")
Back to Journals » Eye and Brain » Volume 8
Anti-VEGF therapy in the management of retinopathy of prematurity: what we learn from representative animal models of oxygen-induced retinopathy
Authors Wang H
Received 1 October 2015
Accepted for publication 8 December 2015
Published 20 May 2016 Volume 2016:8 Pages 81—90
DOI https://doi.org/10.2147/EB.S94449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Margaret Wong-Riley
Haibo Wang
Department of Ophthalmology, John A Moran Eye Center, The University of Utah, Salt Lake City, UT, USA
Abstract: Retinopathy of prematurity (ROP) remains a leading cause of childhood blindness, affecting infants born prematurely. ROP is characterized by the onset of delayed physiological retinal vascular development (PRVD) and followed by pathologic neovascularization into the vitreous instead of the retina, called intravitreal neovascularization (IVNV). Therefore, the therapeutic strategy for treating ROP is to promote PRVD and inhibit or prevent IVNV. Vascular endothelial growth factor (VEGF) plays an important role in the pathogenesis of ROP. There is a growing body of studies testing the use of anti-VEGF agents as a treatment for ROP. Intravitreal anti-VEGF treatment for ROP has potential advantages compared with laser photocoagulation, the gold standard for the treatment of severe ROP; however, intravitreal anti-VEGF treatment has been associated with reactivation of ROP and suppression of systemic VEGF that may affect body growth and organ development in preterm infants. Therefore, it is important to understand the role of VEGF in PRVD and IVNV. This review includes the current knowledge of anti-VEGF treatment for ROP from animal models of oxygen-induced retinopathy (OIR), highlighting the importance of VEGF inhibition by targeting retinal Müller cells, which inhibits IVNV and permits PRVD. The signaling events involved in mediating VEGF expression and promoting VEGF-mediated angiogenesis, including hypoxia-dependent signaling, erythropoietin/erythropoietin receptor-, oxidative stress-, beta-adrenergic receptor-, integrin-, Notch/Delta-like ligand 4- and exon guidance molecules-mediated signaling pathways, are also discussed.
Keywords: vascular endothelial growth factor, retinopathy of prematurity, intravitreal neovascularization, oxygen-induced retinopathy model, physiological retinal vascular development
Introduction
Retinopathy of prematurity (ROP) was first recognized in the late 1940s as a blinding disease affecting only premature infants.1 Now ROP remains a leading cause of childhood blindness worldwide. ROP is characterized by abnormal retinal vascular development with first delayed physiological retinal vascular development (PRVD) with vaso-attenuation centrally, and subsequent vasoproliferation as intravitreal neovascularization (IVNV).2 Clinical studies showed that vascular endothelial growth factor (VEGF) levels were increased in ocular fluid of patients with ROP3 and intravitreal injections of anti-VEGF agents effectively decreased intraocular VEGF levels.4,5 VEGF is required for retinal vascularization and maturation of retinal neurons in preterm infants.6–8 Therefore, inhibition of VEGF might potentially interfere with these processes. A multicenter randomized clinical trial evaluated the efficacy of intravitreal anti-VEGF (bevacizumab) for stage 3 ROP compared with laser photocoagulation of the peripheral avascular retina.9,10 The outcomes showed that intravitreal anti-VEGF permitted regression of ROP; however, there was reduced serum VEGF for up to 2 months5,11 and reactivation of ROP at 14.4 weeks after initial treatment with bevacizumab.12 Studies are needed regarding dosing, systemic safety, optimal treatment window, and long-term effect of anti-VEGF treatment in ROP. To improve the therapeutic effect of anti-VEGF treatment in ROP, it is important to gain insight into the roles of VEGF in PRVD and IVNV. For this purpose, animal models with oxygen-induced retinopathy (OIR) are often used.
Animal models of OIR
Animals develop their retinal vasculature after birth; therefore, most animal OIR models develop some of the pathologic features of human ROP. Of these OIR models, the mouse and rat OIR models are the most studied.
Mouse OIR model
Mouse OIR model was created by Smith et al13 in 1994. In the mouse OIR model, newborn pups at postnatal day 7 (p7) are exposed to a hyperoxic environment with 75% inspired oxygen level for 5 days. Pups develop avascular retina from vaso-obliteration of newly formed capillaries in the center retina at p12. After pups are moved to room air, avascular central retina becomes hypoxic and intravitreal neovascular tufts are formed at the junctions of vascular and avascular retina at p17. When day 7 pups are placed into 75% oxygen environment, they already complete the primary plexus vascularization in the retina, which is different from preterm infants whose retinas are incompletely vascularized. Therefore, the mouse OIR model does not mimic similar pathologic conditions as that seen in human ROP. However, the mouse OIR model is very useful to study the signaling pathways implicated in human ROP using transgenic mice.
Rat OIR model
In 1994, Penn et al14 created a rat OIR model using newborn pups. In this model, rats are placed into a regulated oxygen environment and exposed to repeated fluctuations of oxygen cycled between 50% and 10% oxygen every 24 hours for 14 days. The inspired oxygen produces arterial oxygen extremes in rat pups similar to the levels in human preterm infants who develop severe ROP. The rat ROP model develops delayed PRVD in the peripheral retina at p14 and IVNV at the junction of vascularized and avascularized retina at p18; features similar to those seen in human ROP. The rat OIR model is considered the most representative model of ROP (Figure 1).
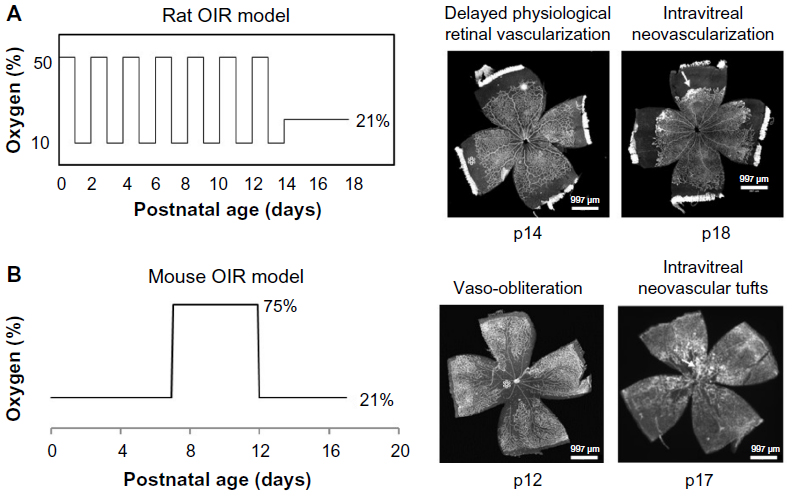 | Figure 1 Models of rat (A) and mouse (B) OIR and retinal flat mounts stained with lectin to visualize the vasculature. |
Other animal OIR models
There are some other animal models of OIR, for example, the beagle OIR model,15 in which newborn dogs on p1 are exposed to 100% oxygen and returned to room air on p5. The beagle OIR model is very useful to test drug doses and translate them into treatment for human preterm infants. Kitten OIR model,16 in which newborn kittens on p3 are exposed to 80% oxygen for up to 112 hours, is also an important animal model in studying the mechanisms of ROP, particularly the effect from hyperoxia.
Anti-VEGF treatment in animal models of OIR
VEGF expression in the retina is increased when IVNV develops in both mouse and rat OIR models. Therefore, these two models are suitable to test the effect of anti-VEGF treatment on IVNV. Tremendous advances have been achieved in understanding the roles of VEGF in ROP from animal studies. Here, different approaches are discussed in inhibiting retinal VEGF in the OIR models.
Inhibition of retinal VEGF
To inhibit retinal VEGF, neutralizing antibody against VEGF is often used. In the mouse model of OIR, intravitreal injection of antibody against mouse VEGF (all the splice variants) at p16 effectively reduced neovascular area in the retina at p17, potentially via a mechanism involving blocking leukocyte infiltration into the retina.17 Another study evaluated the effects of anti-VEGF treatment on retinal development. In that study, intravitreal injections of aflibercept, a VEGF Trap, which prevents VEGF from binding to vascular endothelial growth factor receptors (VEGFRs), at p14 and avascular retinal area (AVA) and neovascular area were analyzed at p17.18 The findings from this study showed that aflibercept treatment not only decreased neovascular tufts, but also increased AVA. Pups treated with aflibercept also showed decreased neuroretinal functions with reduced B-wave amplitude in electroretinography test at p21 and the reduced B-wave persisted at p42 when retinal vascularization recovered. Histologic analysis also showed that aflibercept-treated eyes had thinner retina with disruption of outer nuclear layer and loss of bipolar and amacrine cells at p42.
The effects of intravitreal injection of a neutralizing antibody to rat VEGF164 on PRVD and IVNV has also been evaluated in the rat model of OIR.19 At p12, while rats were still in an environment of fluctuating inspired oxygen, rat pups received intravitreal antibody against rat VEGF164 at several doses or isotype IgG as control, and AVA and IVNV were analyzed in retinal flat mounts. Compared with IgG-injected pups, pups injected with 50 ng anti-VEGF antibody had significantly reduced IVNV and AVA at p18, but increased IVNV and AVA at p25, suggesting anti-VEGF antibody slowed the regression of IVNV and impaired physiological retinal vascularization. At p25, most IVNV regressed in a form of fanning; however, some retinas treated with 50 ng anti-VEGF antibody showed atypical vessel growth in the form of plaques. This comparatively increased and “plaque-like” IVNV at p25 was associated with increased VEGF and VEGFR2 activation and increased levels of the hormone, erythropoietin, which has angiogenic properties.20,21 Pups treated with anti-VEGF antibody also showed reduced weight gain and systemic VEGF levels, suggesting that systemic effects occurred from intravitreal anti-VEGF treatment.
The findings from both OIR models raised concerns about the use of anti-VEGF antibody to inhibit pathologic neovascularization in the developing retina of preterm infants, because retinal VEGF was important for PRVD and systemic VEGF was important for postnatal growth.
Targeting splicing variants of VEGF
The VEGF gene consists of eight exons, which are spaced by seven introns. Through alternative exon splicing, at least five splice variants in human and three in mouse and rat are generated, and each has different biological functions.8 The most studied human splice variants (mouse and rat analogs in parentheses) are soluble VEGF121 (VEGF120), which is soluble protein, cell-associated VEGF189 (VEGF188), which is sequestered in the extracellular matrix (ECM) through its heparin binding domain, and VEGF165 (VEGF164), which has intermediate properties as it is a secreted protein but also it can bind to cell surface and ECM via the heparin binding domain.22 Of the three splice variants, VEGF165 is the predominant one in human and characterizes most of the VEGF properties in promoting angiogenesis and vascular permeability. By in situ mRNA hybridization, mRNA of these three splice variants was detected in rat retina,23 but only VEGF164 mRNA was significantly upregulated in the retina of OIR pups compared with room air raised pups or pups exposed only to hypoxia (10% oxygen) at the same developmental age,24 suggesting that VEGF164 was upregulated by fluctuating oxygen similar to what occurs in premature infants and may be involved in pathologic angiogenesis, IVNV, induced by OIR model. Besides these VEGF splice variants with proangiogenic activity, new splice variants of VEGF, called VEGFXXXb, were identified. Instead of exon 8a in VEGFXXX splice variants, VEGFXXXb has alternate exon 8b, which encodes six different amino acids at C-terminus.25 Therefore, VEGFXXXb has the same length as VEGFXXX with different terminal six amino acids. VEGF165b protein was first identified in retinal cell carcinoma and its downregulation was associated with tumor growth.25 In human umbilical vein endothelial cells, VEGF165b inhibited VEGF165-mediated endothelial cell proliferation and migration,26 suggesting that VEGF165b has antiangiogenic activity. Another study also reported that VEGF165b had neuroprotective effect in vivo and in vitro through inhibition of caspase 3.27 Therefore, modulating VEGF165b expression level or the ratio between VEGF165 and VEGF165b has been considered for diseases that involve pathologic angiogenesis and neuronal damages, such as diabetic retinopathy,28 age-related macular degeneration,29 and cancer.30,31
In the retina, VEGF165b was detected in the developing vasculature of human fetal eyes,32 suggesting the important role of VEGF165b in PRVD. A study also showed that VEGF165b was not only antiangiogenic, but it also protected the retina from ischemia-induced damage and showed cytoprotective effect for retinal pigment epithelium.33 In the rat model of OIR, VEGF165, but not VEGF165b, increased in the retina,34 which led to comparatively reduced expression level of retinal VEGF165b. Administration of recombinant human VEGF165b by intravitreal injections significantly reduced neovascular tufts without delaying PRVD. Compared with retina treated with anti-VEGF antibody, VEGF165b treatment also reduced retinal vessel tortuosity in the mouse OIR model. In addition, intravitreal injection of a serine arginine protein kinase inhibitor, SRPIN340, which was shown to inhibit splicing of VEGF gene into VEGF165 without affecting splicing of VEGF165b, significantly reduced IVNV in both mouse35 and rat OIR models.34 These studies provide evidence that modulating VEGF splicing from proangiogenic factor VEGF165 to antiangiogenic factor VEGF165b may be an effective and less neurotoxic approach to treat ROP, though more studies are needed to understand the molecular mechanisms.
Cell-specific inhibition of VEGF by gene therapy vector
VEGF is an important factor in regulating the development of retinal blood vessels,36,37 and it has also been recognized as a survival factor for a number of cells in the retina, including retinal pigment epithelium and retinal neurons.38 In the rat model of OIR, anti-VEGF antibody introduced into the vitreous reduced vascular density in the inner and deep plexi,39 indicating broad cell inhibition of VEGF in the retina affected PRVD. Therefore, reducing VEGF from pathologic levels to the levels required for PRVD would be a safer way to inhibit IVNV without interfering with PRVD.
In the rat OIR model, VEGF mRNA signal was detected in the retinal inner nuclear layer at the places where cellular retinaldehyde binding protein-labeled Müller cells were located, and this mRNA signal was increased in OIR-treated pups,23 suggesting that Müller cell-derived VEGF contributed to IVNV. To knock down Müller cell-derived VEGF, a lentivector with Müller cell-specific promoter CD44 (pFmCD44.1GW) was used to deliver a short hairpin RNA (shRNA) targeting rat VEGF (VEGFA) (Lenti-CD44-VEGFA shRNA). The shRNA was embedded in a microRNA30 context, which allowed shRNA to be expressed in specific cells. Lentivirus was delivered by subretinal injections at p8. Compared with control lentivector, which expressed a shRNA to luciferase (Lenti-CD44-luciferase shRNA), Lenti-CD44-VEGFA shRNA reduced retinal VEGF protein to the levels in retinas of room air raised pups at p18 and inhibited VEGFR2 activation in retinal vascular endothelial cells. Compared with control lentivector, IVNV was significantly reduced at p18 in retinas of VEGFA shRNA-treated pups. At p25, VEGFA shRNA-treated pups did not show increased IVNV. Another study evaluated the effect of anti-VEGF antibody and Lenti-CD44-VEGFA shRNA on physiological retinal vascularization.39 Intravitreal injection of rat VEGF164 antibody and Lenti-CD44-VEGFA shRNA resulted in a similar fold reduction of IVNV; however, retinas treated with Lenti-CD44-VEGFA shRNA had increased vascular density in the deep plexus, in contrast with the retinas treated with VEGF164 antibody with reduced vascular density in both inner and deep plexi. Knockdown of pathologic Müller cell-derived VEGF by Lenti-CD44-VEGFA shRNA reduced the number of mitotic retinal vascular cells labeled by phosphohistone H3 and promoted cells dividing with cleaved angles at 60°–90°, which predicted ordered angiogenesis involved in PRVD.40,41 In contrast, the retinas treated with VEGF164 antibody showed pathologic angiogenic features with mitotic retinal vascular cells at 0°–60° cleavage angles, which predicted disordered angiogenesis as seen in IVNV. Lenti-CD44-VEGFA shRNA treatment did not reduce serum VEGF level and pup body weight gain at both p18 and p25, as the anti-VEGF treatment did. Altogether, these findings provide evidence that Lenti-CD44-VEGFA shRNA permits physiological retinal vascularization by restoring normal orientation of dividing vascular cells without causing systemic safety concerns.
In the mouse model of OIR, Müller cell-specific expression of endostatin using a self-complementary adeno-associated virus vector with a hypoxia-regulated glial fibrillary acidic protein promoter,42 significantly reduced IVNV and vaso-obliteration in the central retina by a mechanism associated with reduction of VEGF in the retina.43 Therefore, targeted inhibition of Müller cell-derived VEGF may be a safer and better approach to treat ROP compared with broad inhibition of retinal VEGF protein by VEGF antibody, even though more studies are needed to evaluate the long-term effect on retinal functions.
Signaling pathways involved in VEGF expression and VEGF-mediated retinal angiogenesis in the OIR models
VEGF is a key regulator of blood vessels growth in both physiological development6 and pathologic neovascularization.44 In the rat model of OIR, increased retinal VEGF was seen at p14 when PRVD is delayed, suggesting that increased VEGF alone may not be able to promote retinal angiogenesis, and other signaling events might be involved in retinal angiogenesis.21 ROP is a complex disease, and a number of signaling pathways contribute to the development of ROP pathogenesis, including hypoxia, oxidative stresses, and inflammation-mediated signaling. Recent studies also showed that erythropoietin/erythropoietin receptor, beta-adrenergic receptor (β-AR), integrin, Notch/Delta-like-ligand 4 (Dll4) and exon guidance molecule-mediated signaling pathways are also implicated in retinal angiogenesis. The effects of these signaling pathways in VEGF expression and VEGF-mediated retinal angiogenesis are discussed.
Hypoxia-dependent signaling
Oxygen plays important roles in the development of ROP pathogenesis. VEGF expression in retina is regulated by hypoxia-dependent signaling.45 Hypoxia-inducible factor-1 (HIF-1), a transcriptional factor, is a heterodimer composed of subunits alpha (HIF-1α) and beta (HIF-1β). HIF-1α is an inducible subunit and its activity and stability are regulated by prolyl hydroxylases (PHD). In normoxia, PHD promote hydroxylation on proline residues located within the oxygen degradation domain of HIF-1α, leading to the interaction of the von Hippel-Lindau protein with oxygen degradation domain to cause degradation of HIF-1α. Hypoxia prevents proline hydroxylation from PHD and therefore increases the stability of HIF-1α, which allows HIF-1α nuclear translocation. By dimerizing with β-subunit and binding to hypoxia-responsive element in the promoter of VEGF gene, HIF-1α/β regulates the transcription of VEGF in response to hypoxia.46,47
In the mouse OIR model, when mice were exposed to a hyperoxic condition with 75% inspired oxygen, retinal VEGF levels were reduced compared with room air raised mice. Stabilizing HIF-1/2α by intraperitoneal injection of PHD inhibitor, dimethyloxalylglycine, at p6 and p8, significantly increased VEGF expression in the retina during hyperoxia and reduced vaso-obliteration at p12 and vascular tortuosity and neovascular tufts at p17.48 Hyperoxia treatment by exposing OIR mice to 75% oxygen for 3 or 24 hours at p17 caused a significant reduction in VEGF expression and VEGFR2 activation, and subsequent regression of neovascular tufts in the retina.49
In the rat OIR model, supplemental oxygen treatment by moving OIR rats to 28% oxygen instead of room air (21%) reduced retinal VEGF levels and IVNV.50 Another study reported that inhibition of PHD by intraperitoneal injections of dimethyloxalylglycine at p3, p5, and p7 during 50% oxygen cycle significantly improved retinal vascularization at p14 and reduced IVNV at p21.51
Erythropoietin/erythropoietin receptor signaling
Erythropoietin (EPO) is another oxygen-regulated growth factor. It is mainly produced in kidney in response to anemia and hypoxia and plays important roles in hematopoiesis.52 In the mouse OIR model, like VEGF, EPO in the retina was downregulated during hyperoxia and upregulated in hypoxic retina at p17. Intravitreal injection of exogenous EPO reduced hyperoxia-induced vessel dropout and subsequent hypoxia-induced neovascular tufts53; knockdown of retinal EPO by intravitreal injection of a small interference RNA targeting mouse EPO reduced neovascular area.54
In the rat model of OIR, EPO was downregulated in retina with elevated VEGF at p14 when delayed PRVD developed.21 Reduction in retinal EPO was regulated via a mechanism involving VEGF-activated signaling transducer and activator of transcription 3 (STAT3). Rescuing EPO expression in the retina by giving STAT3 inhibitor or administering exogenous EPO at p2, p4, and p6 reduced AVA.21 In cultured human retinal microvascular endothelial cells (hRMVECs), EPO and VEGF synergistically induced cell proliferation through activation of STAT3.19 These findings provide the first evidence for the interaction of VEGF with EPO in retinal angiogenesis, and targeting Janus kinase/STAT3 signaling can rescue endogenous EPO expression level, therefore, facilitating VEGF to promote PRVD and protecting neuroretina from hypoxia-induced dysfunction.55
The function of EPO is to signal through its receptor (EPOR). EPOR exists as homodimer and upon EPO binding, EPORs transduce the signaling through Janus kinase/STAT5.56 Using EPOR–/–-rescued mice that have EPOR expression only in erythroid lineage, the role of EPO/EPOR in angiogenesis in response to hind limb ischemia has been established.57 In this study, the investigators identified a mechanism by which EPO/EPOR regulated ischemia-induced angiogenesis via a mechanism involving upregulation of VEGF and activation of VEGF/VEGFR signaling. In the rat OIR model and cultured hRMVECs, a different signaling was found that VEGFA upregulated and activated EPOR through activated VEGFR2, and activated EPOR interacted with activated VEGFR2 to enhance VEGF-induced proliferation of hRMVEC through exacerbating STAT3 activation. EPOR expression and activation was increased and activated EPOR by phosphorylation of EPOR in retinal vascular endothelial cells of pups at p18 when IVNV occurred,58 providing evidence that EPO/EPOR interacted with VEGF/VEGFR2 in mediating pathologic angiogenesis in the retina. Therefore, modulating EPO/EPOR signaling may be a therapeutic target to inhibit the pathologic effect of VEGF in the development of IVNV without affecting the role of VEGF in PRVD.
Oxidative stress
Cumulative evidence indicates that oxidative stress plays an important role in the development of pathogenesis of ROP.45 Oxidative stress represents an imbalance between reactive oxygen species (ROS) production and antioxidants.59 Overproduction of ROS can cause cell damage or induce apoptosis, but intracellular ROS can also function as second messengers to transduce signals involved in physiological60 and pathological processes,61 including regulation of VEGF expression in the retina.62 Both hyperoxia and hypoxia can induce ROS generation.45 In the mouse OIR model, mice deficient in glutathione peroxidase, one of the antioxidant enzymes, showed increased ROS generation, VEGF expression, and neovascular area in the retina.63 Reduction of ROS production in aldose reductase-deficient mice reduced vaso-obliteration and neovascular tufts by downregulating VEGF expression in the retina.64 Inhibition of ROS production by intravitreal injection of luteolin reduced neovascular tufts and the transcription of vegf gene.65 Increasing Nrf2-regulated antioxidant gene expression by intraperitoneal injection of Ebselen, a mimic of glutathione peroxidase, protected retinal Müller cells and vessels from oxidative stress-induced injury and reduced oxidative stress-mediated retinal VEGF expression.66 In the rat OIR model, hypoxia-induced accumulation of hydrogen peroxide in the choroid increased IVNV,67 which was reduced by intraperitoneal injection of apocynin to quench retinal ROS.
NADPH oxidase is the main source of intracellular ROS generation in vascular endothelial cells.68 Endothelial cell generated ROS are involved in promoting cell proliferation and migration.69 Isoforms of NADPH oxidase, NOX1-5 and Doux1 and -2, are differentially expressed in tissues and cells. In rat and mouse OIR models, NOX2 colocalized with retina endothelial cells and macrophages and regulated physiologic and pathologic angiogenesis in the retina, through mechanisms involving apoptosis and inflammation.70,71 In the rat model of OIR, NOX4 was colocalized in retinal vascular endothelial cells and its expression was increased at p18 when IVNV developed, but not at p14 when PRVD occurred; in hRMVECs, NOX4 interacted with VEGF-activated VEGFR2 to induce cell proliferation via activation of STAT3,68 supporting the role of NOX4 in the development of IVNV.
Altogether, these studies highlight the importance of oxidative stress in regulating VEGF expression and interacting with VEGF/VEGFR2 signaling in retinal angiogenesis.
Beta-adrenergic receptor
Retinal hypoxia is a stimulator for the development of IVNV in Phase II of ROP. Hypoxia increases norepinephrine levels in the rat brain and retina. β-ARs are the targets of norepinephrine and were found to be expressed in rat and mouse retina.72–74 β-ARs belong to the superfamily of transmembrane G-protein coupled receptor. Upon agonist binding, β-ARs are activated to couple with heterotrimeric G proteins, Gαs and Gβγ, leading to disassociation of GS from Gβγ. The GS activates adenylyl cyclase to increase intracellular cyclic adenosine monophosphate levels, which activates protein kinase A. Recently, the role of β-ARs in the regulation of VEGF expression and pathologic neovascularization has gained more attention. Of three isoforms of β-AR, β1-AR and β2-AR were found to be mainly expressed in Müller cells, while β3-AR was detected in retinal capillaries. In the mouse model of OIR, in response to hypoxia, β-AR isoforms expressed differently with upregulation of β3-AR in the retina at p17 when neovascular tufts develop, while the expression of β1-AR and β2-AR in the retina was unaffected by hypoxia.75 Deletion of β1/2-AR in mice abolished OIR-induced vascular abnormalities in the superficial plexus, improved vascular development in the deep plexus, and reduced retinal VEGFR2 activation, suggesting β1/2-AR may promote the development of OIR via activation of VEGF signaling. This was supported by a study,76 in which administration of eye drop propranolol to inhibit β-ARs activity reduced neovascular area, VEGF expression, and STAT3 activation. However, these findings conflicted with the findings from another study,75 in which subcutaneous injection of β-AR agonist, isoproterenol, decreased retinal VEGF and reduced neovascular tufts through a mechanism involving reduction of β2-AR expression and protein kinase A activity. Activation of β3-AR by subcutaneous injection of selective β3-AR agonist, BRL 37344, increased neovascular tufts and VEGF expression in the retina in both wild-type and β1/2-AR deleted mice, suggesting that β3-AR in the retina was not activated in the OIR model, but once activated, it may have the potential effect in promoting pathologic angiogenesis.
Taken together, these studies established the role of β-ARs in OIR and hypoxia-induced VEGF expression and VEGFR2 activation in the retina; however, more studies are needed to discriminate isoform-specific effect.
Integrin signaling
Initially, in angiogenesis, endothelial cells sprout from existing blood vessels that adhere to an ECM rich in vitronectin and fibrinogen via integrin. Integrins are the most important family of ECM receptors and function as heterodimers consisting of transmembrane α- and β-subunits.76 The major integrins on endothelial cells are αvβ3, αvβ5, α5β1, and α6β4. VEGF-induced endothelial cell migration is dependent on integrin-mediated communication between endothelial cells and the ECM.77 A number of studies have shown that integrins are implicated in pathologic angiogenesis in the retina. Integrin αvβ3 has been found localized in retinal neovascular tissue removed from patients with proliferative diabetic retinopathy.78 In the mouse model of OIR, inhibition of αvβ3 and αvβ5 using a nonpeptidic antagonist reduced neovascular area, and VEGF and VEGFR2 expression in the retina.79 Intraperitoneal or intravitreal injection of αvβ3 integrin antagonist, tetraiodothyroacetic acid, from p12 to p15 inhibited neovascularization at p18 and angiogenic effects of EPO and VEGF in retinal endothelial cells.80 In the retina, integrin α2β1 was strongly expressed in activated Müller cells of OIR mice and growth factor-stimulated endothelial cells. Mice deficient in α2β1 integrin showed reduced vaso-obliteration, neovascularization, and VEGF expression, suggesting α2β1 integrin was implicated in regulating hypoxia-induced VEGF production from Müller cells.
Notch/Dll4 signaling pathways in VEGF-mediated angiogenesis
Angiogenic sprouting from a preexisting vessel is a process involving at least two different types of endothelial cells, stalk cells and tip cells. During angiogenic growth, a fraction of endothelial cells have tip cell behavior to initiate sprouting, and others, called stalk cells, stay quiescent to maintain the structural and functional integrity of the vessels and tissue perfusion. Both tip cells and stalk cells are regulated by VEGF and VEGFR signaling. A balance between tip cell migration and stalk cell proliferation in response to extracellular VEGF gradients is believed to be important for normal vascular patterning.81 VEGF regulates tip cell formation by inducing Dll4 expression in endothelial cells. Dll4 is the ligand of Notch receptors. Notch signaling pathways play critical roles in cell fate determination and differentiation. Dll4 gene expression was found to exclusively express in developing arteries and the tips of vascular sprouts. The roles of Notch receptors and ligand Dll4 in angiogenesis have been established based on findings from in vitro and in vivo studies. Activation of Notch signaling inhibited branching at the tip cell sprout and maintained a mature and quiescent phenotype of cultured human umbilical vein endothelial cells. In the retina, Dll4 expression was dynamically regulated in actively growing retinal vessels by VEGF.82 Pharmacological inhibition of Dll4 by intravitreal injection of Dll4 antibody increased angiogenic sprouting in mouse retina with abnormally increased vascular density, which resulted in poor vascular network functions. Exposed to the OIR model, Dll4+/lacZ mice with single allele deletion of dll4 gene developed less avascular area in the center of retina at p12 and less neovascular tufts at p17. A hypothetic mechanism by which Notch/Dll4 regulates VEGF-mediated angiogenesis is well accepted. In angiogenic sprouting, VEGF induces Dll4 expression in tip cells, and increased Dll4 in turn activates Notch receptor and reduces VEGFR2 activation in an adjacent stalk cell, in which a strong expression of Jagged 1, another ligand for Notch receptor, antagonizes Notch activation on neighboring tip cells, which leads to tip cell growing toward VEGF gradient. Therefore, a feedback loop between Notch/Dll4 and VEGF/VEGFR2 signaling regulates tip cell migration and stalk cell proliferation in a polarized pattern, which is important for vascular patterning. All these together suggested that Notch signaling negatively regulated VEGF-mediated angiogenic sprouting involved in both physiological and pathological angiogenesis.
Axon guidance molecules and vascular patterning
Capillary tip cells and axon growth cones use the same signaling cues to regulate polarized direction. Angiogenic endothelial cells express some axon guidance receptors including Roundabout 4 (Robo4), Eph family receptor (Ephrin), UNC5B, and PlexinD1.81 Genetic deletion of these receptors results in defects in vascular patterning, suggesting exon guidance receptor signaling is involved in vascular development. Several studies have reported that Slits and Robos signaling was implicated in VEGF-mediated angiogenesis. Robos are single transmembrane receptors for Slits. Activation of Robo4 by Slit2 inhibited VEGF-induced angiogenic effects including cell migration, tube formation, and permeability in vitro.83 In mouse retina, Robo4 showed endothelial-specific and stalk cell-centric expression. Robo4AP/AP mice, in which exons encoding the immunoglobulin repeats required for the interaction with Slits were deleted, showed increased VEGF-induced vessel permeability, suggesting that Robo4 was important in maintaining vessel integrity by antagonizing VEGF-induced angiogenesis and permeability.84 In the mouse model of OIR, Robo4AP/AP mice showed increased neovascular tufts and intravitreal injection of Slit2 reduced neovascular tufts in wild-type mice, but not in Robo4AP/AP mice, suggesting Robo4/Slits contributed to pathological angiogenesis in the retina.83 However, a recent study has also reported that Slit2 was essential for retinal angiogenesis signaling through Robo1 and Robo2, not Robo4.85 In this study, by in situ hybridization, Slit2 mRNA signal was detected in retinal bipolar neurons underneath the retinal vasculature. Mice with slit2 deletion in the retina showed reduced vessel branching and outgrowth, suggesting that Slit2 was selectively required for retinal angiogenesis of postnatal mice. By analyzing postnatal retinal vascular development in Robo1–/– mice and Robo1–/–Robo2ΔEC, which have global deletion of Robo1 and conditional knockout Robo2 in endothelial cells, the effects of Slit2 in retinal angiogenesis were found signaling through Robo1, and in the absence of Robo1, endothelial Robo2, but not Robo4, was found to function as Slit2 receptor in endothelial cells. Interestingly, Robo1 and Robo2 were also implicated in VEGF-induced Rac1 activation and endothelial migration. All these findings provided evidence of Robos/Slits signaling in regulating vessel patterning and guidance, which are important in both PRVD and IVNV.
In summary, VEGF plays a fundamental role in the pathogenesis of ROP. However, ROP is a multifactorial disease. Better understanding the mechanisms and signaling events involved in VEGF-mediated retinal angiogenesis would help us to develop a safer and effective therapy for ROP.
Acknowledgments
This work was supported by Career Starter Grant to Haibo Wang from Knights Templar Eye Foundation and an Unrestricted Grant from Research to Prevent Blindness, Inc., New York, NY, to the Department of Ophthalmology and Visual Sciences, University of Utah.
Disclosure
The author reports no conflicts of interest in this work.
References
Ashton N. Editorial: retrolental fibroplasia now retinopathy of prematurity. Br J Ophthalmol. 1984;68:689. | |
Hartnett ME, Penn JS. Mechanisms and management of retinopathy of prematurity. N Engl J Med. 2012;367:2515–2526. | |
Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. New Engl J Med. 1994, 331:1480–1487. | |
Davidovic SP, Nikolic SV, Curic NJ, et al. Changes of serum VEGF concentration after intravitreal injection of avastin in treatment of diabetic retinopathy. Eur J Ophthalmol. 2012;22:792–798. | |
Wu WC, Lien R, Liao PJ, et al. Serum levels of vascular endothelial growth factor and related factors after intravitreous bevacizumab injection for retinopathy of prematurity. JAMA Ophthalmol. 2015;133:391–397. | |
Achen MG, Stacker SA. The vascular endothelial growth factor family; proteins which guide the development of the vasculature. Int J Exp Pathol. 1998;79:255–265. | |
Gerber HP, Hillan KJ, Ryan AM, et al. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. | |
Ferrara N, Gerber HP, Lecouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. | |
Mintz-Hittner HA, Kennedy KA, Chuang AZ. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N Engl J Med. 2011;364:603–615. | |
Geloneck MM, Chuang AZ, Clark WL, et al. Refractive outcomes following bevacizumab monotherapy compared with conventional laser treatment: a randomized clinical trial. JAMA Ophthalmol. 2014;132:1327–1333. | |
Kong L, Fry M, Al-Samarraie M, Gilbert C, Steinkuller PG. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J AAPOS. 2012;16:501–507. | |
Hu J. REactivation of retinopathy of prematurity after bevacizumab injection. Arch Opthalmol. 2012;130:1000–1006. | |
Smith LEH, Wesolowski E, McLellan A, et al. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci. 1994;35:101–111. | |
Penn JS, Henry MM, Tolman BL. Exposure to alternating hypoxia and hyperoxia causes severe proliferative retinopathy in the newborn rat. Pediatr Res. 1994;36:724–731. | |
Flower RW, Blake DA. Retrolental fibroplasia: evidence for a role of the prostaglandin cascade in the pathogenesis of oxygen-induced retinopathy in the newborn beagle. Pediatr Res. 1981;15: 1293–1302. | |
Ashton N, Garner A, Knight G. Intermittent oxygen in retrolental fibroplasia. Am J Ophthalmol. 1971;71:153–160. | |
Nakao S, Arima M, Ishikawa K, et al. Intravitreal anti-VEGF therapy blocks inflammatory cell infiltration and re-entry into the circulation in retinal angiogenesis. Invest Ophthalmol Vis Sci. 2012;53: 4323–4328. | |
Tokunaga CC, Mitton KP, Dailey W, et al. Effects of anti-VEGF treatment on the recovery of the developing retina following oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2014;55:1884–1892. | |
McCloskey M, Wang H, Jiang Y, Smith GW, Strange J, Hartnett ME. Anti-VEGF antibody leads to later atypical intravitreous neovascularization and activation of angiogenic pathways in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2013;54:2020–2026. | |
Kertesz N, Wu J, Chen THP, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Dev Biol. 2004;276:101–110. | |
Wang H, Byfield G, Jiang Y, Smith GW, McCloskey M, Hartnett ME. VEGF-mediated STAT3 activation inhibits retinal vascularization by downregulating erythropoietin expression. Am J Pathol. 2012;180:1243–1253. | |
Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. | |
Wang H, Smith GW, Yang Z, et al. Short hairpin RNA-mediated knockdown of VEGFA in Muller cells reduces intravitreal neovascularization in a rat model of retinopathy of prematurity. Am J Pathol. 2013;183:964–974. | |
Budd SJ, Hartnett ME. Increased angiogenic factors associated with peripheral avascular retina and intravitreous neovascularization: a model of retinopathy of prematurity. Arch Ophthalmol. 2010;128: 589–595. | |
Bates DO, Cui TG, Doughty JM, et al. VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 2002;62:4123–4131. | |
Gu F, Li X, Kong J, et al. VEGF111b, a new member of VEGFxxxb isoforms and induced by mitomycin C, inhibits angiogenesis. Biochem Biophys Res Commun. 2013;441:18–24. | |
Beazley-Long N, Hua J, Jehle T, et al. VEGF-A165b is an endogenous neuroprotective splice isoform of vascular endothelial growth factor A in vivo and in vitro. Am J Pathol. 2013;183:918–929. | |
Perrin RM, Konopatskaya O, Qiu Y, Harper S, Bates DO, Churchill AJ. Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia. 2005;48:2422–2427. | |
Hua J, Spee C, Kase S, et al. Recombinant human VEGF165b inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci. 2010;51:4282–4288. | |
Woolard J, Wang WY, Bevan HS, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. | |
Rennel ES, Hamdollah-Zadeh MA, Wheatley ER, et al. Recombinant human VEGF165b protein is an effective anti-cancer agent in mice. Eur J Cancer. 2008;44:1883–1894. | |
Baba T, McLeod DS, Edwards MM, et al. VEGF 165 b in the developing vasculatures of the fetal human eye. Dev Dyn. 2012;241:595–607. | |
Magnussen AL, Rennel ES, Hua J, et al. VEGF-A165b is cytoprotective and antiangiogenic in the retina. Invest Ophthalmol Vis Sci. 2010;51:4273–4281. | |
Gammons MV, Dick AD, Harper SJ, Bates DO. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2013;54:5797–5806. | |
Nowak DG, Amin EM, Rennel ES, et al. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J Biol Chem. 2010;285:5532–5540. | |
Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001;280:C1358–C1366. | |
Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25. | |
Foxton RH, Finkelstein A, Vijay S, et al. VEGF-A is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am J Pathol. 2013;182:1379–1390. | |
Wang H, Yang Z, Jiang Y, et al. Quantitative analyses of retinal vascular area and density after different methods to reduce VEGF in a rat model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2014;55:737–744. | |
Hartnett ME, Martiniuk D, Byfield G, Geisen P, Zeng G, Bautch VL. Neutralizing VEGF decreases tortuosity and alters endothelial cell division orientation in arterioles and veins in a rat model of ROP: relevance to plus disease. Invest Ophthalmol Vis Sci. 2008;49: 3107–3114. | |
Zeng G, Taylor SM, McColm JR, et al. Orientation of endothelial cell division is regulated by VEGF signaling during blood vessel formation. Blood. 2007;109:1345–1352. | |
Prentice HM, Biswal MR, Dorey CK, Blanks JC. Hypoxia-regulated retinal glial cell-specific promoter for potential gene therapy in disease. Invest Ophthalmol Vis Sci. 2011;52:8562–8570. | |
Biswal MR, Prentice HM, Dorey CK, Blanks JC. A hypoxia-responsive glial cell-specific gene therapy vector for targeting retinal neovascularization. Invest Ophthalmol Vis Sci. 2014;55:8044–8053. | |
Aiello LP, Pierce EA, Foley ED, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci U S A. 1995;92:10457–10461. | |
Hartnett ME. Pathophysiology and mechanisms of severe retinopathy of prematurity. Ophthalmology. 2015;122:200–210. | |
Ahn GO, Seita J, Hong BJ, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci U S A. 2014;111:2698–2703. | |
Linden T, Katschinski DM, Eckhardt K, Scheid A, Pagel H, Wenger RH. The antimycotic ciclopirox olamine induces HIF-1α stability, VEGF expression, and angiogenesis. FASEB J. 2003;17:761–763. | |
Sears JE, Hoppe G, Ebrahem Q, Anand-Apte B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc Natl Acad Sci. 2008;105:19898–19903. | |
Liu H, Zhang W, Xu Z, Caldwell RW, Caldwell RB, Brooks SE. Hyperoxia causes regression of vitreous neovascularization by downregulating VEGF/VEGFR2 pathway. Invest Ophthalmol Vis Sci. 2013;54:918–931. | |
Saito Y, Uppal A, Byfield G, Budd S, Hartnett ME. Activated NAD(P)H oxidase from supplemental oxygen induces neovascularization independent of VEGF in retinopathy of prematurity model. Invest Ophthalmol Vis Sci. 2008;49:1591–1598. | |
Trichonas G, Lee TJ, Hoppe G, Au J, Sears JE. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy in the rat 50/10 model. Invest Ophthalmol Vis Sci. 2013;54:4919–4926. | |
Haase VH. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev. 2013;27:41–53. | |
Chen J, Connor KM, Aderman CM, Smith LE. Erythropoietin deficiency decreases vascular stability in mice. J Clin Invest. 2008;118:526–533. | |
Chen J, Connor KM, Aderman CM, Willett KL, Aspegren OP, Smith LEH. Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest Ophthalmol Vis Sci. 2009;50:1329–1335. | |
Mowat FM, Gonzalez F, Luhmann UF, et al. Endogenous erythropoietin protects neuroretinal function in ischemic retinopathy. Am J Pathol. 2012;180:1726–1739. | |
Mannello F, Tonti GAM. Erythropoietin and Its receptor in breast cancer: putting together the pieces of the puzzle. Oncologist. 2008;13:761–768. | |
Nakano M, Satoh K, Fukumoto Y, et al. Important role of erythropoietin receptor to promote VEGF expression and angiogenesis in peripheral ischemia in mice. Circ Res. 2007;100:662–669. | |
Yang Z, Wang H, Jiang Y, Hartnett ME. VEGFA activates erythropoietin receptor and enhances VEGFR2-mediated pathological angiogenesis. Am J Pathol. 2014;184:1230–1239. | |
Blokhina O, Virolainen E, Fagerstedt KV. Antioxidants, oxidative damage and oxygen deprivation stress: a review. Ann Bot. 2002;91:179–194. | |
Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. | |
Wang H, Zhang SX, Hartnett ME. Signaling pathways triggered by oxidative stress that mediate features of severe retinopathy of prematurity. JAMA Ophthalmol. 2013;131:80–85. | |
Al Shabrawey M, Bartoli M, El Remessy AB, et al. Inhibition of NAD(P)H Oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. | |
Tan SM, Stefanovic N, Tan G, Wilkinson-Berka JL, de Haan JB. Lack of the antioxidant glutathione peroxidase-1 (GPx1) exacerbates retinopathy of prematurity in mice. Invest Ophthalmol Vis Sci. 2013;54:555–562. | |
Fu ZJ, Li SY, Kociok N, Wong D, Chung SK, Lo AC. Aldose reductase deficiency reduced vascular changes in neonatal mouse retina in oxygen-induced retinopathy. Invest Ophthalmol Vis Sci. 2012;53:5698–5712. | |
Park SW, Cho CS, Jun HO, et al. Anti-angiogenic effect of luteolin on retinal neovascularization via blockade of reactive oxygen species production. Invest Ophthalmol Vis Sci. 2012;53:7718–7726. | |
Tan SM, Deliyanti D, Figgett WA, Talia DM, de Haan JB, Wilkinson-Berka JL. Ebselen by modulating oxidative stress improves hypoxia-induced macroglial Muller cell and vascular injury in the retina. Exp Eye Res. 2015;136:1–8. | |
Beharry KD, Cai CL, Sharma P, et al. Hydrogen peroxide accumulation in the choroid during intermittent hypoxia increases risk of severe oxygen-induced retinopathy in neonatal rats. Invest Ophthalmol Vis Sci. 2013;54:7644–7657. | |
Wang H, Yang Z, Jiang Y, Hartnett ME. Endothelial NADPH oxidase 4 mediates vascular endothelial growth factor receptor 2-induced intravitreal neovascularization in a rat model of retinopathy of prematurity. Mol Vis. 2014;20:231–241. | |
Datla SR, Peshavariya H, Dusting GJ, Mahadev K, Goldstein BJ, Jiang F. Important role of Nox4 type nADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol. 2007:27:2319–2324. | |
Stevenson L, Matesanz N, Colhoun L, et al. Reduced nitro-oxidative stress and neural cell death suggests a protective role for microglial cells in TNFalpha-/- mice in ischemic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:3291–3299. | |
Saito Y, Geisen P, Uppal A, Hartnett ME. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853. | |
Ristori C, Filippi L, Dal Monte M, et al. Role of the adrenergic system in a mouse model of oxygen-induced retinopathy: antiangiogenic effects of beta-adrenoreceptor blockade. Invest Ophthalmol Vis Sci. 2011;52:155–170. | |
Smith CP, Sharma S, Steinle JJ. Age-related changes in sympathetic neurotransmission in rat retina and choroid. Exp Eye Res. 2007;84:75–81. | |
Martini D, Monte MD, Ristori C, et al. Antiangiogenic effects of beta2 -adrenergic receptor blockade in a mouse model of oxygen-induced retinopathy. J Neurochem. 2011;119:1317–1329. | |
Dal Monte M, Martini D, Latina V, Pavan B, Filippi L, Bagnoli P. Beta-adrenoreceptor agonism influences retinal responses to hypoxia in a model of retinopathy of prematurity. Invest Ophthalmol Vis Sci. 2012;53:2181–2192. | |
Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285: 1028–1033. | |
Byzova TV, Goldman CK, Pampori N, et al. A Mechanism for modulation of cellular responses to VEGF: activation of the integrins. Molecular Cell. 2000;6:851–860. | |
Ning A, Cui J, Maberley D, Ma P, Matsubara J. Expression of integrins in human proliferative diabetic retinopathy membranes. Can J Ophthalmol. 2008;43:683–688. | |
Wilkinson-Berka JL, Jones D, Taylor G, et al. SB-267268, a nonpeptidic antagonist of alpha(v)beta3 and alpha(v)beta5 integrins, reduces angiogenesis and VEGF expression in a mouse model of retinopathy of prematurity. Investigative Ophthalmol Vis Sci. 2006;47:1600–1605. | |
Yoshida T, Gong J, Xu Z, Wei Y, Duh EJ. Inhibition of pathological retinal angiogenesis by the integrin alphavbeta3 antagonist tetraiodothyroacetic acid (tetrac). Exp Eye Res. 2012;94:41–48. | |
Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. 2010;2:a001875. | |
Lobov IB, Renard RA, Papadopoulos N, et al. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci U S A. 2007;104:3219–3224. | |
Jones CA, Nishiya N, London NR, et al. Slit2-Robo4 signalling promotes vascular stability by blocking Arf6 activity. Nat Cell Biol. 2009;11:1325–1331. | |
Jones CA, London NR, Chen H, et al. Robo4 stabilizes the vascular network by inhibiting pathologic angiogenesis and endothelial hyperpermeability. Nat Med. 2008;14:448–453. | |
Rama N, Dubrac A, Mathivet T, et al. Slit2 signaling through Robo1 and Robo2 is required for retinal neovascularization. Nat Med. 2015;21:483–491. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.