Back to Journals » Cancer Management and Research » Volume 12
Anticancer Effects of Antihypertensive L-Type Calcium Channel Blockers on Chemoresistant Lung Cancer Cells via Autophagy and Apoptosis
Authors Wong BS, Chiu LY, Tu DG, Sheu GT ![]() , Chan TT
, Chan TT
Received 26 August 2019
Accepted for publication 13 February 2020
Published 13 March 2020 Volume 2020:12 Pages 1913—1927
DOI https://doi.org/10.2147/CMAR.S228718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Bing-Sang Wong,1 Ling-Yen Chiu,2 Dom-Gene Tu,2,3 Gwo-Tarng Sheu,4– 6,* Ting-Tat Chan7,*
1Division of Neurosurgery, Antai Medical Care Corporation Antai Tian-Sheng Memorial Hospital, Pingtung County, Taiwan; 2Department of Nuclear Medicine, Ditmanson Medical Foundation, Chia-Yi Christian Hospital, Chiayi City, Taiwan; 3Department of Biomedical Sciences, National Chung Cheng University, Chiayi 62102, Taiwan; 4Institute of Medicine, Chung Shan Medical University, Taichung, Taiwan; 5Immunology Research Center, Chung Shan Medical University, Taichung, Taiwan; 6Department of Medical Oncology and Chest Medicine, Chung Shan Medical University Hospital, Taichung, Taiwan; 7Palliative Care Unit, Department of Family Medicine, Ditmanson Medical Foundation, Chia-Yi Christian Hospital, Chiayi City, Taiwan
*These authors contributed equally to this work
Correspondence: Gwo-Tarng Sheu
Institute of Medicine, Chung Shan Medical University, No. 110, Sec. 1, Jianguo N. Road, Taichung City 402, Taiwan
Tel +886-4-24730022 Ext 11692
Fax +886-4-24751101
Email [email protected]
Purpose: Hypertension and cancer are frequently found comorbidity occurring in same individual. This study was intended to evaluate the anticancer effects of commonly used antihypertensive medications and chemotherapy on chemoresistant lung cancer cells.
Methods: Calcium channel blockers (CCBs), including Verapamil, Diltiazem, and Nifedipine, either alone or combined with docetaxel (DOC) or vincristine (VCR) were used to treat A549 lung adenocarcinoma chemoresistant sublines. Cell viability was determined by MTT assay, and colony formation assay was used to demonstrate the long-term effect of CCBs on proliferation of the sublines. Apoptosis was evaluated by Annexin V assay and autophagy intensity was quantitated from acidic vesicular organelle formation. Pan-caspase inhibitor, shATG5 interference and chloroquine were applied to study the roles of Verapamil on apoptosis and autophagy, with related proteins verified by Western blot analysis.
Results: Results show that 10 μM of Verapamil and Diltiazem, but not Nifedipine, differentially induce autophagy in DOC-resistant or VCR-resistant A549 cells, respectively. When CCBs are combined with DOC or VCR to treat the sublines, 10 μM of Verapamil induces autophagy more significantly than Diltiazem and Nifedipine, respectively, in DOC-resistant (54.91± 0.76, 18.03± 0.69, 7.05± 0.30) or VCR-resistant A549 (32.41± 1.04, 21.51± 0.63, 7.14± 0.24) cells. Inhibition of apoptosis by pan-caspase inhibitor partly reduced cell death indicates association of caspase-dependent cell death but with persistence of autophagy. Inhibition of autophagy by interfering ATG5 expression reduced c-PARP level and apoptotic cells suggest a pro-death role of autophagy. Chloroquine treatment enhanced autophagosome accumulation and cell death but with reduced c-PARP level suggests that mechanism of caspase-independent cell death also contributes to Verapamil/chemotherapy-induced anticancer effects.
Conclusion: Verapamil combined with DOC or VCR induces chemoresistant lung cancer cells to death through autophagy burst and apoptosis more strongly than Diltiazem and Nifedipine. Administering Verapamil or Diltiazem individually with chemotherapy, but not Nifedipine, can be considered in lung cancer patients with hypertension.
Keywords: hypertension, calcium channel blockers, lung cancer, Verapamil, Diltiazem, Nifedipine, chemoresistance
Introduction
Eighty percent of lung cancer cases are non-small cell lung cancer (NSCLC). The 5-year survival rate for lung cancer (18%) is next to pancreas cancer (8%), the lowest of all cancers;1 and chemotherapy is generally suggested for treatment of advanced-stage cancers. However, initial chemotherapy often leaves residual disease, from which tumors recur, and this multidrug resistance (MDR) limits the efficacy of chemotherapy.2 In addition to the resistance caused by regulation of drug transporters, such as ABCB1, the mechanisms of resistance to “classical” cytotoxic chemotherapeutics share many features, such as alterations in the target of drug, activation of prosurvival pathways and ineffective induction of cell death.3
Docetaxel (DOC) has anti-mitotic properties through the binding to microtubules (MTs) and preventing of depolymerization and stabilization of MTs.4 Vincristine (VCR) is a classic anti-tubulin agent that induces disruption of MTs by binding to tubulin and inhibits tubulin polymerization/MT formation.5 The action of VCR differs from that of DOC, which destabilizes MTs. Both DOC6,7 and VCR8,9 have been applied clinically as part of various cancer chemotherapy regimens. However, both drugs are a substrate of the ABCB1 transporter P-gp, so overexpression of ABCB1 in cancer cells is considered the major phenotype of multidrug resistance to DOC and VCR.10,11
There are many classes of antihypertensive, which lower blood pressure by different means. Among the most important and most widely used drugs are calcium channel blockers (CCBs), thiazide diuretics (TD), angiotensin-converting enzyme inhibitors (ACEi), angiotensin II receptor antagonists (ARBs), and beta blockers (BBs).12 L-type CCBs block the transmembrane flow of calcium, resulting in antagonism of vascular smooth muscle, contraction of myocardial smooth muscle, reduction of blood pressure, and coronary artery dilation.13,14 CCBs have assumed a major role in the treatment of patients with hypertension or coronary artery disease. CCBs can be broadly classified into 2 groups: dihydropyridine (DHP), such as Nifedipine (a 1,4-dihydropyridine, NIF); and non-dihydropyridine (non-DHP) groups. The prototypical agents of non-DHP group are Verapamil (a phenylalkylamine, VER), and Diltiazem (a benzothiazepinone, DIL). CCBs were the ninth most commonly prescribed class of drugs in the United States in 2009, with over 90 million prescriptions filled.15
To overcome the high prevalence of MDR, researchers have developed ABC transporter inhibitors to increase the intracellular concentration of chemotherapy drugs.16 In the early 1980s, it was found that CCBs are inhibitors of MDR in leukemia cells,17,18 and VER was the first compound to reach clinical trial for its ability to reverse MDR.16 Though some clinical trials failed due to high toxicity of VER or absence of improvement in the clinical outcome,19 others showed that VER improves patient survival,20–23 including a randomized study of NSCLC.24
The association between antihypertensive medications and the survival in cancer patients remains unclear. A recent report has documented survival in patients with common cancers who had been prescribed BBs, ACEi/ARBs, CCBs or TD for a minimum of one year prior to diagnosis of cancer, based on the provincial Drug Program Information Network.25 Cancer patients who were users of each class of antihypertensive agent were compared to cancer patients who were non-users of antihypertensive drugs. The survival of patients using only one drug class was compared, with BBs serving as the reference class. Compared to the antihypertensive drug non-user cohort, BBs had no effect on survival for any of the cancers. ACEi/ARBs use was weakly associated with increased deaths for breast cancer and lung cancer patients. Deaths were also increased with CCB use in patients with breast cancer and with TD use in lung cancer patients. When including only antihypertensive drug users prescribed with one drug class, lung cancer patients receiving CCBs had improved survival compared to BBs. This may be explained by a previous report that CCBs promote the sensitivity of multidrug-resistant lung cancer cells to chemotherapy.3
Autophagy is a cellular process responsible for delivering proteins or organelles to lysosomes.26 It not only helps maintain cellular homeostasis but also promotes survival during cellular stress situations. In contrast, excessive autophagy also promotes death of cancer cells.27 Therefore, autophagy seems to be a double-edged sword in the context of cancer therapies. It has been found that VER induces autophagy that reveals a cyclical mTOR-independent pathway regulating autophagy.28 Increased calcium influx is found in mononuclear cells (MNC) of patients with systemic lupus erythematosus (SLE), and NIF can promote SLE-MNC apoptosis.29 In addition, DIL may induce prostate cancer cell death, which could make it a drug of choice for treating cancer associated with hypertension.30 Interestingly, the sodium channels expressed in highly invasive breast cancer cell line MDA-MB-231 are sensitive to CCBs such as VER and DIL, but not to NIF with reduced cell proliferation.31 Currently, there is no conclusive cellular mechanism that explains the observed association between CCB use and lung cancer survival/outcome. Therefore, this study demonstrate the anticancer effects of CCBs, either alone or combined with chemotherapy, in chemoresistant A549 cells. It provides suggestions for using CCBs differentially in hypertension with lung cancer comorbidity. This investigation thus offers new proposals for appropriate application of CCBs in lung chemotherapy when patients also under hypertension management.
Methods
Drugs and Chemicals
DOC was obtained from Aventis Pharmaceuticals Inc. (Bridgewater, NJ). VCR, VER, NIF, DIL, and Chloroquine diphosphate salt were obtained from Sigma-Aldrich (St. Louis, MO). Z-VAD-FMK was purchased from Bachem (Torrance, CA). U0126 was purchased from Cell Signaling (Danvers, MA).
Chemoresistant Subline and Viability MTT Assay
Human adenocarcinoma A549 cells (ATCC, Manassas, VA, USA) were maintained as previously described.3 DOC- and VCR-resistant sublines were established from parental cells by exposure to increasing concentrations of DOC or VCR in a stepwise manner. The DOC-resistant subline maintained at 16 nM DOC is denoted A549/D16 and identified as P-gp overexpressing cells. A similar designation, A549/V16, was given to a VCR stably resistant subline and this subline is P-gp-independent.3 Cell viabilities were determined on MTT colorimetric assay. Briefly, cells (2×104/per well) were seeded onto 24-well plates. After 24 hrs incubation, the cells were exposed to various concentrations of DOC or VCR or CBCs in fresh medium for 48 hrs. Cells were washed with PBS, and MTT (300 μL/well, 1 mg/mL; Sigma) was added before further incubation at 37°C for 2.5 hrs. Cells were washed with PBS, and 2-propanol solution (300 μL/well) was added to dissolve the water-insoluble formazan salt with shaking at 70 rpm for 10 min at room temperature. Finally, the absorbance (570 nm) was measured using an ELISA plate reader (Molecular Devices SPECTRA max 340 PC).
Western Blot Analysis
Western blot analysis for apoptosis, autophagy and signaling pathway regulated proteins. The relevant procedures have been previously described.3 Proteins were reacted with one of the followings: anti-LC3B (#3868), anti-cleavaged Poly(ADP-ribose) Polymerase (cPARP) (#5625) purchased from Cell Signaling (Danvers, MA), anti-p62 (GTX100685) obtained from GeneTex (Irvine, CA), or monoclonal anti-β-actin (Sigma, AC-40). Chloroquine (CQ, 10 μM) was added with DOC or VCR or VER in fresh medium for 48 hrs treatment and cells were harvested for analysis.
Annexin V Assay for Apoptosis Characterization
Cells were trypsinized and incubated for 30 mins in binding buffer with propidium iodide (PI) and Annexin V (FITC Annexin V Apoptosis Detection Kit 1, BD Biosciences, San Jose, CA), followed by analysis with flow cytometry.
Detection and Quantification of Acidic Vesicular Organelles (AVOs) for Autophagy Formation
The detailed steps of AVO analysis have been previously described.32 Briefly, after treatment, cells were washed with HBSS twice, followed by staining with acridine orange (1 g/mL, Sigma, A 6014). After staining, cells were washed with HBSS and suspended in HBSS containing 5% FBS. The cells were observed under a red filter fluorescence microscope.
Clonogenic Cell Proliferation Assay
Cells were seeded in 6-well plates (150 cells/well). After 24 hrs of incubation, cells were treated with respective doses of CCBs, DOC or VCR for 48 hrs only and eventually cultured for 10 days with fresh medium. The colonies formed were fixed with ice-cold methanol for 30 mins then stained with 20% Giemsa. Survival fractions were calculated with microscope and normalization to appropriate control groups.
Inhibition of ATG5 by VSV-G Pseudo Lentivirus-shRNA
All RNAi reagents were obtained from the National RNAi Core Facility at the Institute of Molecular Biology/Genomic Research Center, Academia Sinica. Individual clones were identified by their unique TRC number: shLuc TRCN0000072246 targeted to luciferase for vector control; clones of shATG5-394 (TRCN0000330394) and shATG5-474 (TRCN0000151474) targeted to ATG5. Lung cancer cells (5 × 105/plate) were seeded onto 60 mm plates and incubated at 37°C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% non-essential amino acids solution, 1% sodium pyruvate, 1% L-glutamine, 100 IU penicillin mL−1 and 100 mg streptomycin mL−1 for 16 hrs. Then, cells were infected with lentivirus vectors at a multiplicity of infection of 1. The next day, the cells were selected by 2 μg/mL puromycin (Sigma, P8833) with a fresh medium for 2 days. The stable cell lines of shLUC, shATG5-394 and shATG5-474 were used for drug treatment and analysis.
Statistical Analysis
All values are presented as mean ± SD. Data were compared between groups using t-test and *p<0.05 was considered statistically significant.
Results
Differential Effects of L-Type Calcium Blockers on Chemoresistant Lung Cancer A549/D16 Cells That Overexpressing P-gp
We have previously reported that A549/D16 cells are P-gp overexpressing chemoresistant lung cancer cells.3 When A549/D16 cells were treated with VER for 48 hrs, the viability was not markedly reduced. In contrast, when A549/D16 cells were co-treated with VER and DOC, cytotoxicity was increased significantly on A549/D16 cells, reducing the survival of cells in a dose-dependent manner (Figure 1A). Interestingly, VER alone induces autophagy, also in a dose-dependent manner, as shown by the increased levels of LC3BII, and co-treatment of VER/DOC preserved both autophagy induction and increased c-PARP production (Figure 1B). PARP cleavage has active roles in apoptosis.33 Surprisingly, large amounts of AVO formations were detected when VER/DOC was co-treated on A549/D16 cells (Figure 1C and Supplemental Figure 1). Although VER alone has little effect on viability (Figure 1A) and apoptosis (Figure 1B), the DOC-enhanced autophagy may upregulate apoptosis, as evidenced by increased c-PARP levels (Figure 1B). It is known the formation of LC3BII and significant breakdown of p62 indicate the initiation and completion of autophagic flux,34 but our data shown p62 expression was not reduced when LC3BII increased. A previous report has demonstrated that this phenomenon represents autophagosome accumulation and results in autophagic death in A549 cells.35 Thus, VER induces autophagy but not apoptosis, and strongly enhances DOC sensitivity on A549/D16 cells by excess autophagy and apoptosis.
 |
Figure 1 Investigation of the individual CCB-related sensitivity, apoptosis and autophagy in A549/D16 cells with P-gp-overexpression. Viabilities of cells treated with (A) VER or VER combined with DOC (D) DIL or DIL combined with DOC (G) NIF or NIF/DOC analyzed by MTT assay. (B), (E) and (H) are Western blot analyses of the proteins of PARP and LC3B levels for apoptosis and autophagy. Analysis of AVO formation for autophagy when A549/D16 cells treated with (C) VER or VER/DOC co-treatment, (F) DIL or DIL/DOC co-treatment, and (I) NIF or NIF/DOC co-treatment. *p<0.05 was considered statistically significant. |
The results shown in Figure 1D indicate that DIL plus DOC has less effect than VER combined with DOC for reducing A549/D16 cells survival (Figure 1A). DIL alone induces a moderate level of autophagy (Figure 1E and F) and only a high dose (10 μM) of DIL combined with DOC induces apoptosis, as indicated by increased c-PARP (Figure 1E). When combined with DOC, DIL moderately upregulates autophagy (Figure 1F and Supplemental Figure 1).
The results of Figure 1G and H indicate that NIF alone or combined with DOC has no effect on A549/D16 cell viability, and only background level of AVO formation was detected (Figure 1I and Supplemental Figure 1).
Effects of CBCs on P-gp Independent Chemoresistant A549/V16 Lung Cancer Cells
The effects of VER on A549/V16 cells were very similar to VER on A549/D16 cells, indicating that VER co-treatment with VCR sensitized A549/V16 cells to death (Figure 2A and B). Apoptotic A549/V16 cells (Figure 2B) were also associated with a high level of autophagy when VER was combined with VCR (Figure 2B, C and Supplemental Figure 2).
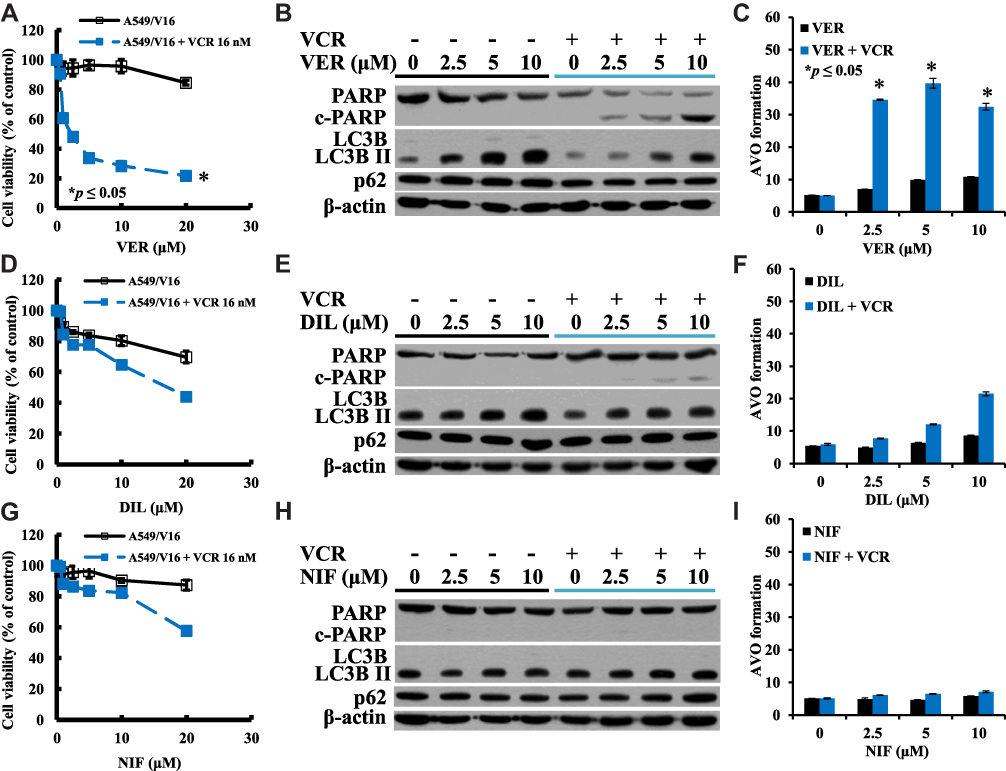 |
Figure 2 Investigation of the individual CCB-related sensitivity, apoptosis and autophagy in A549/V16 chemoresistant cells lacking P-gp-overexpression. Viabilities of cells treated with (A) VER or VER combined with VCR (D) DIL or DIL combined with VCR (G) NIF or NIF/VCR analyzed by MTT assay. (B), (E), and (H) are Western blot analyses of the proteins of PARP and LC3B levels for apoptosis and autophagy. Analysis of AVO formation for autophagy when A549/V16 cells treated with (C) VER or VER/VCR co-treatment, (F) DIL or DIL/VCR co-treatment, and (I) NIF or NIF/VCR co-treatment. *p<0.05 was considered statistically significant. |
When A549/V16 cells were treated with DIL, cell survival was downregulated (Figure 2D) with steady levels of autophagy (Figure 2F). Only when DIL was combined with VCR, autophagy levels were significantly increased (Figure 2F and Supplemental Figure 2), but only minimal c-PARP proteins were detected under the same conditions (Figure 2E). The results demonstrate that DIL and VCR sensitized A549/V16 cells to death less effectively than VER combined with VCR (Figure 2A and B). These results indicate that in addition to apoptosis, an autophagy burst with VER/DOC- or VER/VCR-induced autophagosome accumulation may lead to the death of chemoresistant lung cancer cells.
NIF alone has little effect on the sensitivity of A549/V16 cells (Figure 2G) and has little effect on autophagy (Figure 2H and I). When A549/V16 cells were treated with NIF combined with VCR, although cell survival has been reduced (Figure 2G), apoptotic markers of c-PARP were not detected (Figure 2H) and AVO formation did not increase significantly (Figure 2I and Supplemental Figure 2).
Anti-Proliferation Effects of CCBs Analyzed by Clonogenic Cell Survival Assay
To characterize the long-term effect of individual CCB on chemoresistant A549 sublines proliferation, the cells were divided into two sets, treated for 48 hrs with either CCB alone or CCB combined with DOC or VCR. Then, cells were feeded with fresh medium and incubated for 10 days, the numbers of colonies were counted as shown in Figure 3. For the A549/D16 cells treated with VER (Figure 3A) or DIL alone (Figure 3B), the numbers of colonies were not higher than cells without CCB (0, VER or DIL). In contrast, there were more colonies in NIF-treated A549/D16 cells (Figure 3C). For the A549/D16 cells treated with individual CCB combined with DOC, VER (2.5 μM) strongly enhances DOC toxicity and reduces colony numbers (Figure 3A), and DIL (5 μM) moderately enhances DOC toxicity (Figure 3B). NIF (10 μM) has less effect on reversing DOC resistance when combined with DOC on A549/D16 cells (Figure 3C). For A549/V16 cells treated with VER alone, more colonies were detected (Figure 3D). DIL (Figure 3E) and NIF alone (Figure 3F) have little effect on survival of A549/V16 cells. When CCB was combined with VCR, VER significantly sensitized VCR-resistant cells resulting in fewer colonies (Figure 3D). DIL moderately enhances DOC toxicity (Figure 3E) and NIF has little effect on enhancing VER toxicity on A549/V16 cells (Figure 3F). The results of MTT assay showed CCBs by themselves may repress proliferation of chemoresistant cells (Figures 1 and 2) but similar data were not obtained from the clonogenic assay. The difference may result from those cells only treated with CCBs for 48 hrs but not continuously exposed to CCBs in the clonogenic assay. These results indicate that CCB medication with chemotherapy suppresses the proliferation of chemoresistant lung cancer cells.
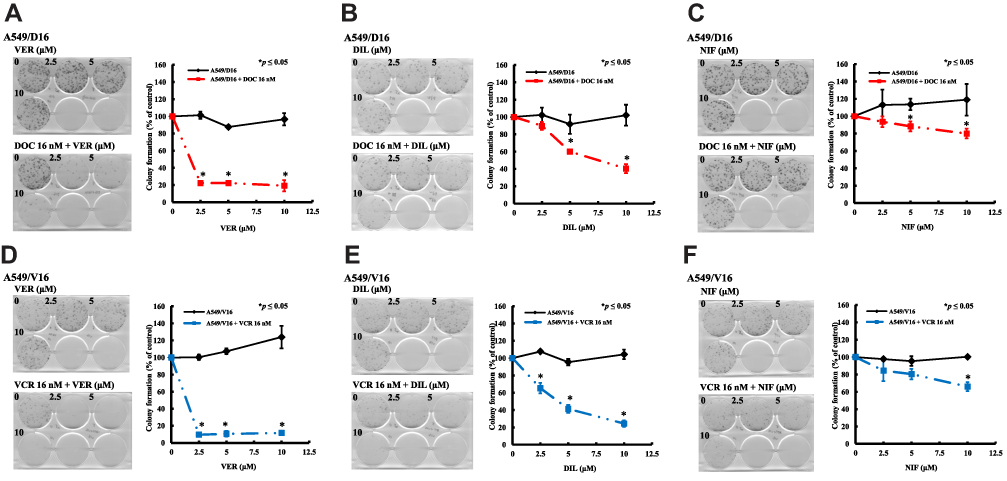 |
Figure 3 Antiproliferation effect of CCBs on A549 chemoresistant sublines analyzed by colony formation assay. A549/D16 and A549/V16 sublines were treated with VER (A, D), DIL (B, E), and NIF (C, F) alone, respectively, or co-treated with DOC (A, B, and C) or VCR (D, E, and F) for 48 hrs and incubated with fresh medium for 10 days and the colonies were calculated. The data were collected and normalized to the control set that without CCBs. *p<0.05 was considered statistically significant. |
Individual CCB-Sensitizing DOC/VCR Chemotherapy on Chemoresistant Sublines with Different Efficacy
To further analyze the anticancer effect of CCBs on chemotherapy, the growth kinetics of A549/D16 cell was compared in three different groups. The first group compared chemoresistant A549/D16 cells treated with DOC (16 nM) to blank control cells, the second group compared cells treated with CCB (5 μM) only with cells treated with CCB (5 μM) combined with DOC (16 nM). The third group compared cells treated with CCB (10 μM) with cells treated with CCB (10 μM) and DOC (16 nM). The data were analyzed by MTT assay for three-day periods (Figure 4A). Similar conditions for the A549/V16 subline were also studied (Figure 4B). Results obtained from the group-1 demonstrate that these two sublines were resistant to DOC or VCR, and that they have no response to chemotherapy with a high dose of cytotoxic DOC or VCR. Groups 2 and 3 show growth inhibition effects of individual CCB on A549 sublines with dose-responsiveness. Clearly, VER is a better candidate to suppress chemoresistance than DIL and NIF for both A549/D16 and A549/V16 cells when combined with chemotherapy.
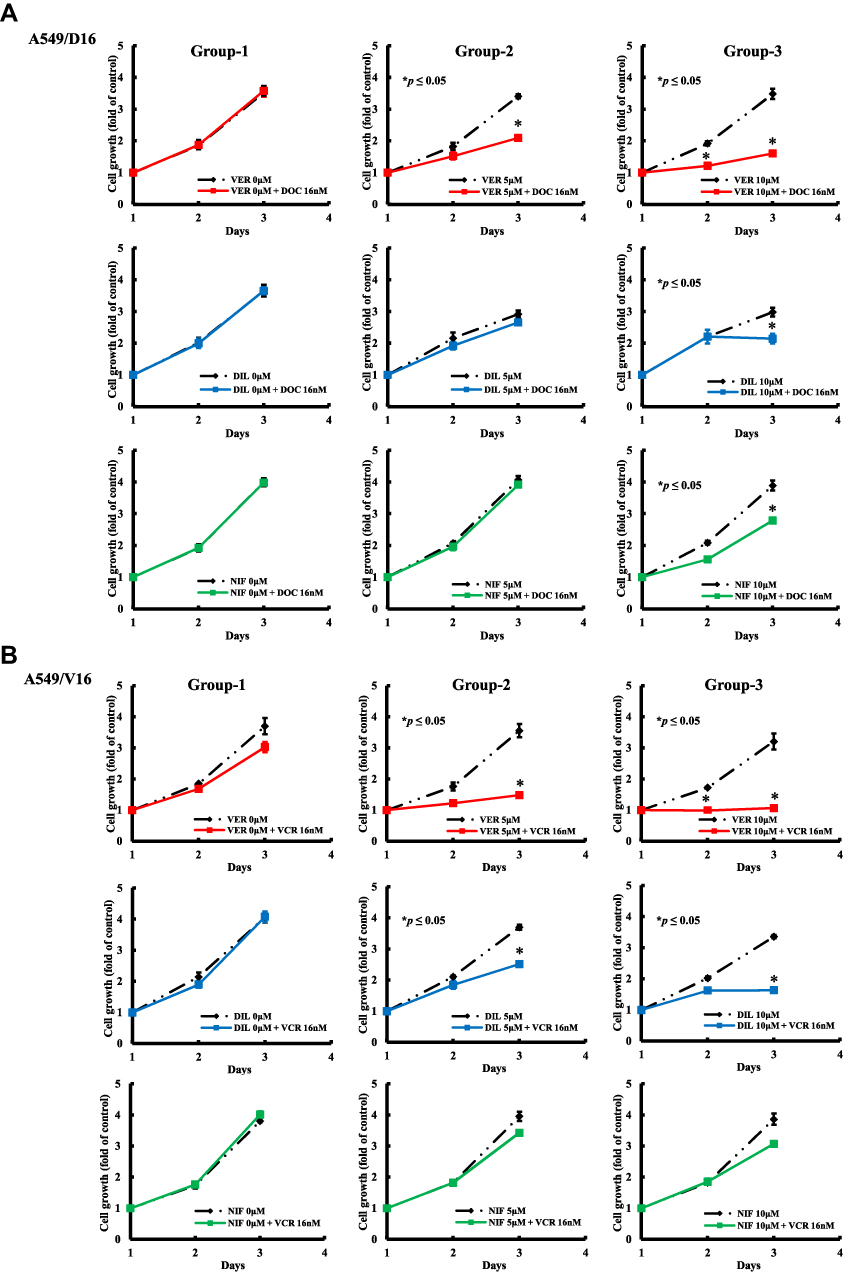 |
Figure 4 Analysis of the growth inhibition effect of individual CCBs on A549 chemoresistant sublines analyzed by MTT assay. Group-1 compared chemoresistant A549/D16 cells (A) treated with DOC (16 nM) to blank control cells, and group-2 compared cells treated with CCB (5 μM) only with cells treated with CCB (5 μM) combined with DOC (16 nM). Group-3 compared cells treated with CCB (10 μM) with cells treated with CCB (10 μM) and DOC (16 nM). The data were analyzed by MTT assay for three-day periods. Similar conditions for A549/V16 subline were applied and studied (B). *p<0.05 was considered statistically significant. |
Pan-Caspase Inhibitor Z-VAD-FMK Reduces VER/DOC-Induced Cell Death with Persistent Autophagy
The associations of apoptosis in CCB and chemotherapy-induced chemoresistant lung cancer cell death were further investigated by applying the pan-caspase inhibitor (Z-VAD, 50 μM) followed by Annexin V assay of flow cytometry (Figure 5A and B) and Western blot analysis (Figure 5C and D). Pre-treatment of Z-VAD, significantly reduced the VER/DOC- or VER/VCR-induced death of A549 sublines (Figure 5A and B, respectively). At the same time, the levels of cleavage product of PARP were reduced by Z-VAD but LC3BII remained at higher levels in response to VER/DOC or VER/VCR treatment (Figure 5C and D, respectively). Therefore, VER/DOC- or VER/VCR-induced autophagy were not been affected when caspase-dependent apoptosis was blocked. The data indicate that CCB- and chemotherapy-induced chemoresistant lung cancer cell death is associated with caspase-dependent apoptosis.
 |
Figure 5 Effects of caspases inhibition on VER/DOC- and VER/VCR-induced apoptosis and autophagy. (A) Pre-treatment of pan-caspase inhibitor (Z-VAD, 50 μM) for 1 hr followed by VER/DOC or (B) VER/VCR co-treatment for 48 hrs and analyzed by Annexin V assay of flow cytometry on A549/D16 and A549/V16 cells, respectively. (C and D) Expressions of PARP, cPARP LC3B and LC3B-II were detected by Western blot analysis with the same treatment conditions as (A and B). *p<0.05 was considered statistically significant. |
Autophagy Inhibition by shATG5 Also Reduces VER/DOC-Induced Cell Death and Represses Caspases Activation
To investigate the role of CCB-induced autophagy in the death of chemoresistant lung cancer cells, we induced shRNA interference by introducing two individual clones of shATG5-394 and shATG5-474 to reduce ATG5 expression in A549/D16 cells that can block the autophagosome formation.36 The effect of autophagy inhibition on cell apoptosis was analyzed by Annexin V assay of flow cytometry (Figure 6A). After autophagy inhibition, VER/DOC combinations have less cell death than the shLUC control. Compared with the shLUC infection control, shATG5-394, and shATG5-474 significantly reduced ATG5 expression, resulting in LC3B-II down-regulation without VER treatment. When VER was combined with DOC, shATG5-394 infected cells still lacked LC3B-II expression corresponding to autophagy inhibition (Figure 6B). Similar responses were also observed when a clone of shATG5-474 was used. The c-PARP levels were also reduced when autophagy was inhibited. These results indicate a pro-death role for VER/chemotherapy-induced autophagy.
 |
Figure 6 Continued. |
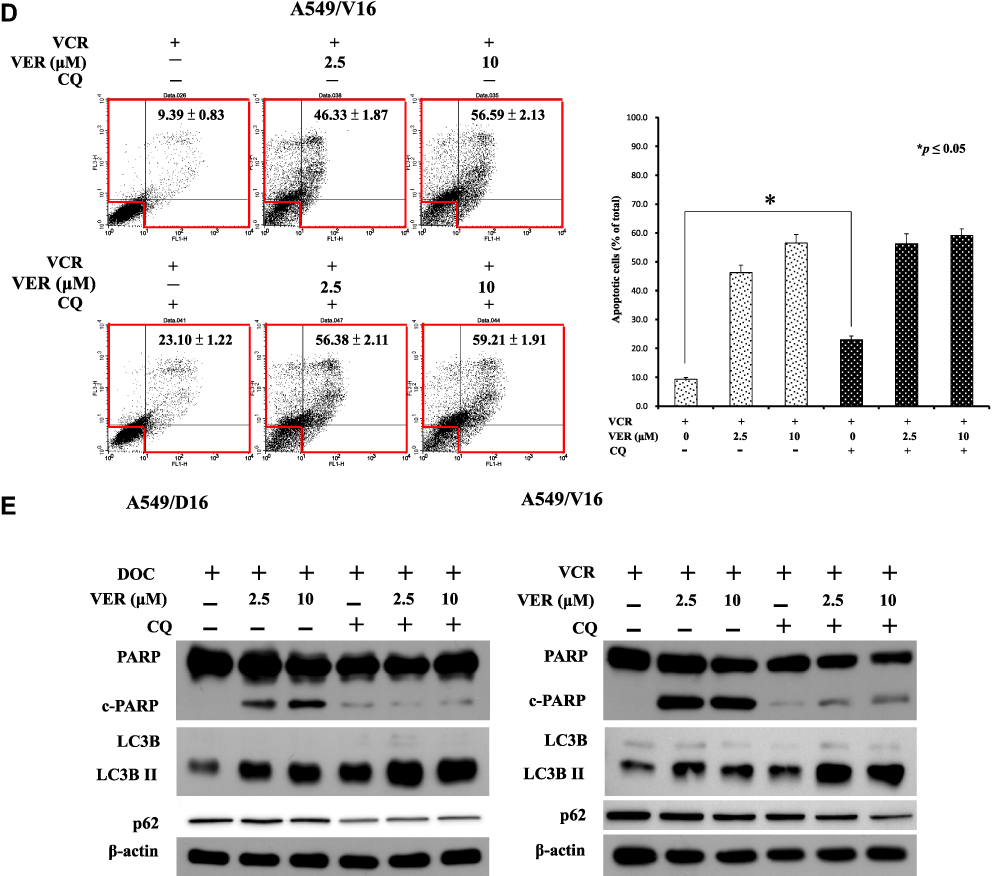 |
Figure 6 Effects of autophagy inhibition by shATG5 infection and chloroquine on VER/DOC-induced apoptosis and autophagy. (A) Individual shATG5-394 and shATG5-474 were used to inhibit ATG5 protein expression with shLUC as the infection control. Collected cells were further analyzed by Annexin V assay of flow cytometry on A549/D16 cells in the presence of DOC (16 nM) with or without VER for 48 hrs, followed by (B) Western blot analysis of protein obtained under same experimental conditions. (C) Chloroquine (CQ, 10 μM) was combined with DOC or DOC/VER (D) CQ was combined with VCR or VCR/VER to treat the cells for 48 hrs and analyzed by Annexin V assay of flow cytometry on A549/D16 and A549/V16 cells, respectively. (E) Proteins obtained under the same experimental conditions were analyzed by Western blot assay. *p<0.05 was considered statistically significant. |
Chloroquine Blocks Autophagic Flux and Enhance Caspase-Independent Cell Death
Chloroquine (CQ), which is an inhibitor of autophagy that blocks the binding of autophagosomes to lysosomes.37 To address the VER/chemotherapy mediated autophagy burst resulted in cell death, CQ (10 μM) was added with DOC or combined with DOC/VCR to treat the cells. The data obtained from Annexin V assay without CQ and only with DOC was 8.99±0.57 of total apoptotic cells that versus 36.81±1.17 when CQ was added. Increased c-PARP and LC3BII levels were detected (Figure 6E left panel) when CQ co-treated with DOC. The data suggest that CQ treatment associated with apoptosis and autophagy. In the present of VER (10 μM), CQ and DOC, more annexin V positive cells were detected (89.10±2.19) that reflecting large amount of cell death was occurred (Figure 6C). Although CQ augment LCBII level; reduced c-PARP expression on cells that treated with CQ, VER (10 μM) and DOC when compared with VER (10 μM) and DOC treatment (Figure 6E left panel). The expression of p62 was not significantly decreased in Figure 6E. Similar results were also obtained from the A549/V16 cells that investigated with same conditions (Figure 6D). These results support that CQ block autophagic flux and enhance VER/chemotherapy-induced autophagy burst that resulted in caspase-independent cell death.
A Proposed Model for CCBs and Chemotherapy-Induced Cell Death on Chemoresistant Lung Cancer Cell
We have tested the anticancer effect of DHP (NIF) and Non-DHP (VER, DIL) with chemotherapy on chemoresistant lung cancer cells (Figure 7). The caspase dependent cell death is associated with anticancer effect of Non-DHP type CCBs. Autophagy induced by CCBs/chemotherapy (autophagy burst) plays a pro-death role that were supported by (1) the shATG5 knockdown that reduced autophagy and cell death; (2) CQ blocks autolysosome formation and enhances VER/chemotherapy-induced autophagy burst and resulted in caspase independent cell death. Verapamil combined with chemotherapy provides more strongly anticancer effect than Diltiazem, and Nifedipine, respectively.
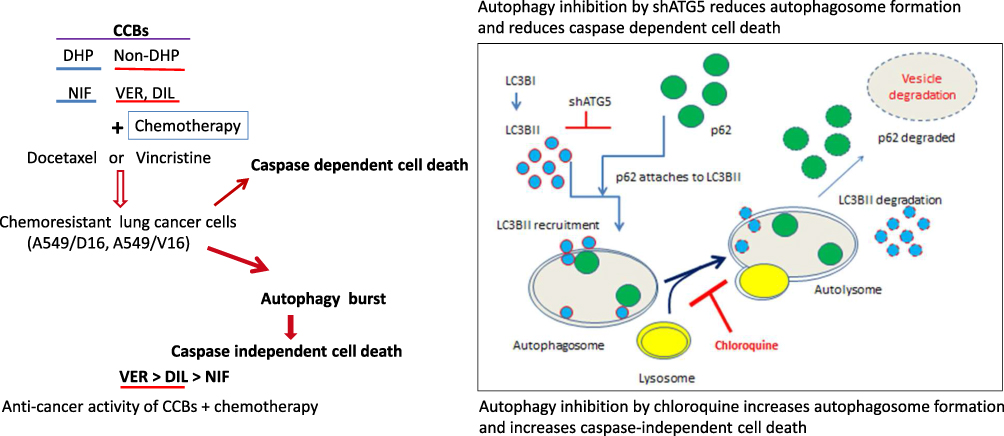 |
Figure 7 A proposed model of anticancer effects using CCBs combined with chemotherapy to induce cell death on chemoresistant lung cancer cells. Administration of VER or DIL induces autophagy; and death ratios of chemoresistant cells were significantly increased when VER combined with chemotherapy. Both caspase-dependent and independent cell death mechanisms contribute to the anticancer effects of VER/chemotherapy. Knockdown of ATG5 by shATG5 reduced caspase-dependent cell death. Whereas, accumulation of autophagosomes by VER/chemotherapy or CQ results in cells to be burst in a caspase-independent pathway of cell death. |
Discussion
Hypertension is known to have significant effects on cardiovascular (CVD) outcomes, which is the leading cause of death and disability in the world.38 In 2012–2013, the prevalence of hypertension in Canadian adults was 22.6%, an increase from 19.6% in 2009–2011.39 It is associated with 7.6 million premature deaths (about 13.5% of the global total) per year worldwide, making it the leading risk factor for cardiovascular disease (CVD).40 In Europe, hypertension is one of the most common causes of primary care interventions, and CCBs are a first-line treatment for this symptom.41 There has, however, been a long debate regarding the potential association of CCBs with increased cancer risk, especially for breast cancer.42–44 Several studies and randomized controlled trials suggest a positive association of CCBs with breast cancer incidence.45,46 There has been little research on the possible effects of CCBs on cancer survival. It was reported that there had no evidence that CCB use was associated with a better or worse survival in cancer patients in Asian and Caucasian populations.47 On the contrary, association of CCB use with risks of breast cancer outcomes has also been reported,48 and CCBs have been associated with increased cancer recurrence in patients with head and neck squamous cell carcinoma.49 CCBs also have been associated with decreased survival in certain cancers when compared with β-blockers.50 Deaths were also increased with CCBs use in patients with breast cancer; in contrast, CCBs use was associated with improved survival in lung cancer patients.25 To resolve this discrepancy in lung cancer and breast cancer outcomes, this study demonstrates the potential mechanisms of CCB association with improved survival in lung cancer patients.
We show that VER and DIL alone can induce autophagy and co-treatment with DOC or VCR, further enhancing autophagy and apoptosis in typical-MDR A549/D16 and atypical-MDR A549/V16 chemoresistant lung cancer cells. Therefore, the anticancer activities of individual CCBs are not affected by P-gp expression. In contrast, NIF is a member of DHP-group CCBs, and NIF alone or combined with chemotherapy has little anticancer effect. We also address that autophagy induced by CCBs is a pro-death response in chemoresistant lung cancer A549 cells. In the present of chemotherapy, VER further enhance autophagosome formation but not vesicle degradation. The protein of p62 should be efficiently degraded by autophagy,51 therefore, autophagic flux suppression should correlate with an increased p62 level. Logically, CQ is an autophagy flux blocker that should enhance p62 level. Surprisingly, the expression of p62 was not significantly increased in Figure 6E. It has been reported that p62 is degraded by both the autophagy and ubiquitin-proteasome system,52 It is possible that when autophagosomes were accumulated and ready to be burst, p62 can be degraded by proteasome when cells were overwhelmed with death stress. It is presently unclear how VER/Chemotherapy suppress autophagic flux and resulted in autophagy burst.
In contrast to our findings, VER-induced cytoprotective autophagy has been found in a series of human cell lines, including colon adenocarcinoma COLO 205 cells, prostate cancer PC-3 cells and prostate PNT-2 cells, pancreatic cancer KP-4 cells, and pancreatic HPNE cells. Inhibition of autophagy in those Verapamil-treated cells stimulates apoptosis.53 Although without autophagy analysis, it has been demonstrated that DIL in combination with proteasome inhibitors induces apoptosis in proteasome inhibitor-resistant prostate cancer DU145 cells.30 Our and others’ results strongly indicate that DHP- and non-DHP-CCBs may affect autophagy and apoptosis differentially in various cancer cells.
Furthermore, several reports have evaluated the clinical efficacy of targeted arterial perfusion of VER and chemotherapeutic agents in the interventional therapy of various cancers. In order to reach a concentration capable of reversing MDR in tumors and avoid cardiac toxicity exceeding the safe concentration (1.0–2.0 μM), targeted arterial perfusion of VER and chemotherapeutic agents were performed for tumor chemotherapy. It was found that VER via targeted arterial infusion could effectively reverse the MDR in primary hepatocellular carcinoma patients and enhanced the efficacy of chemotherapy.54 In patients with advanced gastric cancer, chemotherapy is more effective when combined with targeted arterial infusion of VER, leading to extended patient survival and improved quality of life.55 Furthermore, targeted arterial perfusion of VER and chemotherapeutic drugs can improve clinical symptoms of patients with advanced lung cancer and increase the efficacy of chemotherapeutic agents.56
Despite chemotherapy, advanced NSCLC is usually associated with poor prognosis. When patients have chemoresistance and hypertension concurrently, the outcome may be affected by drug/drug interactions. Although oral administration of non-DHP CCBs may not reach a concentration capable of reversing MDR in tumors, the metastatic lung cancer cells in systemic circulation may be sensitized when combined with chemotherapy, leading to improved survival,25 and targeted arterial perfusion of VER and chemotherapeutic agents may more effectively prolong patient survival.
Conclusions
VER plus DOC/VCR effectively sensitize chemoresistant lung cancer cells to death via autophagy burst and apoptosis, and NIF has little anticancer effect than VER or DIL. Our data suggest that patients with advanced lung cancer and hypertension should be carefully monitored with chemotherapy and Non-DHP type CCBs may provide a better anticancer effect.
Abbreviations
NSCLC; non-small cell lung cancer; ABCB1; ATP-binding cassette sub-family B member 1; DOC; Docetaxel; MTs; microtubules; VCR; vincristine; MDR; multidrug resistance; CCBs; calcium channel blockers; TD; thiazide diuretics; ACEi; angiotensin-converting enzyme inhibitors; ARBs; angiotensin II receptor antagonists; BBs; beta blockers; DHP; dihydropyridine; non-DHP; non-Dihydropyridine; NIF; Nifedipine; VER; Verapamil; DIL; Diltiazem; mTOR; Mammalian target of rapamycin; MNC; mononuclear cells; SLE; lupus erythematosus; P-gp; P-glycoprotein; ELISA; Enzyme-linked immunosorbent assay; PI; propidium iodide; AVOs; acidic vesicular organelles; ATG5; autophagy-related 5; VSV-G; Vesicular stomatitis virus-G protein; shRNA; Small hairpin RNA; shLuc; shRNA specific for luciferase gene; CQ; chloroquine; SD; standard deviation; c-PARP; Cleavage form of Poly (ADP-ribose) polymerase; LC3B II; microtubule-associated protein 1A/1B-light chain 3 phosphatidylethanolamine conjugated B II.
Data Sharing Statement
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We would like to thank the staff of the Instrument Center of Chung Shan Medical University for their technical support.
Author Contributions
BS and LY performed the experiments; interpretation of the results. DG, GT and TT designed the experiments, collected data and analyzed. BS, GT and TT wrote the paper. DG and GT revised the manuscript. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Funding
This work was supported by grant (CSMU-TSMH-105-02) and Ditmanson Medical Foundation Chia-Yi Christian Hospital (Grant No. R106-031).
Disclosure
The authors declare that there are no potential conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Baird RD, Kaye SB. Drug resistance reversal–are we getting closer? Eur J Cancer. 2003;39(17):2450–2461. doi:10.1016/S0959-8049(03)00619-1
3. Chiu LY, Ko JL, Lee YJ, Yang TY, Tee YT, Sheu GT. L-type calcium channel blockers reverse docetaxel and vincristine-induced multidrug resistance independent of ABCB1 expression in human lung cancer cell lines. Toxicol Lett. 2010;192(3):408–418. doi:10.1016/j.toxlet.2009.11.018
4. Fitzpatrick FA, Wheeler R. The immunopharmacology of paclitaxel (Taxol), docetaxel (Taxotere), and related agents. Int Immunopharmacol. 2003;3(13–14):1699–1714. doi:10.1016/j.intimp.2003.08.007
5. Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol. 1999;17(3):1061–1070. doi:10.1200/JCO.1999.17.3.1061
6. Davies AM, Lara Jr PN
7. Green MR. Perspectives and opportunities: docetaxel in the current and future treatment of non-small cell lung cancer. Semin Oncol. 2002;29(3 Suppl 12):17–21. doi:10.1053/sonc.2002.34259
8. Kobayashi S, Okada S, Hasumi T, Sato N, Fujimura S. Combined modality therapy including surgery for stage III small-cell lung cancer on the basis of the sensitivity assay in vitro. Surg Today. 2000;30(2):127–133. doi:10.1007/PL00010060
9. Wood KW, Cornwell WD, Jackson JR. Past and future of the mitotic spindle as an oncology target. Curr Opin Pharmacol. 2001;1(4):370–377. doi:10.1016/S1471-4892(01)00064-9
10. Kartner N, Riordan JR, Ling V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science (New York, NY). 1983;221(4617):1285–1288. doi:10.1126/science.6137059
11. McGrogan BT, Gilmartin B, Carney DN, McCann A. Taxanes, microtubules and chemoresistant breast cancer. Biochim Biophys Acta. 2008;1785(2):96–132. doi:10.1016/j.bbcan.2007.10.004
12. Ogihara T, Kikuchi K, Matsuoka H, et al. The Japanese society of hypertension guidelines for the management of hypertension (JSH 2009). Hypertens Res. 2009;32(1):3–107. doi:10.1038/hr.2009.34
13. Katz AM. Pharmacology and mechanisms of action of calcium-channel blockers. J Clin Hypertens. 1986;2(3 Suppl):28S–37S.
14. Elliott WJ, Ram CV. Calcium channel blockers. J Clin Hypertens (Greenwich). 2011;13(9):687–689. doi:10.1111/j.1751-7176.2011.00513.x
15. Coogan PF. Calcium-channel blockers and breast cancer: a hypothesis revived. JAMA Intern Med. 2013;173(17):1637–1638. doi:10.1001/jamainternmed.2013.9069
16. Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci. 2000;11(4):265–283. doi:10.1016/S0928-0987(00)00114-7
17. Tsuruo T, Iida H, Nojiri M, Tsukagoshi S, Sakurai Y. Circumvention of vincristine and Adriamycin resistance in vitro and in vivo by calcium influx blockers. Cancer Res. 1983;43(6):2905–2910.
18. Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41(5):1967–1972.
19. Ozols RF, Cunnion RE, Klecker RW
20. Cairo MS, Siegel S, Anas N, Sender L. Clinical trial of continuous infusion verapamil, bolus vinblastine, and continuous infusion VP-16 in drug-resistant pediatric tumors. Cancer Res. 1989;49(4):1063–1066.
21. Salmon SE, Dalton WS, Grogan TM, et al. Multidrug-resistant myeloma: laboratory and clinical effects of verapamil as a chemosensitizer. Blood. 1991;78(1):44–50. doi:10.1182/blood.V78.1.44.44
22. Timcheva CV, Todorov DK. Does verapamil help overcome multidrug resistance in tumor cell lines and cancer patients? J Chemother. 1996;8(4):295–299. doi:10.1179/joc.1996.8.4.295
23. Belpomme D, Gauthier S, Pujade-lauraine E, et al. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann Oncol. 2000;11(11):1471–1476. doi:10.1023/A:1026556119020
24. Millward MJ, Cantwell BM, Munro NC, Robinson A, Corris PA, Harris AL. Oral verapamil with chemotherapy for advanced non-small cell lung cancer: a randomised study. Br J Cancer. 1993;67(5):1031–1035. doi:10.1038/bjc.1993.189
25. Holmes S, Griffith EJ, Musto G, Minuk GY. Antihypertensive medications and survival in patients with cancer: a population-based retrospective cohort study. Cancer Epidemiol. 2013;37(6):881–885. doi:10.1016/j.canep.2013.09.001
26. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi:10.1016/j.cell.2011.10.026
27. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. doi:10.1038/s41580-018-0003-4
28. Williams A, Sarkar S, Cuddon P, et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol. 2008;4(5):295–305. doi:10.1038/nchembio.79
29. Lu MC, Lai NS, Yu HC, Hsieh SC, Tung CH, Yu CL. Nifedipine suppresses Th1/Th2 cytokine production and increased apoptosis of anti-CD3 + anti-CD28-activated mononuclear cells from patients with systemic lupus erythematosus via calcineurin pathway. Clin Immunol. 2008;129(3):462–470. doi:10.1016/j.clim.2008.08.001
30. Kaddour-djebbar I, Choudhary V, Lakshmikanthan V, et al. Diltiazem enhances the apoptotic effects of proteasome inhibitors to induce prostate cancer cell death. J Pharmacol Exp Ther. 2012;341(3):646–655. doi:10.1124/jpet.111.188151
31. Roger S, Le Guennec JY, Besson P. Particular sensitivity to calcium channel blockers of the fast inward voltage-dependent sodium current involved in the invasive properties of a metastatic breast cancer cell line. Br J Pharmacol. 2004;141(4):610–615. doi:10.1038/sj.bjp.0705649
32. Chiu LY, Hu ME, Yang TY, et al. Immunomodulatory protein from ganoderma microsporum induces pro-death autophagy through Akt-mTOR-p70S6K pathway inhibition in multidrug resistant lung cancer cells. PLoS One. 2015;10(5):e0125774. doi:10.1371/journal.pone.0125774
33. Boulares AH, Yakovlev AG, Ivanova V, et al. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274(33):22932–22940. doi:10.1074/jbc.274.33.22932
34. Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi:10.4161/auto.5338
35. Hsin IL, Sheu GT, Jan MS, et al. Inhibition of lysosome degradation on autophagosome formation and responses to GMI, an immunomodulatory protein from Ganoderma microsporum. Br J Pharmacol. 2012;167(6):1287–1300. doi:10.1111/j.1476-5381.2012.02073.x
36. Staskiewicz L, Thorburn J, Morgan MJ, Thorburn A. Inhibiting autophagy by shRNA knockdown: cautions and recommendations. Autophagy. 2013;9(10):1449-1450.
37. Mauthe M, Orhon I, Rocchi C, et al. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435–1455. doi:10.1080/15548627.2018.1474314
38. Bundy JD, Li C, Stuchlik P, et al. Systolic blood pressure reduction and risk of cardiovascular disease and mortality: a systematic review and network meta-analysis. JAMA Cardiol. 2017;2(7):775–781. doi:10.1001/jamacardio.2017.1421
39. Padwal RS, Bienek A, McAlister FA, Campbell NR; Outcomes Research Task Force of the Canadian Hypertension Education P. Epidemiology of hypertension in canada: an update. Can J Cardiol. 2016;32(5):687–694. doi:10.1016/j.cjca.2015.07.734
40. Lawes CM, Vander Hoorn S, Rodgers A; International Society of H. Global burden of blood-pressure-related disease, 2001. Lancet. 2008;371(9623):1513–1518. doi:10.1016/S0140-6736(08)60655-8
41. McManus RJ, Caulfield M, Williams B. National Institute for H, Clinical E. NICE hypertension guideline 2011: evidence based evolution. BMJ. 2012;344:e181. doi:10.1136/bmj.e181
42. Grimaldi-bensouda L, Klungel O, Kurz X, et al. Calcium channel blockers and cancer: a risk analysis using the UK Clinical Practice Research Datalink (CPRD). BMJ Open. 2016;6(1):e009147. doi:10.1136/bmjopen-2015-009147
43. Raebel MA, Zeng C, Cheetham TC, et al. Risk of breast cancer with long-term use of calcium channel blockers or angiotensin-converting enzyme inhibitors among older women. Am J Epidemiol. 2017;185(4):264–273. doi:10.1093/aje/kww217
44. Chang CH, Chiang CH, Yen CJ, Wu LC, Lin JW, Lai MS. Antihypertensive agents and the risk of breast cancer in women aged 55 years and older: a nested case-control study. J Hypertens. 2016;34(3):
45. Li W, Shi Q, Wang W, Liu J, Li Q, Hou F. Calcium channel blockers and risk of breast cancer: a meta-analysis of 17 observational studies. PLoS One. 2014;9(9):e105801. doi:10.1371/journal.pone.0105801
46. Li CI, Daling JR, Tang MT, Haugen KL, Porter PL, Malone KE. Use of antihypertensive medications and breast cancer risk among women aged 55 to 74 years. JAMA Intern Med. 2013;173(17):1629–1637. doi:10.1001/jamainternmed.2013.9071
47. Sun H, Zhuang RY, Li T, Zheng YT, Cai WM. No association between calcium channel blockers and survival in patients with cancer: a systematic review and meta-analysis. Asian Pac J Cancer Prev. 2016;17(8):3917–3921.
48. Chen L, Chubak J, Boudreau DM, Barlow WE, Weiss NS, Li CI. Use of antihypertensive medications and risk of adverse breast cancer outcomes in a SEER-medicare population. Cancer Epidemiol Biomarkers Prev. 2017;26:1603–1610. doi:10.1158/1055-9965.EPI-17-0346
49. Kim SA, Moon H, Roh JL, et al. Postdiagnostic use of beta-blockers and other antihypertensive drugs and the risk of recurrence and mortality in head and neck cancer patients: an observational study of 10,414 person-years of follow-up. Clin Transl Oncol. 2017;19(7):826–833. doi:10.1007/s12094-016-1608-8
50. Wong MC, Tam WW, Lao XQ, et al. The incidence of cancer deaths among hypertensive patients in a large Chinese population: a cohort study. Int J Cardiol. 2015;179:178–185. doi:10.1016/j.ijcard.2014.10.028
51. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi:10.1083/jcb.200507002
52. Cohen-kaplan V, Livneh I, Avni N, et al. p62- and ubiquitin-dependent stress-induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci U S A. 2016;113(47):E7490–E7499. doi:10.1073/pnas.1615455113
53. Kania E, Pajak B, O’prey J, et al. Verapamil treatment induces cytoprotective autophagy by modulating cellular metabolism. FEBS J. 2017;284(9):1370–1387. doi:10.1111/febs.2017.284.issue-9
54. Huang J, Duan Q, Fan P, et al. Clinical evaluation of targeted arterial infusion of verapamil in the interventional chemotherapy of primary hepatocellular carcinoma. Cell Biochem Biophys. 2011;59(2):127–132. doi:10.1007/s12013-010-9125-9
55. Ning Z, Chen D, Liu A, et al. Efficacy of chemotherapy combined with targeted arterial infusion of verapamil in patients with advanced gastric cancer. Cell Biochem Biophys. 2014;68(1):195–200. doi:10.1007/s12013-013-9689-2
56. Huang J, Zhang T, Ma K, et al. Clinical evaluation of targeted arterial perfusion of verapamil and chemotherapeutic drugs in interventional therapy of advanced lung cancer. Cancer Chemother Pharmacol. 2013;72(4):889–896. doi:10.1007/s00280-013-2271-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.