vivo. It is noticed that the time needed to reach maximum plasma concentration is the same at either dose of 15 mg/kg or 30 mg/kg, which fits the feature of first-order reaction kinetics. The tissue concentrations versus time after ip administration of ASP-A at doses of 15 mg/kg for 10 minutes, 30 minutes, and 60 minutes to mice are presented in Figure 6. After treatment for the indicated time, different levels of ASP-A were observed in all collected tissuess. Results showed that there was no long-term accumulation, in accordance with the variation trend of plasma concentration. In addition, ASP-A was enriched in cancer tissue, whereas the highest concentration was found in the kidney, followed by liver and lung, which implied that the distribution of ASP-A was also dependent on the blood flow or perfusion rate of the organ. Furthermore, ASP-A in brain was detectable 10 minutes after ip administration, which suggests that ASP-A could penetrate through the blood–brain barrier.
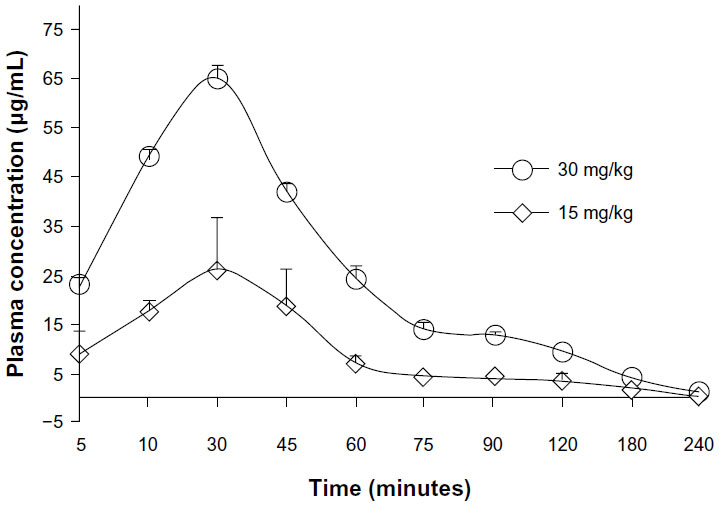 | Figure 5 Mean plasma concentration–time profile of ASP-A after intraperitoneal administration of ASP-A at doses of 15 mg/kg and 30 mg/kg to mice (n=5). |
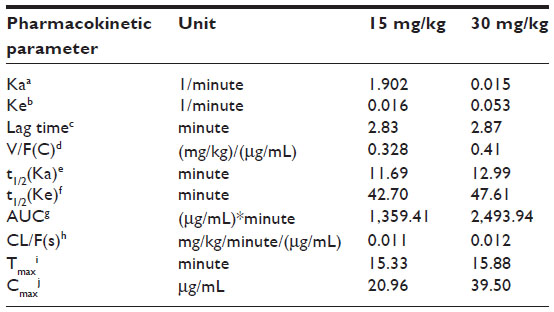 | Table 1 Pharmacokinetic parameters after intraperitoneal administration of ASP-A in mice at doses of 15 mg/kg and 30 mg/kg (n=5, mean ± SD) |
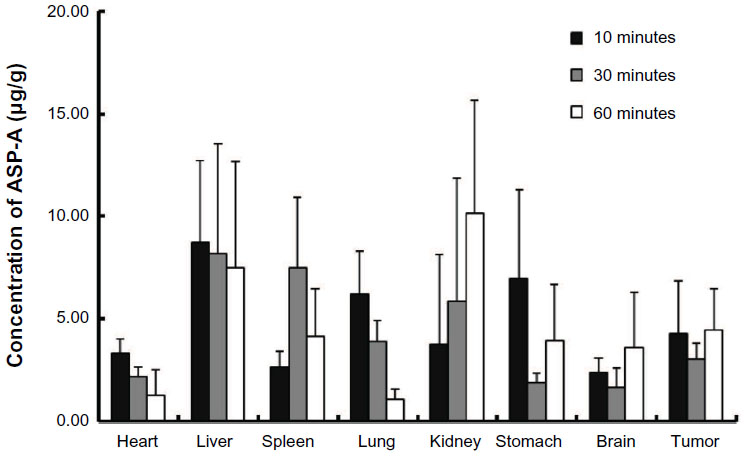 | Figure 6 Concentrations of ASP-A in different tissues 10 minutes, 30 minutes, and 60 minutes after intraperitoneal administration of 15 mg/kg ASP-A to mice (n=5). |
Early safety evaluation for ASP-A
No ASP-A treatment-related mortalities were recorded in animals treated with a single 400 mg/kg dose through the 14-day period. Therefore, the maximal tolerable dose (MTD) of ASP-A in the experimental mice was more than 400 mg/kg. No clinical signs were observed in the skin and fur, eyes and mucus membrane (nasal), respiratory rate, circulatory signs (heart rate and blood pressure), autonomic effects (salivation, perspiration, piloerection, urinary incontinence, and defecation) and central nervous system (ptosis, drowsiness, gait, tremors and convulsion) among mice administered 400 mg/kg of ASP-A, and there were no significant changes in the relative morphology of brain, heart, liver, stomach, and kidney in relation to control groups.
The effects of ASP-A on micronuclei formation are shown in Table 2. It was found that ASP-A did not interfere with normal mitotic cell division under the test condition employed or give any indication of bone marrow inhibition, and ASP-A did not cause any statistically significant increase in micronucleated polychromatic erythrocytes compared with the vehicle control (P<0.05). The negative results of ASP-A in this test suggest that ASP-A is not considered to be a potential genotoxic compound.
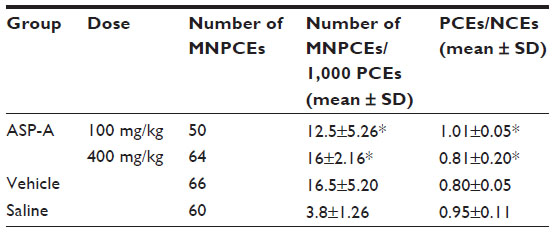 | Table 2 Effects of ASP-A on micronuclei rates in bone marrow polychromatic erythrocytes |
A number of clinically successful drugs in the market have had the tendency to inhibit hERG and to create a concomitant risk of sudden death as a side-effect, which has made hERG inhibition an important antitarget that must be avoided during drug development.10 Concentration and voltage-dependent inhibitions of the hERG channel by ASP-A were further investigated. Figure 7A shows representative current traces in the presence of different concentrations of ASP-A when the membrane potential was depolarized to +50 mV. Figure 7B shows the inhibitory rates of ASP-A (1.25 μM, 2.5 μM, 5 μM, 10 μM) on hERG channel corresponding to current were 0.9%±1.6%, 2.0%±2.3%, 3.8%±4.0% and 10.1%±5.9% (mean ± standard deviation), respectively; however, terfenadine induced nearly 100% inhibition at 100 nM (Figure 7C).
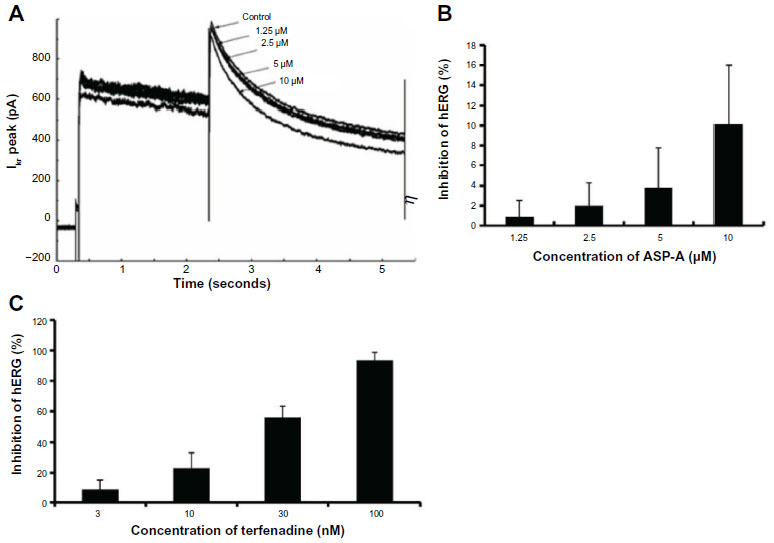 | Figure 7 Concentration-dependent and voltage-dependent inhibition of hERG channels by ASP-A and terfenadine. |
Uptake and transport of ASP-A by monolayers of Caco-2 cells
The Caco-2 cell line, which is derived from human colon adenocarcinoma, exhibits enterocyte-like characteristics and has been used widely as an in vitro model of absorption by intestinal epithelial cells. The time course of the uptake of ASP-A is shown in Figure 8A. The amount of ASP-A in the cells increased linearly at least up to 60 minutes. Similarly, in the concentration range of 25–300 μM, the content increased linearly with increasing concentration of ASP-A, then the uptake gradually became concentration-saturated (Figure 8B), and no further increase of ASP-A occurred.
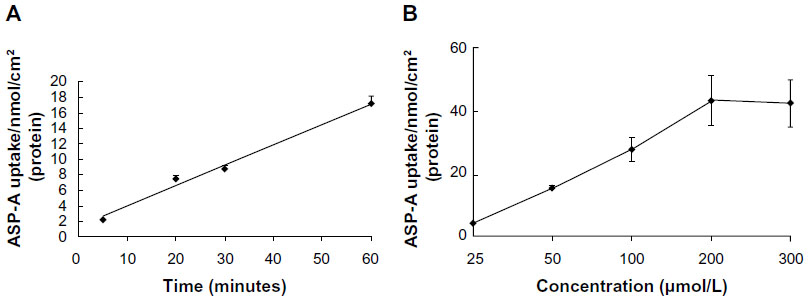 | Figure 8 Time- and concentration-dependent uptake of ASP-A in Caco-2. |
In the transporter experiment, ASP-A was detected for its ability to achieve apical-to-basolateral (AP-to-BL) and basolateral-to-apical (BL-to-AP) transport across Caco-2 monolayers. As shown in Figure 9, ASP-A was efficiently transported across Caco-2 monolayers over the course of a 180-minute experiment. The amounts of transport did not increase with donor concentration but increased with incubation time, which indicated that the transfer rate was not driven by the concentration and ASP-A transport had a feature of saturability. The Papp value in AP-to-BL direction ranged from (5.8–29.1) ×10−7 cm/second; in the reverse direction, BL-to-AP, the Papp ranged only from (0.08–1.38) ×10−7 cm/seconds. The transported level of ASP-A in AP-to-BL direction was much larger than in the BL-to-AP direction; the Pratios (permeability directional ratios, Papp [AP-to-BL]/Papp [BL-to-AP]) of the two sides were calculated to be >20 (Table 3), far above the Pratios of 1.5 for active transport reported by Mahar Doan et al,22 which indicated that the ASP-A transport had directionality and active transport pathway was involved in the process. From the above results, it is concluded that ASP-A can be well absorbed and is expected to be developed as an oral drug.
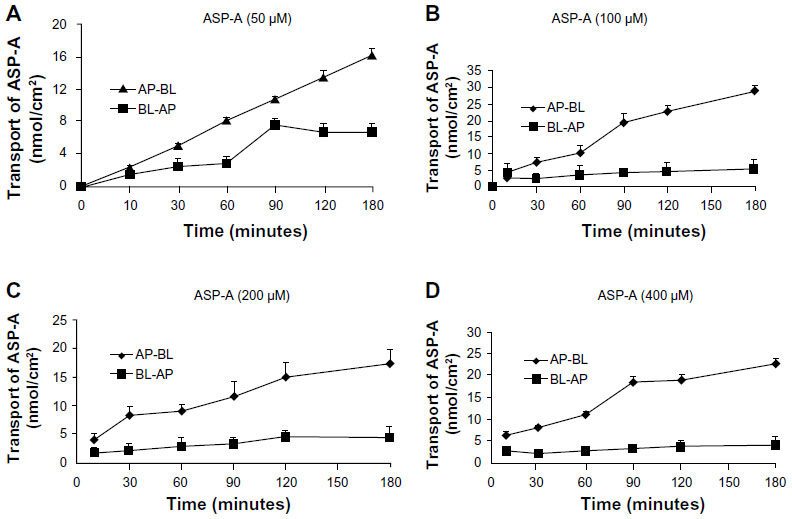 | Figure 9 Apical-to-basolateral and basolateral-to-apical transport of ASP-A. |
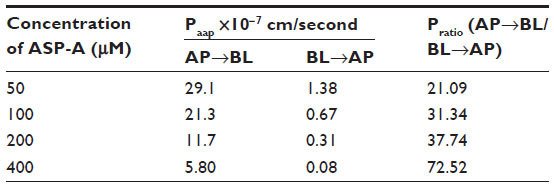 | Table 3 The Papp and Pratios for ASP-A in the apical-to-basolateral and basolateral-to-apical direction at the end of 3 hours of incubation with ASP-A |
Discussion
The plasticity and instability of the cancer genome is impressive and is characterized by gene amplifications and deletions, rearrangements, and many silent and active mutations. Many oncologists believe that targeted therapies are the chemotherapy of the future; however, as most cancers are unlikely to yield to single enzyme-targeted agents, the combination of chemotherapy with broadly active anticancer cytotoxic agents is the best attempt to prevent mutation and resistance.11,12 Therefore, there will continue to be a long-term need for cytotoxic drugs. In the present paper, we systematically assessed the drug activity of ASP-A as a cytotoxic candidate drug in early drug development.
First, ASP-A was estimated for its anticancer activities in vitro and in vivo. The results showed that ASP-A had a broad spectrum of cytotoxic activity in vitro, and significantly inhibited the growth of H22 and BEL-7402 cancer xenografts with less impact on the body weight of mice, whereas ADM inhibited body weight of mice by 25.1% and 33.3%, in H22 and BEL7402 cancer xenografts, respectively, even though the cancer growth inhibitory rate of ADM is higher than that of ASP-A. Pharmacological mechanism studies showed that ASP-A could target against topoisomerase II, just as ADM did.
Nearly half of drug candidates entering into preclinical development will be lost due to lack of safety. Early safety assessment can identify safety risk and recognize poor compounds ahead of entering into preclinical development, reducing attrition effects in the follow-up new drug development.13 We found the MTD of administration in mice was more than 400 mg/kg, which exceeds 10 times the effective dosage, indicating that ASP-A has a larger broad safety scope than ADM, the MTD of which was reported in mice to be 9 mg/kg, and therefore only 3.0–4.5 times higher than the therapeutic dose.14 Furthermore, we measured induction of chromosomal changes after short-term exposures to ASP-A by bone marrow erythrocyte micronucleus assay; no statistically significant distortion was found as compared to vehicle control, while ADM was reported to induce large amounts of micronuclei at 15 mg/kg under similar experiment conditions to ours.15 From these results we believe that ASP-A is much safer than ADM. However, the vehicle itself induced chromosomal changes, which requires us to look for a better solution for ASP-A.
A previously published survey on the causes of failure in drug development indicated that inappropriate pharmacokinetics were a major cause. Inappropriate pharmacokinetic behavior includes such factors as poor absorption characteristics and short elimination half-life leading to short duration of action; this observation has led to an increased emphasis on pharmacokinetic input to the drug discovery process.16 In the present paper, we investigated pharmacokinetic parameters and distribution of ASP-A by intraperitoneally injecting 15 mg/kg and 30 mg/kg of ASP-A at different time points. The results showed that the pharmacokinetics of ASP-A in plasma follow a two-compartment model. The values of the time needed to reach maximum plasma concentration for the two groups are the same, indicating that the clearance of ASP-A fits the first-order kinetic reaction equation. The results also suggest that ASP-A can achieve an effective drug concentration and broadly distribute in vivo; however, it is noticed that the elimination half-life is too short, just around 40 minutes, compared with a reported 23-hour half-life for ADM, which may be responsible for lower anticancer efficacy of ASP-A than of ADM in mice. Furthermore, distribution of drugs in mouse tissues was detected at different time points within the absorption, distribution, and clearance phase; the experimental results showed that ASP-A mainly located in the heart, kidneys, liver, and cancer tissue. ASP-A could penetrate through the blood–brain barrier, unlike ADM, which is excluded from entering the brain,17 suggesting ASP-A may be considered to inhibit brain cancer. Cardiotoxicity is a potential side effect of various chemotherapeutic agents, and the anthracycline class of cytotoxic antibiotics is the most well know to induce this side effect.18 hERG channels are involved in cardiac action potential repolarization, and reduced function of hERG lengthens ventricular action potentials, prolongs the QT interval in an electrocardiogram, and increases the risk for potentially fatal ventricular arrhythmias. It is important to screen compounds for activity on hERG channels early in the lead optimization process.10 We found that the hERG channel was little affected by ASP-A, detected by manual patch clamp technique. We do not believe that the concentration of ASP-A in heart tissue is high enough to induce cardiotoxicity.
A limiting factor for the successful therapeutic application of new drugs is their ability to transport over the gastrointestinal tract toward the target receptor. Caco-2 cell line is most widely used to screen intestinal permeability, to predict drug absorption, and to study passive and active diffusion transport of drug molecules.19 ADM, which is a P-glycoprotein and cytochrome P450 substrate, has poor bioavailability after oral administration.20 In our studies, the amount of ASP-A transported across the Caco-2 cellular monolayer was investigated. It is reported that the overall ranking of compounds with Papp <1×10−6 cm/second, between 1–10×10−5 cm/second, and >10×10−6 cm/second can be classified as poorly (0%–20%), moderately (20%–70%), or well (70%–100%) absorbed compounds, respectively,21 and Pratios >1.5 indicates active transport is involved in drug absorption.22 Therefore, our results revealed that ASP-A could be well uptaken by Caco-2 cells and transported in the apical-to-basolateral direction through the active transport pathway, which suggests a receptor-mediated transport of ASP-A. Oral preparation of ASP-A could be considered for future development.
Conclusion
In our studies, ASP-A was revealed as having both beneficial and harmful effects in early drug development. We found ASP-A had anticancer efficacy targeting topoisomerase II with similar structure and mechanism to ADM, but possessing characteristics of much lower genotoxicity and heart toxicity and better drug oral absorption potential; however, the anticancer efficacy of ASP-A needs to be further improved in vivo. It is reported that the formation and stability of an anthracycline–DNA–topoisomerase II ternary complex rely on defined structural determinants; in particular, the sugar moiety, located in the minor groove, is a critical determinant of the activity of anthracyclines as topoisomerase II poisons. In additional, the sugar moiety was reported to influence pharmacokinetic properties such as half-life,23 suggesting that for better anticancer efficacy, it will be necessary to optimize the structure by conjugating a sugar moiety to ASP-A.
Acknowledgments
This work was supported by NSFC-Shandong Joint Fund (U1406402), National Science and Technology Major Project (2009ZX09103-045), the Natural Science Foundation of China (No 81373323), and the State Key Laboratory of Drug Research.
Disclosure
The authors report no conflicts of interest in this work.
References
Liberra K, Lindequist U. Marine fungi – a prolific resource of biologically active natural products? Pharmazie. 1995;50(9):583–588. | |
Belofsky GN, Jensen PR, Fenical W. Sansalvamide: a new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999;40(15):2913–2916. | |
Du L, Zhu T, Fang Y, Liu H, Gu Q, Zhu W. Aspergiolide A, a novel anthraquinone derivative with naphtho[1,2,3-de]chromene-2,7-dione skeleton isolated from a marine-derived fungus Aspergillus glaucus. Tetrahedron. 2007;63(5):1085–1088. | |
Weiss RB. The anthracyclines: will we ever find a better doxorubicin? Semin Oncol. 1992;19(6):670–686. | |
Hodgson J. ADMET – turning chemicals into drugs. Nat Biotechnol. 2001;19(8):722–726. | |
Wang J. Comprehensive assessment of ADMET risks in drug discovery. Curr Pharm Des. 2009;15(19):2195–2219. | |
LeBeau JE. The role of the LD50 determination in drug safety evaluation. Regul Toxicol Pharmacol. 1983;3(1):71–74. | |
Cook PJ, Ju BG, Telese F, Wang X, Glass CK, Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–596. | |
Zunino F, Capranico G. DNA topoisomerase II as the primary target of anti-tumor anthracyclines. Anticancer Drug Des. 1990;5(4):307–317. | |
Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440(7083):463–469. | |
Vici P, Sergi D, Pizzuti L, et al. [Specificity of action of anticancer agents]. Clin Ter. 2011;162(2):137–149. Italian. | |
Teicher BA. Newer cytotoxic agents: attacking cancer broadly. Clin Cancer Res. 2008;14(16):1610–1617. | |
Stevens JL, Baker TK. The future of drug safety testing: expanding the view and narrowing the focus. Drug Discov Today. 2009;14(3–4):162–167. | |
Colbern GT, Hiller AJ, Musterer RS, Pegg E, Henderson IC, Working PK. Significant increase in anticancer potency of doxorubicin Hc1 by its encapsulation in pegylated liposomes. J Liposome Res. 1999;9(4):523–538. | |
Aleisa AM, Al-Rejaie SS, Bakheet SA, et al. Effect of metformin on clastogenic and biochemical changes induced by adriamycin in Swiss albino mice. Mutat Res. 2007;634(1–2):93–100. | |
Singh SS. Preclinical pharmacokinetics: an approach towards safer and efficacious drugs. Curr Drug Metab. 2006;7(2):165–182. | |
Rousselle C, Clair P, Lefauconnier JM, Kaczorek M, Scherrmann JM, Temsamani J. New advances in the transport of doxorubicin through the blood-brain barrier by a peptide vector-mediated strategy. Mol Pharmacol. 2000;57(4):679–686. | |
Gharib MI, Burnett AK. Chemotherapy-induced cardiotoxicity: current practice and prospects of prophylaxis. Eur J Heart Fail. 2002;4(3):235–242. | |
Fagerholm U. Prediction of human pharmacokinetics – gastrointestinal absorption. J Pharm Pharmacol. 2007;59(7):905–916. | |
Ke W, Zhao Y, Huang R, Jiang C, Pei Y. Enhanced oral bioavailability of doxorubicin in a dendrimer drug delivery system. J Pharm Sci. 2008;97(6):2208–2216. | |
Yee S. In vitro permeability across Caco-2 cells (colonic) can predict in vivo (small intestinal) absorption in man – fact or myth. Pharm Res. 1997;14(6):763–766. | |
Mahar Doan KM, Humphreys JE, Webster LO, et al. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J Pharmacol Exp Ther. 2002;303(3):1029–1037. | |
Zunino F, Pratesi G, Perego P. Role of the sugar moiety in the pharmacological activity of anthracyclines: development of a novel series of disaccharide analogs. Biochem Pharmacol. 2001;61(8):933–938. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.