Back to Journals » Drug Design, Development and Therapy » Volume 8
Anticancer, antioxidant activities, and DNA affinity of novel monocationic bithiophenes and analogues
Authors Ismail M, Arafa R, Youssef M, El-Sayed W ![]()
Received 20 May 2014
Accepted for publication 7 July 2014
Published 29 September 2014 Volume 2014:8 Pages 1659—1672
DOI https://doi.org/10.2147/DDDT.S68016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Mohamed A Ismail,1,2 Reem K Arafa,3 Magdy M Youssef,1,2 Wael M El-Sayed1,4
1Departments of Chemistry and Biological Sciences, College of Science, King Faisal University, Hofuf, Saudi Arabia; 2Department of Chemistry, Faculty of Science, Mansoura University, Mansoura, Egypt; 3Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 4Department of Zoology, Faculty of Science, University of Ain Shams, Abbassia, Cairo, Egypt
Abstract: A series of 15 monocationic bithiophenes and isosteres were prepared and subjected to in vitro antiproliferative screening using the full National Cancer Institute (NCI)-60 cell line panel, representing nine types of cancer. Among the nine types of cancer involved in a five-dose screen, non-small cell lung and breast cancer cell lines were the most responsive to the antiproliferative effect of the tested compounds, especially cell lines A549/ATCC, NCI-H322M, and NCI-H460, whereas compounds 1a, 1c, 1d, and 7 exhibited potent activity, with GI50 values (drug concentration that causes 50% inhibition of cell growth) from less than 10 nM to 102 nM. In addition, compounds 1c and 1d gave GI50 values of 73 nM and 79 nM, respectively, against the MDA-MB-468 breast cancer cell line. Structure–activity relationship findings indicated that the mononitriles were far less active than their corresponding monoamidines and, within the amidines series, the bioisosteric replacement of a thiophene ring by a furan led to a reduction in antiproliferative activity. Also, molecular manipulations, involving substitution on the phenyl ring, or its replacement by a pyridyl, or alteration of the position of the amidine group, led to significant alteration in antiproliferative activity. On the other hand, DNA studies demonstrated that these monoamidine bichalcophenes have promising ability to cleave the genomic DNA. These monoamidines show a wide range of DNA affinities, as judged from their DNA cleavage effect, which are remarkably sensitive to all kinds of structural modifications. Finally, the novel bichalcophenes were tested for their antioxidant property by the ABTS (2,2'-azino- bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt) assay, as well as lipid and nitric oxide scavenging techniques, and were found to exhibit good-to-potent antioxidant abilities.
Keywords: bithiophenes, anticancer, DNA cleavage, antioxidant, Suzuki coupling, Stille coupling
Introduction
According to a recent report by the World Health Organization, there are now more than 10 million cases of cancer per year worldwide. Statistics from the American Cancer Society showed that cancer is the third most lethal disease after cardiovascular diseases and infectious and parasitic diseases.1–4 Cancer refers to a diversity of diseases, characterized by the uncontrolled proliferation of cells into a different form, against the normal complement of the organism.5 The continuous proliferation of cancer cells develops into tumor tissues and may spread across to other organs. The principal need in the chemoprevention of cancer remains the discovery of new agents that are effective and safe.
Sulfur-containing molecules are of particular interest, being widely spread in nature. For example, diallyldisulfide, found in garlic, appears to be potent in protecting against skin, colon, and lung cancer.6–8 Also, biotin, known as vitamin B7, contains a tetrahydrothiazole ring as an integral part of its structure and acts as an essential cofactor in integral biological metabolic pathways, such as gluconeogenesis, fatty acid synthesis, and amino acid anabolism.9 Bithiophenes and oligothiophenes and their derivatives are important synthetic precursors for biologically active materials.10–13 Oligothiophene compound α-terthienyl (I) (Figure 1) is considered an anti-infective, antiviral, and photosensitizing agent.14,15
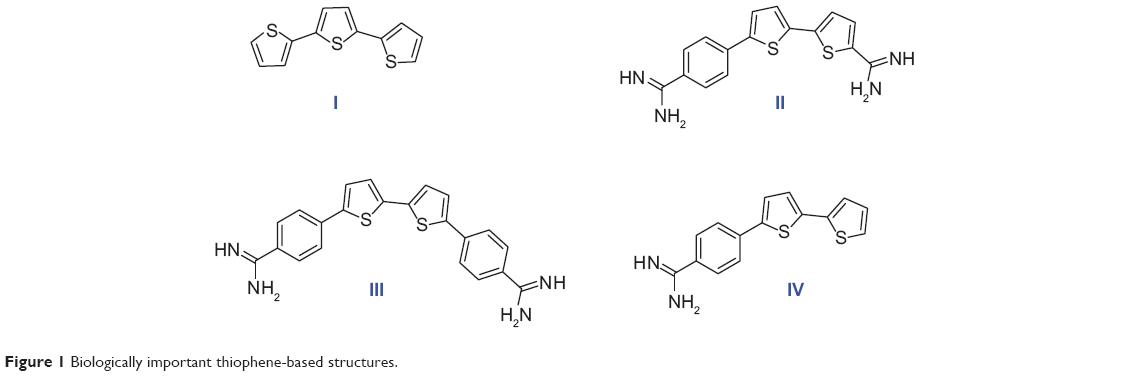 | Figure 1 Biologically important thiophene-based structures. |
A number of compounds in this class of aromatic cationic molecules have been shown to bind to the minor groove of DNA at AT-rich sites, and the details of their interaction with the minor groove have been elucidated from biophysical studies,16–19 including crystal structures.20–22 It is hypothesized that these types of molecules exert their biological activity by first binding to DNA and then by inhibiting one or more of several DNA-dependent enzymes, or perhaps by direct inhibition of transcription.23,24 Recently, bithiophene diamidines (II) showed good DNA binding affinity and potent antiprotozoal activity,10 whereas, a symmetrical diamidinophenylbithiophene (III) exhibited cellular activity by binding to DNA and inhibiting binding of erythroblast transformation-specific (ETS)-related gene, an ETS family transcription factor that is commonly overexpressed or translocated in leukemia and prostate cancer.25,26 Although thiophene and furan rings are known for their broad biological activities,27 we have shown that replacing one or more furan ring with a thiophene one increased the DNA binding affinity,10 antibacterial, and antioxidant activities.11,28
Cancer occurs when genetic mutations accumulate in cells during the replication of genetic material, allowing cells to undergo transformation. Recently, a promising antimutagenic activity against sodium azide and benzo[a]pyrene-induced mutagenicity was shown for many of fluorinated and non-fluorinated bichalcophenes.29,30 In particular, monocationic bithiophene (IV) has the highest antimutagenic activity, which is superior to other tested bichalcophenes.29
As part of a research program directed toward the discovery of anticancer drugs, and due to the importance of bithiophenes and oligothiophenes as therapeutic agents, and their unusual DNA binding affinity, we have decided to synthesize new isosteres of compound IV, which showed potent anticancer activity, by changing the location of the amidine group to be attached directly to the bithiophene moiety. Modifications have also included the replacement of a phenyl ring with 4-methoxyphenyl, 3,5-dimethoxyphenyl, and thiophene units. In this report, DNA cleavage and antioxidant activity testing results of the prepared series of cationic bithiophenes and analogues have also been reported and discussed.
Experimental methods
Chemistry
Melting points (mps) were recorded using a Gallenkamp melting-point apparatus and were uncorrected. Thin-layer chromatography analysis was carried out on silica gel 60 F254 precoated aluminum sheets and detected under ultraviolet (UV) light. Infrared spectra were recorded using a KBr wafer technique on a Shimadzu 5,800 Fourier transform infrared spectrometer. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded using a Varian Mercury VX 300 spectrometer and a Bruker Avance 400 MHz spectrometer, and chemical shifts (δ) were in parts per million, relative to tetramethylsilane as an internal standard. Mass spectra were recorded on a gas chromatography mass spectrometer (Shimadzu GPMS-QP2010 Plus). Elemental analyses were performed using a PerkinElmer 2,400 analyzer at the microanalytical laboratories of the Faculty of Science, Cairo University, and were within ±0.4 of the theoretical values (Table 1). All chemicals and solvents were purchased from Sigma-Aldrich Chemical Company and Thermo Fisher Scientific.
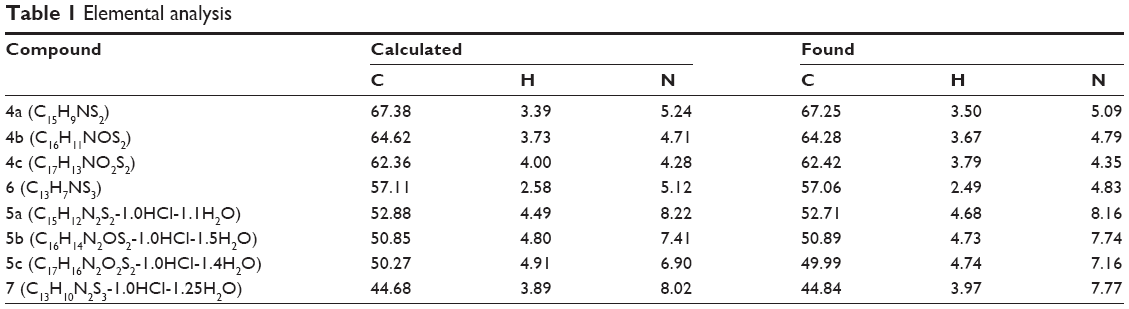 | Table 1 Elemental analysis |
General procedure for preparation of mononitrile bithiophenes 4a–4c
5′-phenyl-2,2′-bithiophene-5-carbonitrile (4a)
To a stirred solution of 5′-bromo-2,2′-bithiophene-5-carbonitrile (2.7 g, 10 mmol) and tetrakis(triphenylphosphine)palladium (190 mg) in toluene (20 mL), 10 mL of a 1.5 M aqueous solution of NaHCO3 was added, followed by phenylboronic acid (1.46 g, 12 mmol) in 5 mL of methanol. The vigorously stirred mixture was warmed to 80°C for 16 hours. The product was extracted with ethyl acetate (250 mL, 3×). The organic layer was passed through celite to remove palladium, dried (Na2SO4), and then concentrated to dryness under reduced pressure, to afford the mononitrile 4a in 78% yield, mp 133°C–134°C (EtOH). Rf =0.69, petroleum ether (60°C–80°C)-EtOAc (8:2). IR (KBr) ν’ 3,093, 3,021 (CH), 2,216 (CN), 1,610, 1,594, 1,581 (C=C) cm−1. 1H NMR (DMSO-d6); δ 7.35–7.59 (m, 6H), 7.69–7.72 (m, 2H), 7.96 (d, J=4.5 Hz, 1H). MS (EI) m/e (relative intensity [rel int]); 267 (M+, 100), 233 (21). 152 (41). Analysis (C15H9NS2) C, H, N.
5′-(4-methoxyphenyl)-2,2′-bithiophene-5-carbonitrile (4b)
A yellow solid in 81% yield, Rf =0.48, petroleum ether (60°C–80°C)-EtOAc (8:2), mp 154°C–155°C (EtOH/DMF). IR (KBr) ν’ 3,089, 2,966, 2,938 (CH), 2,217 (CN), 1,603, 1,594, 1,570 (C=C) cm−1. 1H NMR (DMSO-d6); δ 3.76 (s, 3H), 6.98 (d, J=8.4 Hz, 2H), 7.43–7.46 (m, 2H), 7.51 (d, J=3.8 Hz, 1H), 7.62 (d, J=8.4 Hz, 2H), 7.92 (d, J=4.5 Hz, 1H). MS (EI) m/e ([rel int]); 297 (M+, 100), 282 (73), 254 (25). Analysis (C16H11NOS2) C, H, N.
5′-(3,5-dimethoxyphenyl)-2,2′-bithiophene-5-carbonitrile (4c)
A yellow solid in 76% yield, mp 160°C–161°C (EtOH/ DMF). Rf =0.42, petroleum ether (60°C–80°C)-EtOAc (8:2). IR (KBr) ν’ 3,097, 3,065, 2,993, 2,940 (CH), 2,214 (CN), 1,585, 1,551 (C=C) cm−1. 1H NMR (DMSO-d6); δ 3.76 (s, 6H), 6.47 (s, 1H), 6.78 (s, 2H), 7.47 (d, J=3.8 Hz, 1H), 7.54 (d, J=3.8 Hz, 1H), 7.59 (d, J=3.8 Hz, 1H), 7.92 (d, J=3.8 Hz, 1H). MS (EI) m/e (rel int); 327 (M+, 100), 296 (5). Analysis (C17H13NO2S2) C, H, N.
α-terthienyl-2-carbonitrile (6)
A mixture of 5′-bromo-2,2′-bithiophene-5-carbonitrile (2.7 g, 10 mmol), 2-(tri-n-butylstannyl)thiophene (4.13 g, 11 mmol), and tetrakis(triphenylphosphine)palladium (190 mg) in dry dioxane (20 mL) was heated at reflux (100°C–110°C) for 24 hours. The solvent was evaporated under reduced pressure, the solid was dissolved in ethyl acetate, and the solution was passed through celite to remove palladium. The solution was evaporated, and the solid was filtered and recrystallized from ethanol/pet ether to give compound 6 as a yellow solid in 67% yield, mp 127°C–128°C. Rf =0.67, petroleum ether (60°C–80°C)-EtOAc (8:2). IR (KBr) ν’ 3,087, 3,065 (CH), 2,215 (CN), 1,601, 1,525 (C=C) cm−1. 1H NMR (DMSO-d6); δ 7.12 (d, J=4.8 Hz, 1H), 7.36 (d, J=3.9 Hz, 1H), 7.42–7.60 (m, 4H), 7.96 (d, J=3.9 Hz, 1H). MS (EI) m/e (rel int); 273 (M+, 100). Analysis (C13H7NS3) C, H, N.
General procedure for preparation of monoamidine bithiophenes and isosters 5a–5c and 7
5′-phenyl-2,2′-bithiophene-5-amidine hydrochloride salt (5a)
The mononitrile bithiophene 4a (401 mg, 1.5 mmol), suspended in freshly distilled THF (8 mL), was treated with LiN(TMS)2 (1 M solution in THF, 4 mL, 4 mmol) and the reaction was allowed to stir overnight. The reaction mixture was then cooled to 0°C, to which was added hydrogen chloride ethanolic solution (12 mL, 1.25 M), whereupon a precipitate started forming. The mixture was left to run overnight, whereafter it was diluted with ether, and the resultant solid was collected by filtration. The monoamidine was purified by neutralization with 1N NaOH, followed by filtration of the resultant solid and washing with water. Finally, the free base was stirred with hydrogen chloride ethanolic solution overnight, diluted with ether, and the solid formed was filtered and dried to give the monoamidine 5a hydrochloride salt as a yellow solid in 75% yield, mp 201°C–202°C. IR (KBr) ν’ 3,351, 3,200 (NH, NH2), 3,081 (CH), 1,657, 1,567 (C=C, C=N, NH bending) cm−1. 1H NMR (DMSO-d6); δ 7.37–7.72 (m, 8H), 8.06 (d, J=3.9 Hz, 1H), 9.21 (br s, 4H, exchangeable with D2O). 13C NMR (DMSO-d6); δ 125.4, 125.7, 125.9, 127.0, 128.3, 128.9, 129.7, 133.2, 134.0, 136.0, 144.3, 145.3, 158.8. MS (EI) m/e (rel int); 284 (M+, 100), 268 (46). Analysis (C15H12N2S2-1.0HCl-1.1H2O) C, H, N.
5′-(4-methoxyphenyl)-2,2′-bithiophene-5-amidine hydrochloride salt (5b)
A yellow solid in 70% yield, mp 258°C–260°C. IR (KBr) ν’ 3,351, 3,200 (NH, NH2), 3,081 (CH), 1,657, 1,567 (C=C, C=N, NH bending) cm−1. 1H NMR (DMSO-d6); δ 3.77 (s, 3H), 7.01 (d, J=8.4 Hz, 2H), 7.45–7.66 (m, 5H), 8.04 (d, J=3.8 Hz, 1H), 9.38 (br s, 2H, exchangeable with D2O), 9.08 (br s, 2H, exchangeable with D2O). 13C NMR (DMSO-d6); δ 55.8, 115.1, 124.5, 125.0, 125.9, 126.5, 127.4, 128.4, 132.8, 136.0, 144.6, 145.6, 158.7, 160.0. MS (EI) m/e (rel int); 314 (M+, 100), 297 (33). Analysis (C16H14N2OS2-1.0HCl-1.5H2O) C, H, N.
5′-(3,5-dimethoxyphenyl)-2,2′-bithiophene-5-amidine hydrochloride salt (5c)
A golden-yellow solid in 68% yield, mp 251 5°C–252.5°C. IR (KBr) ν’ 3,321, 3,200, (NH, NH2), 3,093, 2,965 (CH), 1,660, 1,592 (C=C, C=N, NH bending) cm−1. 1H NMR (DMSO-d6); δ 3.78 (s, 6H), 6.49 (s, 1H), 6.80 (s, 2H), 7.54 (d, J=3.8 Hz, 1H), 7.57 (d, J=3.8 Hz, 1H), 7.62 (d, J=3.8 Hz, 1H), 8.03 (d, J=3.8 Hz, 1H), 9.34 (br s, 2H, exchangeable with D2O), 9.07 (br s, 2H, exchangeable with D2O). 13C NMR (DMSO-d6); δ 55.9, 100.7, 104.2, 125.5, 126.4, 127.0, 128.2, 134.1, 135.0, 136.0, 144.3, 145.1, 158.8, 161.5. MS (EI) m/e (rel int); 344 (M+, 100), 327 (76). Analysis (C17H16N2O2S2-1.0HCl-1.4H2O) C, H, N.
α-terthienyl-2-amidine hydrochloride salt (7)
A yellow solid in 71% yield, mp 199°C–200.5°C. IR (KBr) ν’ 3,326, 3,200, 3,079 (NH, NH2, CH), 1,655, 1,568 (C=N, C=C, NH bending) cm−1. 1H NMR (DMSO-d6); δ 7.13 (d, J=3.9 Hz, 1H), 7.38 (d, J=3.9 Hz, 1H), 7.42–7.61 (m, 4H), 8.09 (d, J=3.9 Hz, 1H), 9.15 (br s, 2H, exchangeable with D2O), 9.42 (br s, 2H, exchangeable with D2O). 13C NMR (DMSO-d6); δ 125.5, 125.6, 125.8, 127.0, 127.1, 128.2, 129.1, 133.3, 135.8, 136.0, 138.5, 144.0, 158.8. MS (EI) m/e (rel int); 290 (M+, 100), 274 (40). Analysis (C13H10N2S3-1.0HCl-1.25H2O) C, H, N.
Biology
In vitro antiproliferative screening
Experimental design
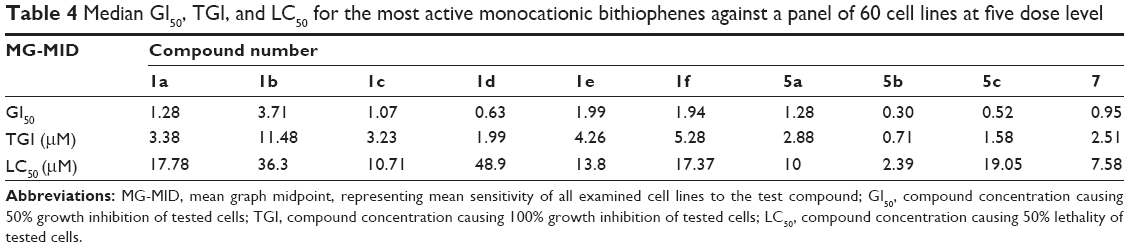
The structures of the synthesized compounds were sent to the National Cancer Institute (NCI) (Bethesda, MA, USA) for evaluation of their anticancer activity, where 15 compounds representing 12 monoamidines and three of their cyano precursors were chosen and subjected to a primary in vitro one-dose (10 μM) anticancer assay using the full NCI 60 cell panel, in accordance with the current protocol of the NCI Drug Evaluation Branch. In this protocol, all compounds submitted to the screen were tested initially at a single high dose (10 μM) in the full NCI panel of 60 cell lines, representing nine types of cancer (leukemia, non-small cell lung, colon, central nervous system [CNS], melanoma, ovarian, renal, prostate, and breast cancers). A 48-hour continuous drug exposure protocol was used, and a sulforhodamine B (SRB) protein assay was employed to estimate cell viability or growth.40–42 The data obtained are a mean graph of the percent growth of treated cells, presented as percentage growth inhibition (GI%) caused by the tested compounds (Table 2). Only compounds showing high GI% against the majority of the tested cell lines and satisfying predetermined threshold inhibition criteria set forth by the Development Therapeutic Program were selected for progression to the five-dose screen study against the panel of 60 cell lines, and the GI50 values against each cell line were determined (Table 3) as well as their mean GI50, TGI, and LC50 values against all tested cell lines, representing the mean graph midpoints (Table 4).
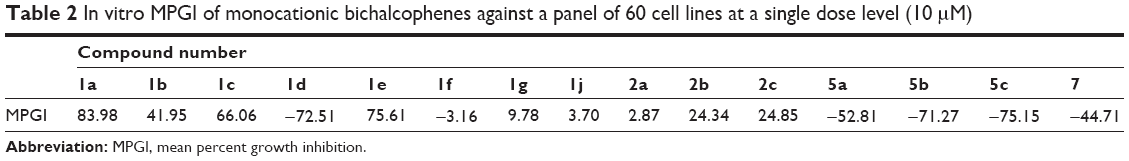 | Table 2 In vitro MPGI of monocationic bichalcophenes against a panel of 60 cell lines at a single dose level (10 μM) |
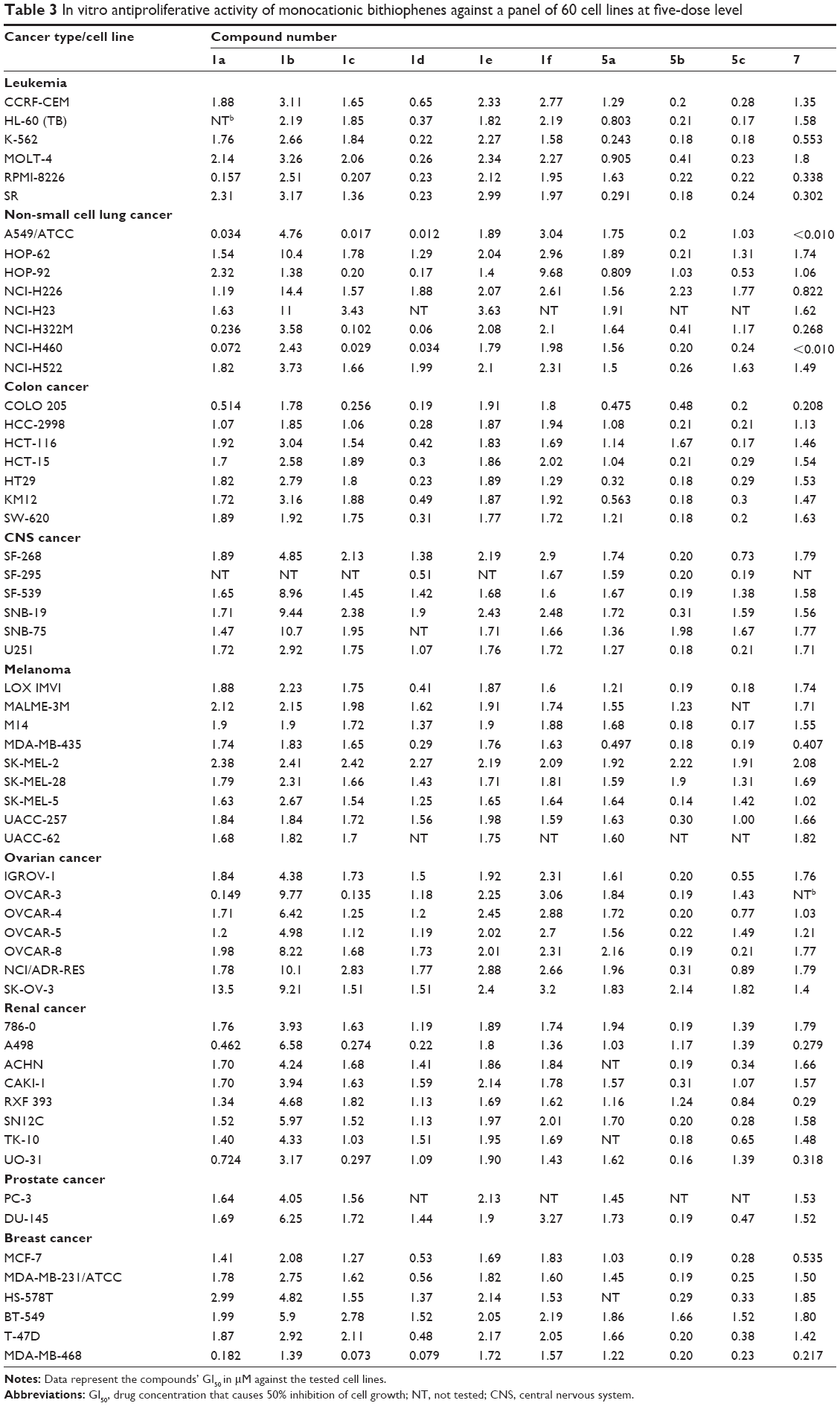 | Table 3 In vitro antiproliferative activity of monocationic bithiophenes against a panel of 60 cell lines at five-dose level |
 | Table 4 Median GI50, TGI, and LC50 for the most active monocationic bithiophenes against a panel of 60 cell lines at five dose level |
Experimental procedure
The experiment was performed under sterile conditions, where cell lines were grown in RPMI 1,640 medium (Gibco, NY, USA), supplemented with 10% fetal bovine serum (Biocell, CA, USA); 5×104 cells/mL were used to test the growth inhibition activity of the tested compounds. Cell culture (1.8 mL) containing a cell population of 6×104 cells/mL was pipetted into each well of a 96-multiwell microtiter plate (104 cells/well) 24 hours before treatment with the test compound, to allow attachment of cells to the wall of the plate. For determination of the GI50, five serial ten-fold dilutions of each compound, with concentrations ranging from 0.01 μM to 100 μM, were prepared in saline buffer and used for the purpose. Each compound was initially solubilized in dimethylsulfoxide (DMSO); however, each final dilution contains less than 1% DMSO. Solutions of the different concentrations (0.2 mL) were pipetted into separate wells of a microtiter tray, in duplicates, followed by an incubation period of 48 hours at 37°C in an atmosphere of 5% CO2. Thereafter, cells were fixed, washed, and stained for 30 minutes with 0.4% (w/v) SRB dissolved in 1% acetic acid. Excess, unbound dye was removed by four washes with 1% acetic acid, and attached stain was recovered with Tris–EDTA buffer. Color intensity was measured in an ELISA reader at a wavelength of 570 nm. Controls, containing only phosphate buffer (PB), saline, and DMSO, at identical dilutions, were also prepared and tested in the same manner.
Nuclease-like activity assay
Genomic DNA extracted from E. coli K91, according to reported methodology,43 was used for the nuclease-like activity assay of the tested bichalcophenes. DNA purity and concentration were examined at 260/280 nm, spectrophotometrically. The bichalcophene compounds were dissolved in DMSO (0.5 mg/mL) and 2 μg, 4 μg, and 6 μg were added individually to 2 μg of the E. coli DNA. DNA alone and DNA with DMSO were used as a control. The reactions were carried out at 37°C for 30 minutes. A solution of 10% w/v ficol 400, 0.06% w/v bromophenol blue, and 0.5% w/v SDS was added to the reaction mixtures prior to running the gel. The DNA was analyzed by using horizontal agarose gel electrophoresis. The electrophoresis was performed using 1.0% agarose gel at a constant voltage of 80 V for 60 minutes in TEA buffer (40 mM Tris–HCl pH 7.9, 5 mM sodium acetate, 1 mM EDTA). The agarose gels were stained with ethidium bromide (0.5 μg/mL), visualized under a UV transilluminator, and photographed using a digital camera.
ABTS radical cation decolorization assay
ABTS [2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) diammonium salt] forms a relatively stable free radical, which decolorizes in its non-radical form. The analysis of ABTS+ scavenging activity was measured spectrophotometrically, according to the method of Re et al.44 A mixture of ABTS (1 mL, 0.1 g/100 mL) and MnO2 (1.5 mL, 25 mg/mL), prepared in PB (pH 7.4, 100 mM) was vortexed, centrifuged, and decanted. Prior to assay, the resulting green-blue solution was diluted in PB to give an absorbance (Acontrol of ABTS radical solution) at 734 nm of 0.600 in a 1 cm cuvette, and equilibrated to 30°C (the temperature at which all the assays were done). Antioxidant activities of the tested bichalcophenes were estimated from the radical cations of ABTS. The absorbance (Atest) was measured upon the addition of 2 μg of the bichalcophenes in DMSO/PB (1:1 v/v) to the ABTS mixture. Ascorbic acid was used as a standard antioxidant (positive control). Blank samples were run by solvent without ABTS. The decrease in absorbance is expressed as % inhibition, which is calculated from the following formula:
% inhibition = ([Acontrol – Atest]/Acontrol) ×100 |
where Acontrol is the absorbance of the control reaction (containing all reagents except the test compound) and Atest is the absorbance of the test compound. All data about total antioxidant activity are the averages of triplicate analyses.
Nitric oxide scavenging assay
A nitric oxide scavenging activity assay was carried out according to the reported methodology,45 where sodium nitroprusside generates NO radicals, which interact with oxygen to produce nitrite ions. These can be estimated using the Greiss reagent [1% sulfanilamide, 2% H3PO4, and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride]. Scavengers of NO compete with oxygen, leading to less production of nitrite ions. For the experiment, sodium nitroprusside (10 mM) in PB saline was mixed with bichalcophenes (2 μg) and standard. The reaction mixtures were incubated at room temperature for 150 minutes at 25°C and, after that, 0.5 mL of Griess reagent was added. The absorbance of the pink color formed during diazotization of nitrite with sulfanilamide and subsequent coupling with naphthylethylenediamine was read at 546 nm and referred to the absorbance of standard solutions of sodium nitrite, treated in the same way with Griess reagent.46 Nitric oxide scavenging activity was calculated by the following equation:
% nitric oxide scavenging activity = [(Acontrol – Atest)/Ablank] ×100 |
where Acontrol is the absorbance of the control reaction (containing all reagents except the test compound) and Atest is the absorbance of the test compound. All data about total antioxidant activity are the average of triplicate analyses.
Total antioxidant activity determination in linoleic acid emulsion
The antioxidant activity of the bichalcophene compounds was determined according to the reported ferric thiocyanate methodology.47 The bichalcophenes (2 μg/mL in DMSO/PB v:v 1:1) was prepared and added to 2.5 mL of linoleic acid emulsion in PB. The linoleic acid emulsion was prepared by homogenizing 15.5 μL of linoleic acid, 17.5 mg of Tween-20 as emulsifier, and 5 mL PB. The control was composed of 2.5 mL of linoleic acid emulsion and 2.5 mL PB. The reaction mixture (5 mL) was incubated at 37°C for 30 minutes. The peroxide levels were determined by recording the absorbance at 500 nm after reaction with FeCl2 and SCN, at intervals during incubation. The peroxides formed during linoleic acid peroxidation oxidize Fe2+ to Fe3+, which forms a complex with SCN that has a maximum absorbance at 500 nm. Therefore, high absorbance indicates high linoleic acid oxidation. The solutions without added bichalcophenes or standards were used as blank samples. The percent inhibition of lipid peroxidation in linoleic acid emulsion was calculated by the following equation:
% inhibition of lipid peroxidation = 100 – (Atest/Acontrol ×100) |
where Acontrol is the absorbance of the control reaction, which contains only linoleic acid emulsion and PB. Atest is the absorbance of the sample in the presence of bichalcophene compounds. All data about total antioxidant activity are the average of triplicate analyses.
Results and discussion
Chemistry
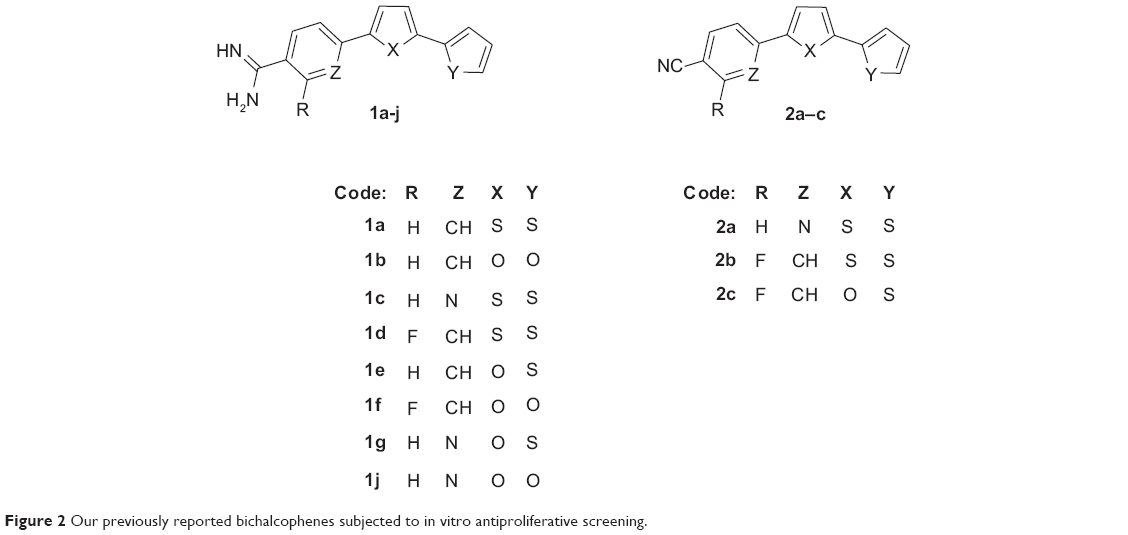
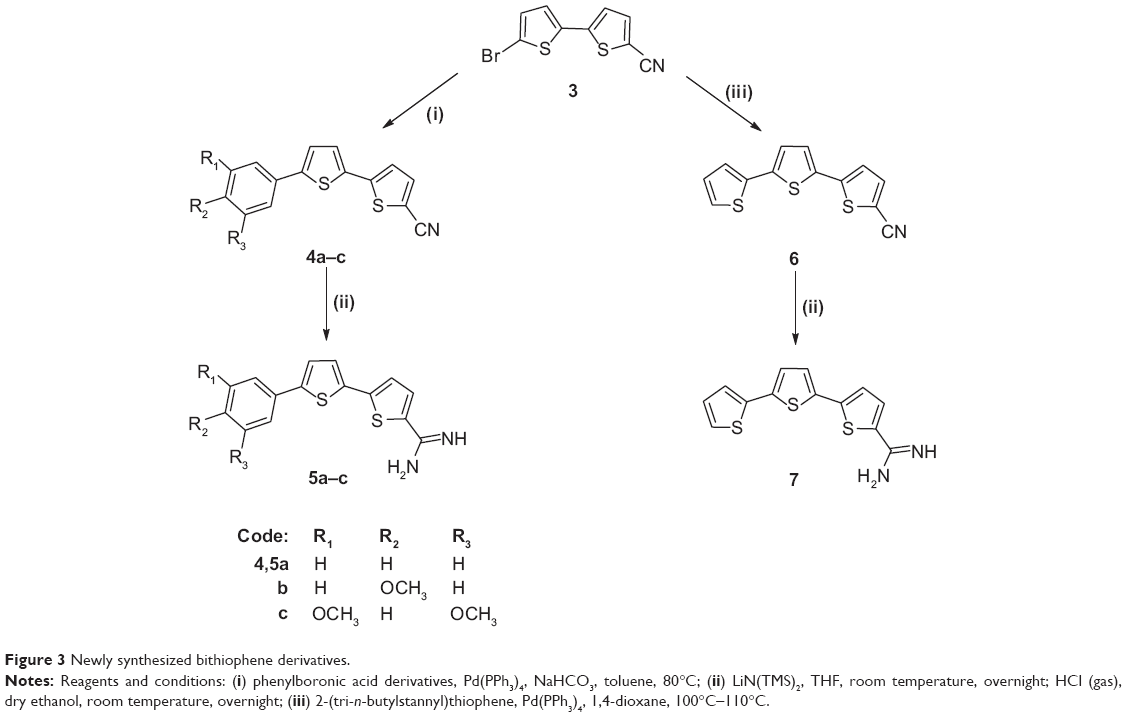
Monocationic bichalcophenes (1a–1j) and mononitrile bichalcophenes (2a–2c) (Figure 2) were prepared according to our previously-reported methodology.11,30 The synthesis of novel monocationic bithiophene derivatives (5a–5c) begins with the Suzuki coupling reaction of 5′-bromo-2,2′-bithiophene-5-carbonitrile with the corresponding phenylboronic acid derivatives to furnish the mononitrile bithiophene derivatives (4a–4c) (Figure 3). The mononitriles were converted to the corresponding monoamidines by the action of lithium trimethylsilylamide [LiN(TMS)2] in tetrahydrofuran (THF). Whereas, the α-terthienyl-2-amidine (7) was synthesized in two steps, starting with Stille coupling reaction of 5′-bromo-2,2′-bithiophene-5-carbonitrile with 2-(tri-n-butylstannyl)thiophene to yield α-terthienyl-2-carbonitrile, followed by direct treatment with LiN(TMS)2. The newly synthesized compounds (4a–4c, 5a–5c, 6, and 7) were assigned based on their spectral and elemental analyses.
 | Figure 2 Our previously reported bichalcophenes subjected to in vitro antiproliferative screening. |
 | Figure 3 Newly synthesized bithiophene derivatives. |
Biology
In vitro antiproliferative screening
A series of 15 monocationic bithiophenes and isosteres was subjected to an in vitro antiproliferative screening against a panel of 60 cell lines, representing nine cancer types (leukemia, non-small cell lung, colon, CNS, melanoma, ovarian, renal, prostate, and breast cancers) at the NCI. The standard practice is testing the chosen compounds at an initial high dose (10 μM). Thereafter, the compounds showing satisfactorily high percent growth inhibition (GI%) of the tested cell lines (ten, in this research work) are subjected to a five-dose screen against the panel of 60 cell lines, and their half maximal inhibitory concentration (IC50) values are determined.
Table 2 shows the mean percent growth inhibition (MPGI) results for the initial, single-dose screen against the panel of 60 cancer cell lines, exhibited by the 12 monoamidines plus three of the mononitrile intermediates that were selected by NCI for a primary evaluation of activity of these derivatives. As a beginning, it was found that the mononitrile precursors exemplified by 2a–2c displayed MPGIs of 2.87, 24.34, and 24.85, respectively. These mononitrile derivatives were far less active than their monoamidine derivatives (1c–1e), which elicited MPGI values of 66.06, 72.51, and 75.61, respectively. For that reason, the mononitrile derivatives were not progressed to the five-dose screen. These primary results clearly indicate that the presence of the amidine group plays a fundamental role in the antiproliferative activity of this class of compounds.
A close look at the structure–activity relationship findings among the 12 monoamidine derivatives gave insight about the structural features associated with enhanced antiproliferative activity of this class of compounds. First, it was shown that the bioisosteric replacement of a thiophene ring by a furan leads to a reduction in antiproliferative activity, the effect being more profound upon substitution of the two thiophene rings by furans. This is evidenced by the witnessed MPGI results of the bithiophene 1a (83.98), the thienofuran 1e (75.61), and the bifuran analogue1b (41.95). A similar pattern was observed for the analogues of 1a, 1e, and 1b, where the corresponding aza derivatives displayed MPGI of 66.06 (for the bithiophene 1c), 9.78 (for the thienofuran 1g), and 3.70 (for the bifuran 1j). Also, the fluorophenylbithiophene 1d and bifuran 1f counterparts followed the same behavior (MPGI: −72.51 and −3.16, respectively).
Studying the effect of substitution on, or isosteric replacement of, the phenyl ring of the phenylbichalcophenes, it was found that the aza analogues having a pyridyl ring in the place of the phenyl, suffered a decrease in MPGI, as evidenced by the isosteric pairs 1a/1c, 1e/1g, and 1b/1j. On the other hand, fluorine substitution on the phenyl ring leads to a profound enhancement of antiproliferative activity, as shown by the isosteric pairs 1a/1b and 1d/1f, even turning these derivatives from only being cytostatic to being cytotoxic (having negative MPGI values). This may be explained in terms of the fluorine substitution leading to the seemingly orthogonal effects of increasing local polarity and molecular hydrophobicity, consequently changing their pharmacokinetic properties.31,32
Attempting to understand the effect of positional isomerism on the antiproliferative effect of these derivatives, a number of compounds (5a–5c and 7), where the amidine moiety was moved from the phenyl to the thiophene side, were prepared. Biological screening data of 5a (MPGI =−52.81), the positional isomer of 1a (MPGI =83.98), demonstrated not only a potentiation of the antiproliferative efficacy but also a great enhancement of cytotoxic activity of this analogue. Indeed, this was the case for the 4-methoxyphenyl derivative 5b (MPGI =−71.27) and the 3,5-dimethoxyphenyl analogue5c (MPGI =−75.15), still having the amidine functionality on the thiophene ring and showing even more enhancement of their cytotoxic abilities compared to 5a. Finally, bioisosteric replacement of the phenyl ring in 5a by a third thiophene ring (in 7) led to a slight decrease in MPGI but still maintained the compound’s cytotoxic power.
On the other hand, Table 3 displays the individual GI50 values (drug concentration that causes 50% inhibition of cell growth) of the ten monoamidine derivatives that were promoted for a 5-dose screen, due to their high antiproliferative profiles demonstrated in the primary single-dose screening assay against a panel of 60 cancer cell lines. Among the nine types of cancer involved in this assay, non-small cell lung cancer cell lines were the most responsive to the antiproliferative effect of the tested monoamidines, especially the A549/ATCC, NCI-H322M, and NCI-H460 cell lines. Particularly, derivatives 1a, 1c, 1d, and 7 showed profound growth-deterring power against these three cell lines, with GI50 values in the nanomolar scale, in a range of less than 10 nM to 102 nM. In addition, compounds 1c and 1d showed good activity against the MDA-MB-468 breast cancer cell line, with GI50, values of 73 nM and 79 nM, respectively.
Finally, Table 4 depicts the median GI50, TGI (concentration causing 100% growth inhibition of tested cells), and LC50 (concentration causing 50% lethality of tested cells) of the compounds tested in the five-dose screen. The most active compounds, in compliance with the primary one-dose cell growth percent inhibition assay, were found to be 5b, 5c, 1d, and 7, displaying sub-micromolar GI50 values of 0.30 μM, 0.52 μM, 0.63 μM, and 0.95 μM, respectively. All the other compounds exhibited GI50 values below 2 μM, except for 1b (GI50 =3.71 μM), which was the least active in the primary single-dose screen assay. The most potent amidine in this study was 5b, showing a submicromolar TGI of 0.71 μM and LC50 of 2.39 μM. The terthienyl derivative 7 comes second, with a TGI of 2.51 μM and LC50 of 7.58 μM. In spite of the fact that 1d showed high potency with regard to its growth inhibitory abilities, reflected in its GI50 and TGI values (0.63 μM and 1.99 μM, respectively), yet it was the least cytotoxic in this series, with an LC50 of 48.9 μM. Finally, derivative 1b, which had the lowest MPGI among the ten monocationic compounds selected for the five-dose screening assay, was still the least potent with regards to the two measured parameters, GI50 and TGI.
DNA affinity and DNA cleavage ability
The nuclease-like and cleavage efficiency of the bichalcophene compounds, compared to that of controls, is due to their efficient DNA binding and degradation ability. The tested bichalcophenes were allowed to interact with genomic DNA isolated from Escherichia coli for cleavage study. In the present investigation, the bichalcophenes have shown increase in nuclease-like activities in a concentration-dependent manner, which illustrates their binding ability with genomic DNA (Figure 4). The nuclease-like activity of the bichalcophenes was investigated under aerobic conditions at 37°C and in the absence of any external additives. The experimental observations demonstrated that the bichalcophene compounds have promising ability toward the cleavage of genomic DNA. The obtained results showed that the examined monocationic bichalcophenes, at 4 μg and 6 μg, are strongly capable of degrading the DNA (Figure 4B and C), while at 2 μg, the monocationic bichalcophenes exhibited weak degradation effect on the DNA (Figure 4A). After 30 minutes of incubation, genomic DNA was strongly degraded at 4 μg of monocationic bithiophene derivatives 1a, 1d, 1e, 5b, 5c, and 7, while at 6 μg of these compounds, the genomic DNA was completely degraded (Figure 4B and C). On the other hand, the observed DNA smear with the monocationic bifuran derivatives 1b, 1f and, 1j, at 6 μg, may be due to their cytostatic binding aptitude. In contrast, the mononitrile bichalcophene compounds 2a, 2b, and 2c, at 2 μg, 4 μg, and 6 μg, did not exhibit any nuclease-like activity toward the genomic DNA, as represented (Figure 4A–C). The weak DNA cleavage ability of the mononitriles 2a–2c, compared against the corresponding monoamidines, may be attributed to the differences in pharmacokinetic properties of the nitrile group and the amidine group. These results indicate that the DNA cleavage activity of bichalcophene compounds depends on the structure and the concentration of the examined bichalcophenes. The monocationic bichalcophenes doses utilized in DNA cleavage were high, compared against cellular doses, because DNA cleavage is a qualitative study, while the cellular assay is quantitative. The data on DNA cleavage by bichalcophenes were in agreement with those previously reported.11 It was noted that replacing one or more of the furan rings with a thiophene ring results in an increase in the DNA cleavage affinity, as witnessed in the pattern displayed by the bifuran 1b, the thienofuran 1e, and the bithiophene 1a (Figure 4A–C). This result is consistent with the increased affinity of other thiophene-based amidines, in comparison to furan counterparts, and this may be attributed to the differences in van der Waals radii between S and O atoms.10,33 The weak DNA cleavage ability of the aza derivatives 1g and 1c, compared against the phenyl counterparts 1e and 1a (Figure 4B), is consistent with the decreased affinity of other aza analogue-based amidines, in comparison to phenyl counterparts.34 The reduction in the cleavage affinity of the aza analogues is consistent with their increased hydrophilic properties, as a result of the presence of nitrogen atoms. This result suggests the importance of the hydrophobic component for minor groove DNA binding affinity.16,34 The monocationic bichalcophene compounds have attracted special attention as endonuclease mimics, due to their interesting structural features and the importance of bichalcophene curvature structure for its interaction with the human telomeric quadruplex sequence, with a recently reported, unique CD exciton-type splitting pattern.35 A previously reported finding suggested that dicationic bifuran molecule DB832 can form a stacked molecular complex35 and can bind with the quadruplex.17–19 Two mechanisms have been hypothesized from the DNA study. The first hypothesis is that monocationic bichalcophenes may bind to the DNA, forming a complex,35 thus preventing DNA from further interacting with ethidium bromide; therefore, the monocationic bichalcophene-DNA adduct was not visualized in the agarose gel. The second postulated mechanism of DNA cleavage is through a hydrolytic pathway.11,36,37 We excluded the oxidative DNA damage mechanism, due to the good antioxidant properties of monocationic bichalcophenes. The present study demonstrates that the monocationic bichalcophenes 1a, 1d, 1f, 1e, 5b, 5c, and 7 have a significant nuclease-like activity toward the cleavage of genomic DNA, in the absence of any external additives, at low concentrations. The DNA cleavage activity without any additive is an appreciated feature of the monocationic bichalcophenes, as promising chemotherapeutic agents in anticancer treatments.
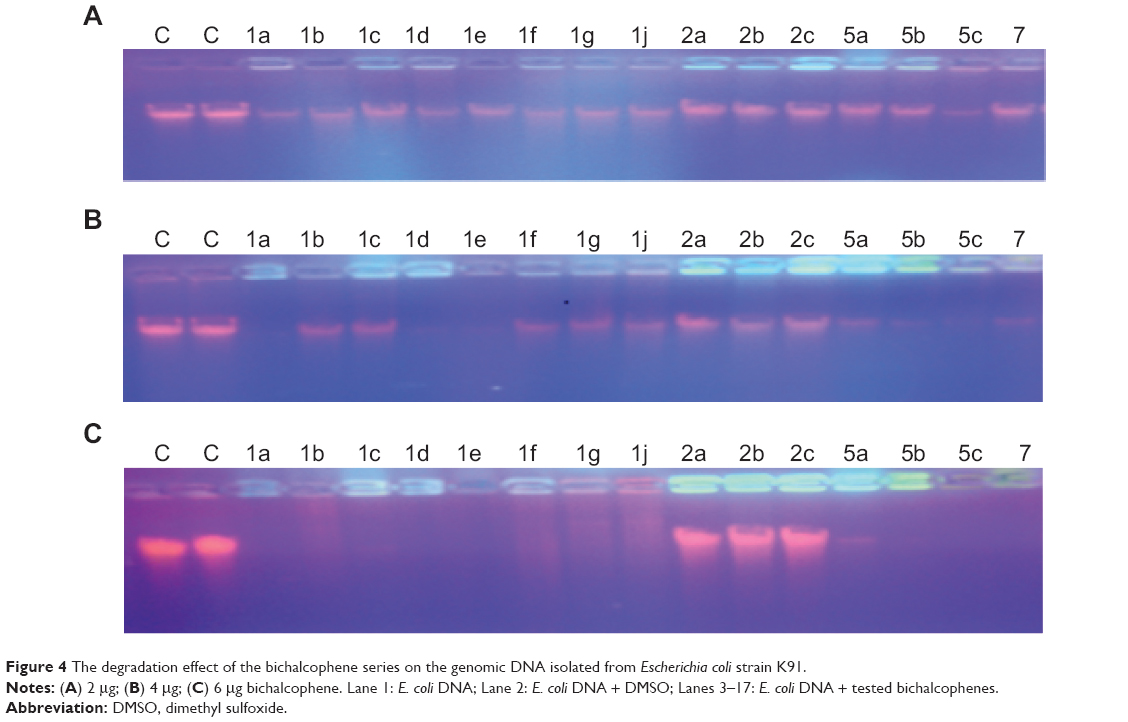 | Figure 4 The degradation effect of the bichalcophene series on the genomic DNA isolated from Escherichiacoli strain K91. |
Antioxidant activity
Since the DNA binding experiments conducted so far have revealed that the monocationic bichalcophenes exhibit good DNA binding affinity, it was considered worthwhile to study the antioxidant activity of this promising type of compound. The ABTS assay is widely used for assessing the ability of ABTS+ radical scavenging activity. Because of the presence of a relatively stable free radical cation, the ABTS assay shows a strong absorption band at 734 nm in the visible spectrum. The antioxidant activities of 15 bichalcophenes were evaluated, and the highest bichalcophenes scavenging effects with ABTS radical were in the order 1a>5a>5c>1d>1e>1b, as represented in Table 5, whereas, compounds 1c, 1f, 1g, and 7 displayed good activity. The monocationic bithiophenes 1a, 5a, 5c, and 1d exhibited better activity, compared against the other bichalcophenes. On the other hand, the mononitrile compounds 2a, 2b, and 2c showed weak scavenging activities with ABTS (Table 5), and this result is consistent with their weak DNA cleavage and antiproliferative activities. Thus, it would appear that introducing an amidine group, or replacing a furan ring with a thiophene unit, enhances the antioxidant properties of bichalcophene moieties.
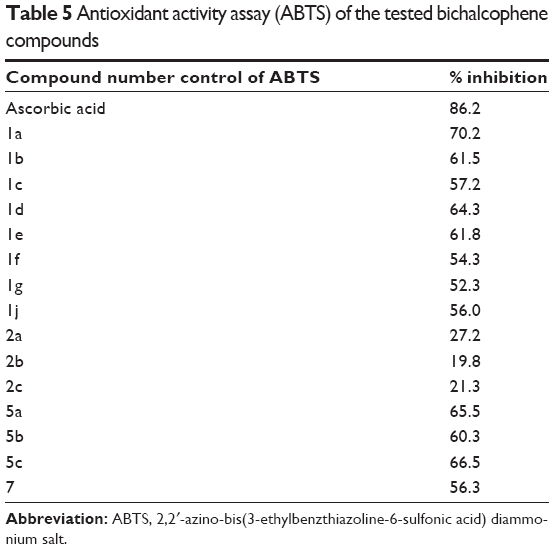 | Table 5 Antioxidant activity assay (ABTS) of the tested bichalcophene compounds |
The bichalcophene compounds were also tested for their antioxidant property by the nitric oxide scavenging activity assay technique (Figure 5). The monocationic bichalcophenes 1a, 1b, 1c, 1d, 1e, 5a, 5b, and 5c showed good radical scavenging activity. The other monocationic bichalcophenes 1f, 1g, 1j, and 7 showed moderate antioxidant activity, whereas, the mononitrile bichalcophenes 2a, 2b, and 2c displayed weak nitric oxide scavenging, compared against the control. The ability of the monocationic bichalcophenes to counteract the effect of NO formation represents an opportunity of considerable interest in preventing the severe effects of excessive NO generation in the biological system. This study demonstrated that monocationic bichalcophenes are potent scavengers for NO. Nitric oxide has been recognized to have a significant functional role in a diversity of physiological systems. Nitric oxide is a short-lived free radical, produced nonenzymatically, that causes damage in most biomolecules, including DNA and protein. Nitric oxide is involved in inflammation, cancer, and other pathological conditions.38 Nitric oxide generated from sodium nitroprusside reacts with oxygen to form nitrite. Therefore, monocationic bichalcophenes may inhibit nitrite formation by competing with oxygen to react with nitric oxide.
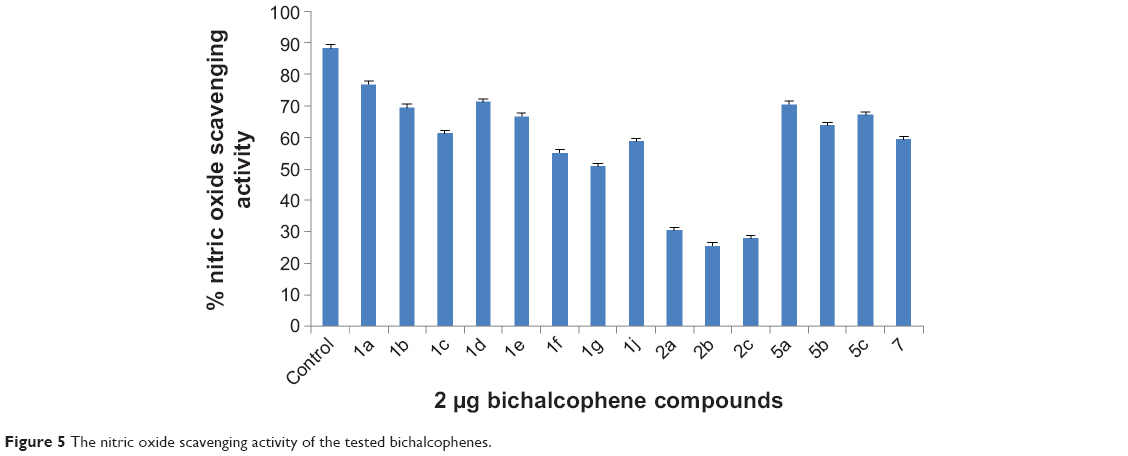 | Figure 5 The nitric oxide scavenging activity of the tested bichalcophenes. |
The presence of antioxidant substances in the antioxidant samples causes the reduction of the Fe3+/ferricyanide complex to the ferrous form. Therefore, Fe2+ can be monitored by measuring the formation of Perl’s Prussian blue at 700 nm.39 The yellow color of the test solution changes to various shades of green and blue, depending on the reducing power of antioxidant samples. The reducing capacity of the bichalcophene compounds may serve as a significant indicator of their potential antioxidant activity. As shown in Figure 6, the bichalcophenes had effective and powerful reducing power, using the potassium ferricyanide reduction method, when compared against the control. For measurement of the reductive ability of the bichalcophenes, the Fe3+−Fe2+ transformation was investigated in the presence of the bichalcophene compounds. The results on reducing power demonstrate the electron donor properties of the monocationic bichalcophenes, thereby scavenging free radicals by forming stable adducts. The outcome of the redox reaction is to terminate radical chain reactions that may otherwise be very damaging.
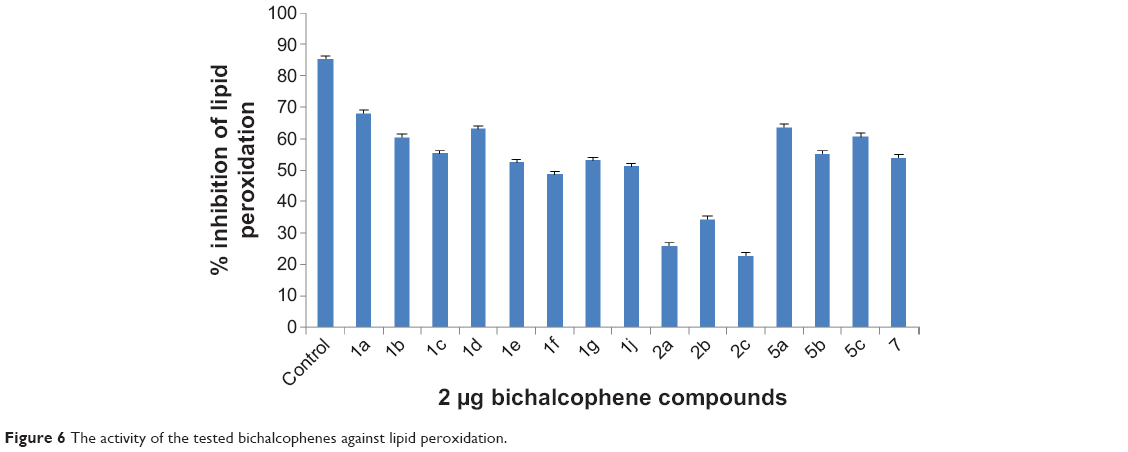 | Figure 6 The activity of the tested bichalcophenes against lipid peroxidation. |
The monocationic bichalcophenes exhibited effective antioxidant activity in the linoleic acid emulsion system. The effect of 2 μg of the bichalcophenes on the lipid peroxidation of linoleic acid emulsion is shown in Figure 6. The autoxidation of linoleic acid emulsion, without bichalcophenes or control, was accompanied by a rapid increase in peroxides. Consequently, these results clearly indicated that the monocationic bichalcophenes 1a, 5a, 1d, 5c, and 1b had good antioxidant activity, compared against the control.
Conclusion
Among the 12 monoamidine derivatives, the results of in vitro antiproliferative screening revealed that the bioisosteric replacement of a thiophene ring by a furan leads to a reduction in activity. Studying the effect of substitution on the phenyl ring of the bichalcophene derivatives, or its isosteric replacement by a pyridyl ring, has shown that the analogues having a pyridyl ring in place of the phenyl suffered a decrease in MPGI. In contrast, fluoro substitution on the phenyl ring led to a profound enhancement of the activity. The effect of positional isomerism on the antiproliferative effect of these derivatives was clearly observed. Interestingly, moving the amidine group from the phenyl to the thiophene side increased the activity. Among the nine types of cancer involved in the five-dose screen, non-small cell lung cancer cell lines were the most responsive to the antiproliferative effect of the tested compounds. The most active compounds were found to be 5b, 5c, 1d, and 7, displaying submicromolar GI50 values of 0.30 μM, 0.52 μM, 0.63 μM, and 0.95 μM, respectively. The monocationic bichalcophenes have demonstrated promising ability toward cleavage of genomic DNA. Moreover, antioxidant study results have indicated that this series of monocationic bichalcophenes have good antioxidant activity. In general, the monocationic bichalcophenes showed potent antiproliferative activity coupled with good antioxidant and nuclease mimic activities, which can, in part, be considered as the mechanism of action of these compounds. Given the potent in vitro anticancer activity of the monocationic bithiophene derivatives, further in vivo studies are merited, to clarify the real anticancer mechanism of this class of compounds.
Acknowledgments
This work was supported by the Deanship of Scientific Research, King Faisal University (Project Number 140058). The in vitro anticancer screening was done by the NCI, Betheseda, MD, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Ten statistical highlights in global public health. In: World Health Statistics 2007. Geneva: World Health Organization; 2007. | ||
Report sees 7.6 million cancer deaths worldwide in 2007. Chin Med J. 2008;121:26. | ||
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–2917. | ||
American Cancer Society. Cancer facts and Figures 2007. Atlanta, GA: American Cancer Society; 2007. | ||
Kufe DW, Pollock RE, Weichselbaum RR, Bast RC Jr, Gansler TS, Holland JF, Frei E III, editors. Holland-Frei Cancer medicine. 6th ed. Hamilton, Canada: BC Decker; 2003. | ||
Singh A, Shukla Y. Antitumor activity of diallyl sulfide in two-stage mouse skin model of carcinogenesis. Biomed Environ Sci. 1998;11(3):258–263. | ||
Sundaram SG, Milner JA. Diallyl disulfide induces apoptosis of human colon tumor cells. Carcinogenesis 1996;17(4):669–673. | ||
Kwon KB, Yoo SJ, Ryu DG, et al. Induction of apoptosis by diallyl disulfide through activation of caspase-3 in human leukemia HL-60 cells. Biochem Pharmacol. 2002;63(1):41–47. | ||
Zempleni J, Mock DM. Biotin biochemistry and human requirements. J Nutr Biochem. 1999;10(3):128–138. | ||
Ismail MA, El Bialy SA, Brun R, et al. Dicationic phenyl-2,2′-bichalcophenes and analogues as antiprotozoal agents. Bioorg Med Chem. 2011;19(2):978–984. | ||
Youssef MM, Al-Omair MA, Ismail MA. Synthesis, DNA affinity, and antimicrobial activity of 4-substituted phenyl-2,2′-bichalcophenes and aza-analogues. Med Chem Res. 2012;21(12):4074–4082. | ||
Batista DG, Pacheco MG, Kumar A, et al. Biological, ultrastructural effect and subcellular localization of aromatic diamidines in Trypanosoma cruzi. Parasitology. 2010;137(2):251–259. | ||
El-Sayed WM, Hussin WA, Ismail MA. Efficacy of two novel 2,2′-bifurans to inhibit methicillin-resistant Staphylococcus aureus infection in male mice in comparison to vancomycin. Drug Des Devel Ther. 2012; 6:279–287. | ||
Hudson JB, Harris L, Marles RJ, Arnason JT. The anti-HIV activities of photoactive terthiophenes. Photochem Photobiol. 1993;58(2):246–250. | ||
Marles RJ, Hudson JB, Graham EA, et al. Structure-activity studies of photoactivated antiviral and cytotoxic tricyclic thiophenes. PhotochemPhotobiol. 1992;56(4):479–487. | ||
Mazur S, Tanious FA, Ding D, et al. A thermodynamic and structural analysis of DNA minor-groove complex formation. J Mol Biol. 2000; 300(2):321–337. | ||
White EW, Tanious F, Ismail MA, et al. Structure-specific recognition of quadruplex DNA by organic cations: influence of shape, substituents and charge. Biophys Chem. 2007;126(1–3):140–153. | ||
Bailly C, Dassonneville L, Carrasco C, et al. Relationships between topoisomerase II inhibition, sequence-specificity and DNA binding mode of dicationic diphenylfuran derivatives. Anticancer Drug Des. 1999;14(1):47–60. | ||
Wilson WD, Tanious F, Ding D, et al. Nucleic acid interactions of unfused aromatic cations: Evaluation of proposed minor-groove, major-groove and intercalation binding modes. J Am Chem Soc. 1998;120(40): 10310–10321. | ||
Liu Y, Kumar A, Depauw S, et al. Water-mediated binding of agents that target the DNA minor groove. J Am Chem Soc. 2011;133(26): 10171–10183. | ||
Guerri A, Simpson IJ, Neidle S. Visualization of extensive water ribbons and network in a DNA minor-groove drug complex. Nucleic Acids Res. 1998;26(12):2873–2878. | ||
Laughton CA, Tanious F, Nunn CM, Boykin DW, Wilson WD, Neidle SA. A crystallographic and spectroscopic study of the complex between d(CGCGAATTCGCG)2 and 2,5-bis(4-guanylphenyl)furan, an analogue of berenil. Structural origins of enhanced DNA-binding affinity. Biochemistry. 1996;35(18):5655–5661. | ||
Fitzgerald DJ, Anderson JN. Selective nucleosome disruption by drugs that bind in the minor groove of DNA. J Biol Chem. 1999;274(38): 27128–27138. | ||
Dykstra CC, McClernon DR, Elwell LP, Tidwell RR. Selective inhibition of topoisomerases from Pneumocystis carinii compared with that of topoisomerases from mammalian cells. Antimicrob Agents Chemother. 1994;38(9):1890–1898. | ||
Nhili R, Peixoto P, Depauw S, et al. Targeting the DNA-binding activity of the human ERG transcription factor using new heterocyclic dithiophene diamidines. Nucleic Acids Res. 2013;41(1):125–138. | ||
Munde M, Kumar A, Peixoto P, et al. The unusual monomer recognition of guanine-containing mixed sequence DNA by a dithiophene heterocyclic diamidine. Biochemistry 2014;53(7):1218–1227. | ||
Boschi D, Guglielmo S, Aiello S, Morace G, Borghi E, Fruttero R. Synthesis and in vitro antimicrobial activities of new (cyano-NNO-azoxy)pyrazole derivatives. Bioorg Med Chem Lett. 2011;21(11): 3431–3434. | ||
Hussin WA, Ismail MA, El-Sayed WM. Novel 4-substituted phenyl-2,2′-bichalcophenes and aza-analogs as antibacterial agents: a structural activity relationship. Drug Des Devel Ther. 2013;7:185–193. | ||
El-Sayed WM, Hussin WA. Antimutagenic and antioxidant activity of novel 4-substituted phenyl-2,2′-bichalcophenes and aza-analogues. Drug Des Devel Ther. 2013;7:73–81. | ||
Ismail MA, Hussin WA, Alzahrani AM, El-Sayed WM. Evaluation of the biological activity of novel monocationic fluoroaryl-2,2′-bichalcophenes and their analogues. Drug Des Devel Ther. 2014;8:963–972. | ||
Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. Chembiochem. 2004;5(5):622–627. | ||
Dimagno SG, Sun H. The strength of weak interactions: aromatic fluorine in drug design. Curr Top Med Chem. 2006;6(14):1473–1482. | ||
Mallena S, Lee MP, Bailly C, et al. Thiophene-based diamidine forms a “super” AT binding minor groove agent. J Am Chem Soc. 2004;126(42): 13659–13669. | ||
Ismail MA, Brun R, Easterbrook JD, Tanious FA, Wilson WD, Boykin DW. Synthesis and antiprotozoal activity of aza-analogues of furamidine. J Med Chem. 2003;46(22):4761–4769. | ||
Nanjunda R, Musetti C, Kumar A, et al. Heterocyclic dications as a new class of telomeric G-quadruplex targeting agents. Curr Pharm Des. 2012;18(14):1934–1947. | ||
Mancin F, Scrimin P, Tecilla P, Tonellato U. Artificial metallonucleases. Chem Commun (Camb). 2005;(20):2540–2548. | ||
He J, Sun J, Mao ZW, Ji LN, Sun H. Phosphodiester hydrolysis and specific DNA binding and cleavage promoted by guanidinium-functionalized zinc complexes. J Inorg Biochem. 2009;103(5):851–858. | ||
Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–142. | ||
Chung YC, Chang CT, Chao WW, Lin CF, Chou ST. Antioxidant activity and safety of the 50% ethanolic extract from red bean fermented by Bacillus subtilus IMR-NK1. J Agr Food Chem. 2002;50:2454–2458. | ||
Skehan P, Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82(13): 1107–1112. | ||
McCaffrey TA, Agarwal LA, Weksler BB. A rapid fluorometric DNA assay for the measurement of cell density and proliferation in vitro. In Vitro Cell Dev Biol. 1988;24(3):247–252. | ||
Monks A, Scudiero D, Skehan P, et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83(11):757–766. | ||
Youssef MM, Al-Omair MA. Cloning, purification, characterization and immobilization of L-asparaginase II from E. coli W3110. Asian J Biochem. 2008;3(6):337–350. | ||
Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic Bio Med. 1999;26(9–10):1231–1237. | ||
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem. 1982;126(1):131–138. | ||
Sreejayan N, Rao MN. Nitric oxide scavenging by curcuminoids. J Pharm Pharmacol. 1997;49(1):105–107. | ||
Gülçin İ, Daştan A. Synthesis of dimeric phenol derivatives and determination of in vitro antioxidant and radical scavenging activities. J Enzyme Inhib Med Chem. 2007;22(6):685-695. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.