Back to Journals » Journal of Inflammation Research » Volume 14
Anti-TWEAK Antibody Alleviates Renal Interstitial Fibrosis by Increasing PGC-1α Expression in Lupus Nephritis
Authors Xue L ![]() , Zhang Y, Xu J, Lu W, Wang Q, Fu J, Liu Z
, Zhang Y, Xu J, Lu W, Wang Q, Fu J, Liu Z
Received 12 January 2021
Accepted for publication 11 March 2021
Published 26 March 2021 Volume 2021:14 Pages 1173—1184
DOI https://doi.org/10.2147/JIR.S301356
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Leixi Xue,1,* Yi Zhang,1,* Jiajun Xu,1 Wentian Lu,1 Qing Wang,1 Jinxiang Fu,2 Zhichun Liu1
1Department of Rheumatology and Immunology, The Second Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China; 2Department of Hematology, The Second Affiliated Hospital of Soochow University, Suzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhichun Liu
Department of Rheumatology and Immunology, The Second Affiliated Hospital of Soochow University, Sanxiang Road No. 1055, Suzhou, Jiangsu, 215004, People’s Republic of China
Tel +86 13771994276
Email [email protected]
Jinxiang Fu
Department of Hematology, The Second Affiliated Hospital of Soochow University, Sanxiang Road No. 1055, Suzhou, Jiangsu, 215004, People’s Republic of China
Tel +86 13962525217
Email [email protected]
Purpose: Current studies on the mechanism of tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in lupus nephritis (LN) mainly focus on the inflammatory pathway. Herein, we aimed to determine whether TWEAK could promote the progression of renal interstitial fibrosis by regulating peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) expression and intervening in lipid metabolism in LN.
Materials and Methods: MRL/lpr mice, an animal model of lupus, were treated with the anti-TWEAK antibody or co-treated with adeno-associated virus-mediated PGC-1α short hairpin RNA (shRNA). In addition, human proximal tubular epithelial cells (HK2 cells) were treated with recombinant human TWEAK (rhTWEAK) or ammonium pyrrolidine dithiocarbamate (PDTC) in vitro.
Results: The renal contents of free fatty acids and triglycerides were higher in MRL/lpr mice than in MRL/MpJ mice; however, these contents were decreased by treatment with the anti-TWEAK antibody. Based on immunofluorescence staining, the expression of PGC-1α was markedly more in the renal tubules of MRL/MpJ mice than in the glomeruli. However, treatment with anti-TWEAK antibody increased the levels of PGC-1α and its downstream target genes, which were remarkably lower in MRL/lpr mice than in MRL/MpJ mice. Anti-TWEAK antibody effectively eased renal interstitial fibrosis, which manifested as a decrease in the deposition of collagen fibers and the inhibition of type I collagen and fibronectin expression. However, the therapeutic effects of the anti-TWEAK antibody were abolished by PGC-1α shRNA. Treatment with rhTWEAK decreased PGC-1α expression in both dose- and time-dependent manners in HK2 cells in vitro. PDTC, an inhibitor of IκBα phosphorylation, suppressed the decrease in the PGC-1α protein level induced by rhTWEAK treatment.
Conclusion: Our results suggest that TWEAK prevents renal tubular PGC-1α expression by promoting NF-κB activation, resulting in a deficiency in lipid metabolism and the progress of renal interstitial fibrosis. The upregulation of renal tubular PGC-1α expression to improve lipid metabolism is one of the mechanisms employed by the anti-TWEAK antibody to treat renal interstitial fibrosis.
Keywords: TWEAK, lupus nephritis, renal interstitial fibrosis, PGC-1α, lipid metabolism
Introduction
Lipid metabolic deficiency in the renal tubular epithelium has received extensive attention, especially in the context of acute and chronic kidney disease.1–4 A higher intracellular lipid accumulation induces cellular lipotoxicity, potentially contributing to the development of renal fibrosis.5,6 Renal proximal tubular epithelial cells exhibit high levels of baseline energy consumption and are abundant in mitochondria. Further, these cells mainly depend on fatty acid oxidation as their energy source; this is because fatty acid oxidation generates more ATP than glucose oxidation.7 It has been reported that a reduction in fatty acid oxidation results in lipid accumulation and is the major inciting factor in renal fibrosis development.7
As a transcription regulator, peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC-1α) can activate many types of intranuclear receptors and regulate the expression of downstream target genes, such as fatty acid translocase (FAT), carnitine palmitoyl-transferase 1 (CPT1), long-chain acyl dehydrogenase (LCAD), cytochrome oxidase II (COX II), etc., which are involved in lipid transport, fatty acid oxidation, oxidative phosphorylation, energy homeostasis, and mitochondrial biogenesis.8–10 Under normal conditions, the uptake, utilization, and synthesis of lipids exist in a strict equilibrium state to avoid lipid accumulation.
Lupus nephritis (LN) is one of the most common manifestations of systemic lupus erythematosus (SLE), where kidney involvement is significantly associated with mortality.11 Despite substantial progress, the renal damage mechanisms in LN have not been fully elucidated, which poses a real challenge for the improvement of treatment strategies. Tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK), a TNF superfamily member, is mainly distributed in the glomerular area, tubules, and inflammatory cells of the kidneys in MRL/lpr mice (a spontaneous LN model), but is weakly observed in the kidneys of normal mice.12 TWEAK can induce resident kidney cells to express a variety of inflammatory factors, including interleukin-6, interferon-γ inducible protein 10 (IP-10), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1α, intercellular adhesion molecule-1, regulated upon activation normal T cell expressed and secreted factor (RANTES), and human vascular endothelial cell adhesion molecule-1 (VCAM-1), thereby stimulating lymphocytes and monocytes/macrophages to migrate to the kidney.13–15 TWEAK receptor knockout in MRL/lpr mice significantly improves renal disease by inhibiting the direct injurious effects of TWEAK on the filtration barrier and/or modulating cytokine production by resident kidney cells.16 Anti-TWEAK neutralizing antibody has also been shown to ease kidney disease by blocking local inflammatory events in a chronic graft-versus-host SLE model.14 In nephrotoxic serum nephritis mice treated with anti-TWEAK antibody, a significant decrease in glomerular immunoglobulin deposition, as well as macrophage infiltration and tubulointerstitial fibrosis, which may be related to reduced renal expression of MCP-1, VCAM-1, IP-10, RANTES, and other molecules associated with fibrosis, was observed.17 Thus, studies on the mechanism of TWEAK in LN mainly focus on the inflammatory pathway. Currently, a clinical trial using anti-TWEAK antibody is underway for the treatment of human LN. However, the molecular mechanism employed in TWEAK neutralizing therapy to protect the kidneys remains to be further elucidated; this clarification will aid in the development of novel nephroprotective therapeutic strategies.
Treatment with TWEAK is reported to promote the progress of cardiac dysfunction by reducing the expression of PGC-1α.18 The PGC-1α level is significantly higher in the skeletal muscle of TWEAK-knockout mice than that in wild-type mice. Further, soluble TWEAK treatment inhibits the expression of PGC-1α in primary myotubes.19 Therefore, we hypothesized that TWEAK could downregulate PGC-1α expression and interfere with lipid metabolism in LN, resulting in the progress of renal interstitial fibrosis, and the upregulation of the level of PGC-1α is one of the kidney protection mechanisms of the anti-TWEAK antibody. In the present study, mice with lupus had an abnormal lipid accumulation and a low expression of PGC-1α in their kidneys; however, these features were improved by treatment with the anti-TWEAK antibody. In vitro, TWEAK could suppress renal tubular PGC-1α expression by activating the NF-κB pathway.
Materials and Methods
Mouse Models
MRL/lpr and MRL/MpJ mice were purchased from Shanghai Slack Laboratory Animal Co. Ltd (Shanghai, China), and bred and maintained in the animal facility of the Second Affiliated Hospital of Soochow University. All animal study protocols were approved by the Ethics Committee of Soochow University (Approval No. ECSU-2019000213), Suzhou, China, and were performed in accordance with the Laboratory Guidelines for Animal Use and Care.20 For the experiments involving the inhibition of PGC-1α expression, MRL/lpr mice were treated with adeno-associated virus (AAV)-mediated PGC-1α short hairpin RNA (shRNA) or control shRNA via a renal pelvis injection at 11 weeks old. To determine the effects of the TWEAK neutralizing antibody, 13-week-old MRL/lpr mice were treated via intraperitoneal injections of either 2 mg/kg anti-TWEAK antibody (1 μg/μL, Biolegend, USA) or 2 mg/kg isotype control antibody (1 μg/μL, Biolegend, USA) every other day for 56 days. The volume of each injection administered to each mouse was 100 μL; this final volume was achieved with normal saline. After 48 h of the final injection, mice were sacrificed.
Vector Delivery
AAV-mediated PGC-1α shRNA and control shRNA were designed and synthesized by GeneChem (Shanghai, China). AAV was administered at a dose of 1.5 × 1011 vg per animal via renal pelvis injection (100 μL of total volume) using a 29-gauge insulin syringe (Becton, Dickinson and Company, USA).
Serum Analysis
Serum creatinine and blood urea nitrogen levels were determined using an automatic biochemical analyzer (Rayto, China) manufactured by Wuhan Servicebio Technology Co., LTD.
Cell Culture
Human proximal tubular epithelial cells (HK2 cells) from Cell Bank of Chinese Academy of Sciences were cultured in a 5% CO2 atmosphere at 37°C in DMEM/F-12 medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA), 100 units/mL penicillin G sodium, and 100 μg/mL streptomycin sulfate (Beyotime, China). When the cells reached 70%–80% confluence, the intervention was carried out. For treatment with recombinant human TWEAK (rhTWEAK) (R&D Systems, USA) alone, cells were treated with rhTWEAK at the indicated concentration for the indicated time. For the co-treatment with rhTWEAK and ammonium pyrrolidine dithiocarbamate (PDTC) (Selleck, USA), HK2 cells were preincubated with PDTC for 1 h, and then incubated with rhTWEAK for 48 h.
Quantitative Reverse Transcription PCR
Total RNA was extracted from the kidney tissues or HK2 cells using Trizol (Invitrogen, USA) according to the manufacturer’s instructions. First strand cDNA was subsequently synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo, USA). Quantitative reverse transcription PCR was performed in triplicate using the FastStart Universal SYBR Green Master (Rox) (Servicebio, China). The following primers were used in the study: mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH), forward 5′-CCT CGT CCC GTA GAC AAA ATG-3′ and reverse 5′-TGA GGT CAA TGA AGG GGT CGT-3′ (133 bp); mouse type I collagen (COL-I), forward 5′-AAG AAG CAC GTC TGG TTT GGA G-3′ and reverse 5′-GGT CCA TGT AGG CTA CGC TGT T-3′ (175 bp); mouse fibronectin, forward 5′-AAT CAG CAG TGG GAA CGG AC-3′ and reverse 5′-CAC TTG TCG CCA ATC TTG TAG G-3′ (278 bp); mouse PGC-1α, forward 5′-GTC GTG TTC CCG ATC ACC ATA T-3′ and reverse 5′-CTT TGC GGT ATT CAT CCC TCT T-3′ (230 bp); mouse FAT, forward 5′-AGA ACA GCA GCA AAA TCA AGG TT-3′ and reverse 5′-AGG ATA TGG AAC CAA ACT GAG GA-3′ (347 bp); mouse CPT1, forward 5′-ACC ATC CAC GCC ATA CTG CT-3′ and reverse 5′-TAC ACG ACA ATG TGC CTG CTG-3′ (188 bp); mouse LCAD, forward 5′-TTA AGT GAT CTC GTG ATC GTC GTG-3′ and reverse 5′-CAC AGG CAG AAA TCG CCA AC-3′ (286 bp); mouse COX II, forward 5′-ATA GAC GAA ATC AAC AAC CCC G-3′ and reverse 5′-GGA TTG GAA GTT CTA TTG GCA G-3′ (193 bp); Human GAPDH, forward 5′-CTT TGG TAT CGT GGA AGG ACT C-3′ and reverse 5′-GTA GAG GCA GGG ATG ATG TTC T-3′ (132 bp); and Human PGC-1α, forward 5′-GGG AGA GGC AGA GGC AGA AGG-3′ and reverse 5′-CTG TCC GTG TTG TGT CAG GTC TG-3′ (81 bp); The GAPDH gene was used as an endogenous control to normalize the differences in the amount of total RNA in each sample. Relative gene expression changes were calculated as fold differences using the 2−∆∆Ct method.
Western Blotting
HK-2 cells or mouse kidneys were lysed in RIPA buffer containing protease and phosphatase inhibitors. Protein concentrations were determined using the BCA Protein Assay Kit (Servicebio, China). The proteins were separated using SDS-PAGE and electrophoretically transferred onto polyvinylidene fluoride membranes. The membranes were incubated overnight at 4 °C with primary antibodies against GAPDH, PGC-1α (Abcam, UK), IκBα, p-IκBα, p65, and p-p65 (Cell Signaling Technology, USA), and then with the secondary antibodies at room temperature for 1 h. Detection was performed using a LumiGLO chemiluminescent substrate system (Cell Signaling Technology, USA). The ImageJ quantitated intensity of bands was normalized to that of GAPDH.
Renal Histopathology
We used 4% paraformaldehyde-fixed, paraffin-embedded kidney sections stained with Masson’s trichrome and Sirius red as well as OCT-embedded frozen sections for oil red O staining. Interstitial fibrosis was graded on a scale of 0–3 (0, < 5%; 1, 5% – 10%; 2, 11% – 25%; 3, > 25%) and evaluated blindly by the same pathologist, as previously described.16,17,21
Immunohistochemical Staining
Immunohistochemical staining of the kidney sections was performed with an established protocol.22 Rabbit polyclonal antibodies specific for COL-I and fibronectin (Servicebio, China) were used separately, followed by incubation with horseradish peroxidase-conjugated goat anti-rabbit IgG (Servicebio, China).
Immunofluorescence Staining
Indirect immunofluorescence staining was carried out according to established procedures. Kidney sections were incubated with rabbit polyclonal antibodies specific for PGC-1α (Abcam, UK), followed by staining with Cy3-conjugated secondary antibody (Servicebio, China). Thereafter, the slides were stained with a DAPI solution. Slides were viewed using a confocal laser scanning microscope.
Gas Chromatograph System Coupled with a Mass Spectrometer
A total of 44 free fatty acids in mice kidneys were measured using a gas chromatograph system coupled with a mass spectrometer (GC-MS) (GeneChem, China).
Statistical Analysis
Data are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism 5. Comparison among groups was carried out using one-way ANOVA, followed by Bonferroni’s multiple comparison test. Estimates of renal interstitial fibrosis were compared using a non-parametric test (Kruskal–Wallis). Differences were considered significant at p < 0.05.
Results
Neutralizing Anti-TWEAK Antibody Alleviated Renal Interstitial Fibrosis
The MRL/lpr mice that presented evident renal injury and produced a large number of autoantibodies were employed as a lupus model in this study,23 and age- and sex-matched MRL/MpJ mice were used as normal controls. First, we sought to determine whether the blockade of TWEAK activity using a rat anti-mouse TWEAK-neutralizing antibody could alleviate renal fibrosis in mice with lupus. Based on the results, treatment with the neutralizing anti-TWEAK antibody reduced the renal interstitial deposition of collagen fibers, as determined using Masson’s trichrome and Sirius red staining (Figure 1A and B). By examining the expression of COL-I and fibronectin, which are the main components of the extracellular matrix (ECM),24 the neutralizing anti-TWEAK antibody was found to considerably decrease the protein and gene levels of these ECM components, as determined using immunohistochemical staining and quantitative reverse transcription PCR (Figure 1C and D). In addition, the anti-TWEAK antibody significantly inhibited the increase in the levels of serum creatinine and blood urea nitrogen, which are important indicators of renal function (Figure 1E).
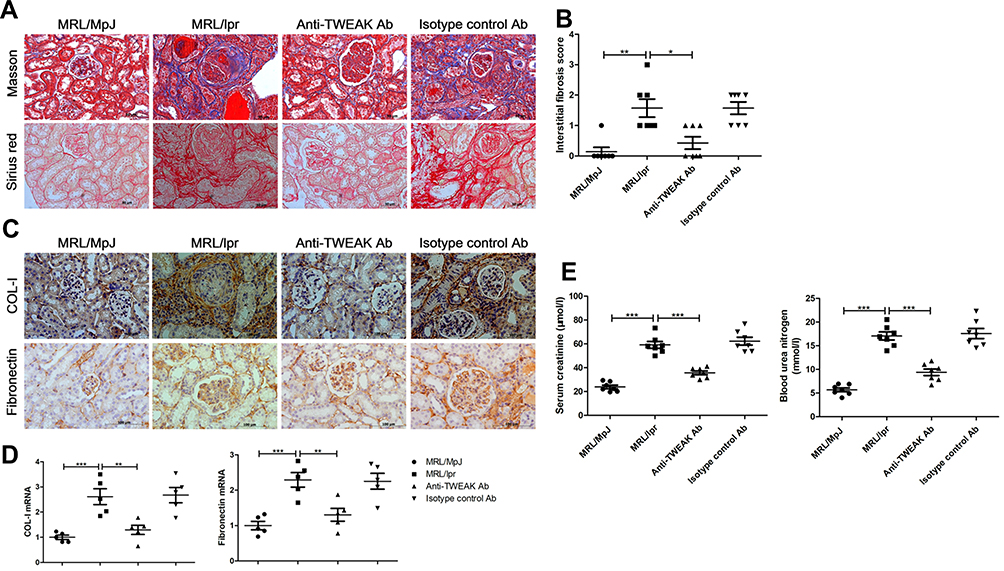 |
Figure 1 Neutralizing the anti-TWEAK antibody alleviated renal interstitial fibrosis. Thirteen-week-old MRL/lpr mice were treated via intraperitoneal injections of either 2 mg/kg anti-TWEAK antibody or 2 mg/kg isotype control antibody every other day for 56 days. (A) Masson’s trichrome and Sirius red staining were used to measure the deposition of collagen fibers in the renal interstitium (n = 7/group) (scale bar, 50 μm). (B) Interstitial fibrosis scores were evaluated blindly by the same pathologist (n = 7/group). The expression levels of COL-I and fibronectin in the kidneys were detected using immunohistochemical staining (C) (scale bar, 100 μm) and quantitative reverse transcription PCR (D), respectively (n = 5/group). (E) The levels of blood urea nitrogen and serum creatinine were also determined (n = 7/group). The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. |
Anti-TWEAK Antibody Alleviated Renal Lipid Accumulation in Mice with Lupus
GC-MS was used to detect the levels of 44 free fatty acids in the kidneys of mice. Docosahexaenoic acid, arachidonic acid, linoleic acid, erucic acid, oleic acid, palmitic acid, and stearic acid were identified as the main free fatty acids in MRL/MpJ mice (Figure 2A-H), accounting for 84.8% of the total free fatty acids. The concentration of arachidonic acid, oleic acid, palmitic acid, and stearic acid was significantly higher in MRL/lpr mice than that in MRL/MpJ mice; the concentration of total free fatty acids and other free fatty acids was also mildly increased in mice with lupus; however, the differences were not statistically significant (Figure 2A-H). Treatment with the anti-TWEAK antibody evidently reduced the concentrations of arachidonic acid, oleic acid, and stearic acid in MRL/lpr mice, and slightly decreased the concentration of total free fatty acid and other free fatty acids (Figure 2A-H). In MRL/lpr mice, oil red O staining revealed an obvious accumulation of triglycerides in the renal tubules; this accumulation was improved by treatment with the anti-TWEAK antibody (Figure 2I). Thus, there is an abnormal lipid accumulation in the kidneys of mice with lupus; however, this accumulation was alleviated by treatment with the anti-TWEAK antibody.
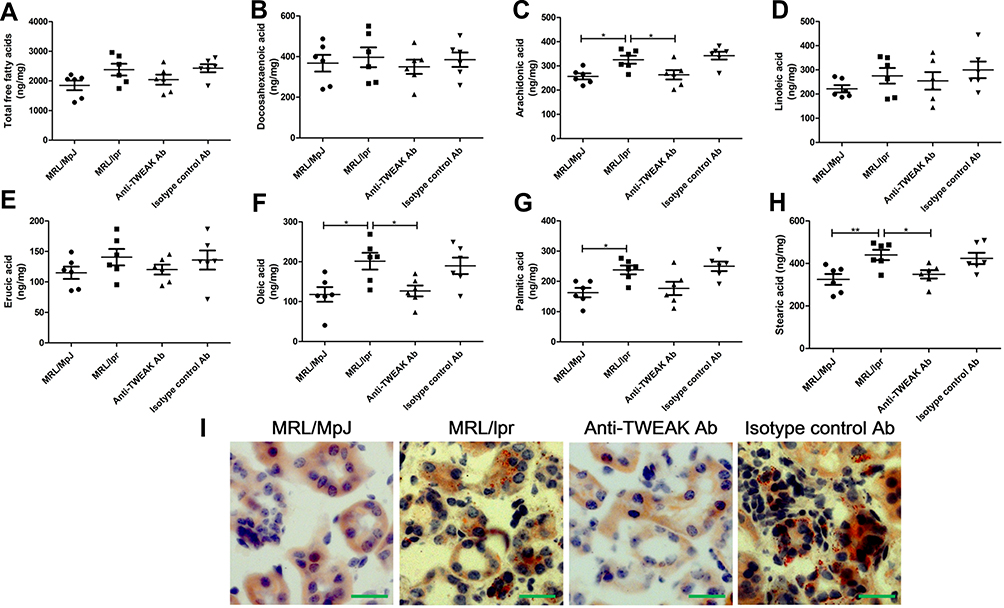 |
Figure 2 The anti-TWEAK antibody decreased the concentration of free fatty acids and alleviated triglyceride accumulation in the kidneys. Thirteen-week-old MRL/lpr mice were treated via intraperitoneal injections of either 2 mg/kg anti-TWEAK antibody or 2 mg/kg isotype control antibody every other day for 56 days. (A–H) Renal free fatty acids were measured using GC-MS. (I) Triglyceride accumulation was identified using oil red O staining (scale bar, 50 μm). The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01; n = 6/group. |
Anti-TWEAK Antibody Promoted Renal PGC-1α Expression in Mice with Lupus
PGC-1α is a key transcription factor that regulates the expression of proteins involved in fatty acid transport and oxidation.9 Thus, we proceeded to determine whether TWEAK could regulate the expression of PGC-1α in the kidneys of mice. Quantitative reverse transcription PCR revealed that the mRNA levels of PGC-1α and its downstream target genes were markedly lower in MRL/lpr mice than in MRL/MpJ mice (Figure 3A-E). However, treatment with the anti-TWEAK antibody increased the mRNA levels of these genes (Figure 3A-E). The protein expression level of PGC-1α was also decreased in MRL/lpr mice, as verified by Western blotting; however, its expression level was increased by treatment with the anti-TWEAK antibody (Figure 3F). Immunofluorescence staining revealed that the expression of PGC-1α was remarkably more abundant in the renal tubules than in the glomeruli of MRL/MpJ mice (Figure 4A). In addition, the renal tubular PGC-1α expression was decreased in MRL/lpr mice and increased in mice treated with the anti-TWEAK antibody (Figure 4B). Therefore, TWEAK negatively modulates PGC-1α expression in the renal tubules of mice with lupus.
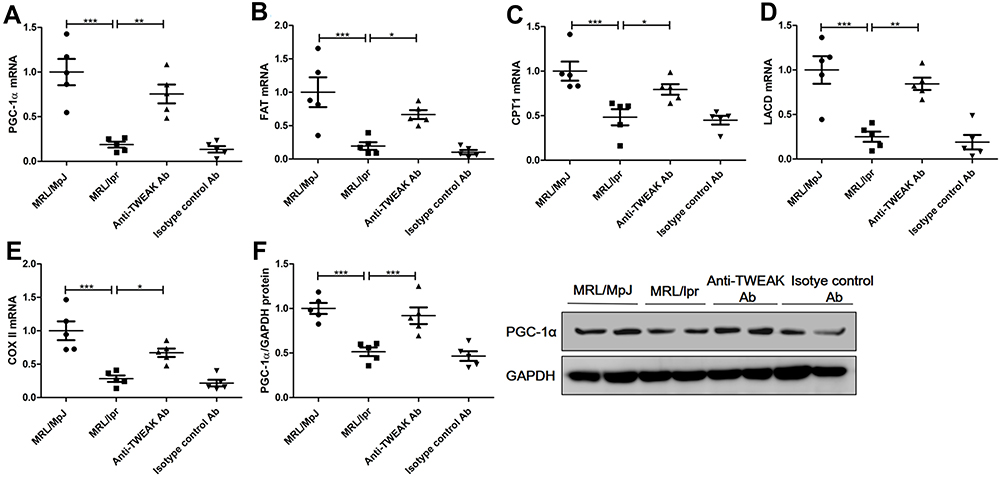 |
Figure 3 The anti-TWEAK antibody upregulated the expression levels of PGC-1α and its downstream target genes in the kidneys. Thirteen-week-old MRL/lpr mice were treated via intraperitoneal injections of either 2 mg/kg anti-TWEAK antibody or 2 mg/kg isotype control antibody every other day for 56 days. (A–E) PGC-1α and its downstream target genes were detected using quantitative reverse transcription PCR. (F) PGC-1α protein expression was examined using Western blotting; the representative band is displayed. The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001; n = 5/group. |
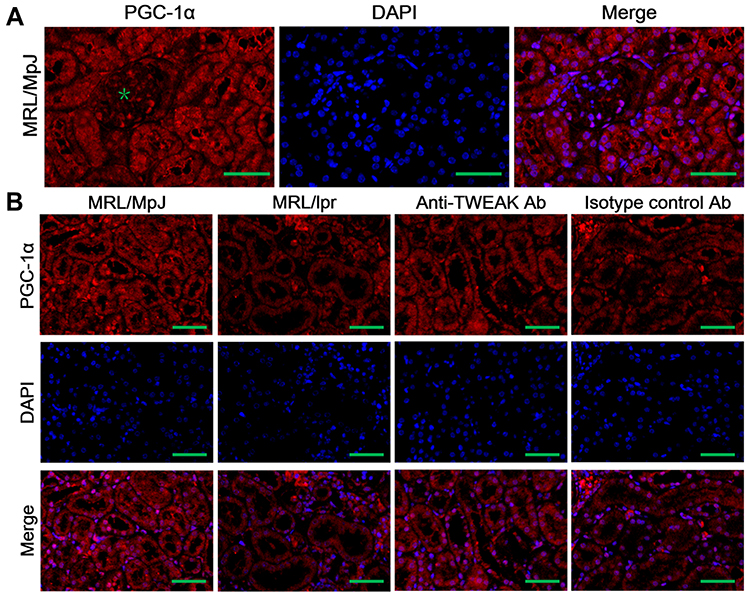 |
Figure 4 The expression of PGC-1α was detected using immunofluorescence staining. (A) The distribution of PGC-1α in the kidneys of MRL/lpr mice was evaluated; the green asterisk indicates the glomeruli. (B) The expression of PGC-1α was detected in mouse renal tubules. Scale bar, 100 μm; n = 5/group. |
TWEAK Downregulated PGC-1α Expression in Cultured Renal Proximal Tubular Epithelial Cells
To verify the negative effect of TWEAK in renal tubular cells, recombinant human TWEAK (rhTWEAK) was used to culture human renal proximal tubular epithelial cells (HK2 cells) in vitro. Treatment with rhTWEAK decreased PGC-1α mRNA expression in both dose- and time-dependent manners (Figure 5A and B). Moreover, rhTWEAK was found to reduce the protein levels of PGC-1α in HK2 cells, as determined by Western blotting (Figure 5C).
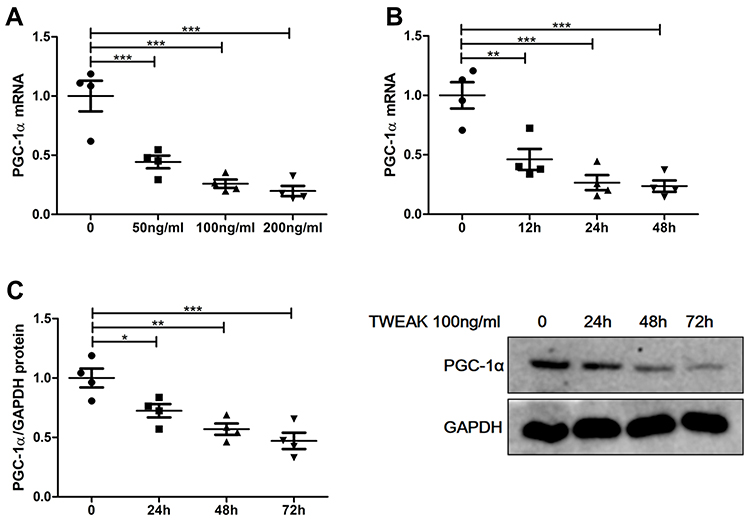 |
Figure 5 Recombinant human TWEAK (rhTWEAK) downregulated PGC-1α expression in HK2 cells. (A) HK2 cells were treated with rhTWEAK at the indicated concentration for 24h, then PGC-1α mRNA were detected by quantitative reverse transcription PCR. 100ng/mL rhTWEAK cultured HK2 cells for the indicated time, and PGC-1α were detected by quantitative reverse transcription PCR (B) and Western blotting (C), respectively; the representative Western blotting band is shown in (C). The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. |
Therapeutic Effects of the Anti-TWEAK Antibody on Renal Interstitial Fibrosis Were Reversed by PGC-1α shRNA
To clarify whether the upregulation of renal tubular PGC-1α expression after treatment with the anti-TWEAK antibody is an accessory change or is involved in the control of renal interstitial fibrosis, we administered renal pelvis injections of PGC-1α shRNA to inhibit the expression of PGC-1α in the kidneys, and observed the changes in renal interstitial fibrosis. In MRL/lpr mice treated with the anti-TWEAK antibody, PGC-1α shRNA not only reduced the PGC-1α mRNA expression in the kidneys (Figure 6A), but also aggravated renal interstitial fibrosis (Figure 6B and C). Moreover, PGC-1α shRNA prevented the decrease in COL-I and fibronectin expression (Figure 6D and E). The levels of serum creatinine and blood urea nitrogen were obviously elevated by treatment with the PGC-1α shRNA (Figure 6F). Therefore, PGC-1α helps to regulate the therapeutic effects of the anti-TWEAK antibody on renal interstitial fibrosis in LN.
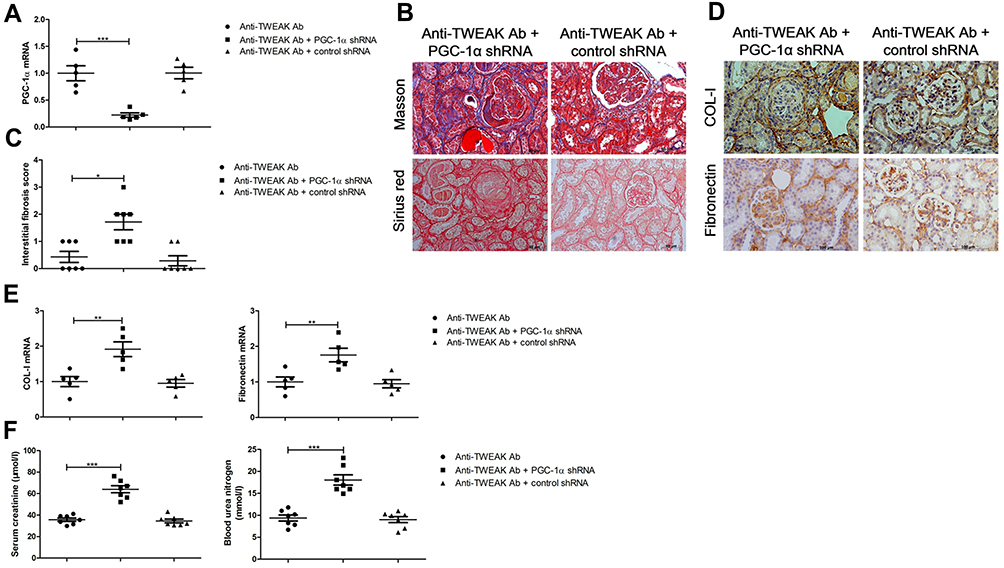 |
Figure 6 PGC-1α shRNA abolished the therapeutic effects of the anti-TWEAK antibody on renal interstitial fibrosis. For experiments involving the inhibition of PGC-1α expression, MRL/lpr mice were treated with PGC-1α shRNA or control shRNA via renal pelvis injection at 11 weeks old; after 2 weeks, MRL/lpr mice were treated with 2 mg/kg anti-TWEAK antibody every other day for 56 days. (A) The mRNA levels of PGC-1α were detected using quantitative reverse transcription PCR (n = 5/group). (B) Masson’s trichrome and Sirius red staining were used to measure the deposition of collagen fibers in the renal interstitium (scale bar, 50 μm), and (C) the interstitial fibrosis scores were evaluated blindly by the same pathologist (n = 7/group). The expression levels of COL-I and fibronectin were measured using immunohistochemical staining (D) and quantitative reverse transcription PCR (E) (scale bar, 100 μm) (n = 5/group). (F) The levels of blood urea nitrogen and serum creatinine were determined (n = 7/group). The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. |
NF-κB Mediates TWEAK-Induced PGC-1α Downregulation in Renal Proximal Tubular Epithelial Cells
Studies have found that TWEAK activates the canonical NF-κB pathway that mediates kidney damage.24,25 Indeed, rhTWEAK treatment stimulated the phosphorylation of IκBα and p65 in HK2 cells, indicating that TWEAK could induce the activation of NF-κB pathway in renal tubular cells (Figure 7A). PDTC suppresses the role of NF-κB by inhibiting IκBα phosphorylation.26,27 In HK2 cells, PDTC inhibited the decrease in PGC-1 α protein levels induced by rhTWEAK treatment (Figure 7B), indicating that the canonical NF-κB pathway participated in the mediation of the TWEAK-induced downregulation of PGC-1α expression.
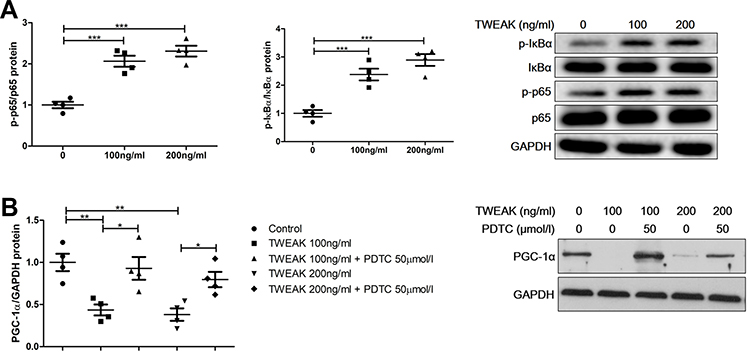 |
Figure 7 PDTC attenuated TWEAK regulation of PGC-1α expression. (A) HK2 cells were treated with rhTWEAK at the indicated concentration for 30 minutes. Thereafter, p-p65, p65, p-IκBα, and IκBα levels were detected using Western blotting; the representative band is displayed. (B) HK2 cells were preincubated with 50 μM PDTC for 1 h, and then incubated with rhTWEAK at the indicated concentration for 48 h. PGC-1α level was detected using Western blotting; the representative band is displayed. The results are expressed as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
The purposes of this study were to elucidate the new mechanism of renal interstitial fibrosis induced by TWEAK and provide a new theoretical basis for the application of the anti-TWEAK antibody in LN. In mice with lupus, an accumulation of lipids and the downregulation of PGC-1α expression were found; treatment with the anti-TWEAK antibody not only inhibited the progress of renal interstitial fibrosis, but also alleviated lipid accumulation and upregulated PGC-1α expression; and the role of the anti-TWEAK antibody in renal interstitial fibrosis was abolished by PGC-1α shRNA. In vitro, TWEAK decreased the expression of PGC-1α in cultured renal proximal tubular epithelial cells through NF-κB activation.
In a study that analyzed human kidney samples from 95 cases, a high-level model analysis was carried out, which revealed two obvious abnormal key pathways in the pathogenesis of chronic kidney disease: inflammation and metabolism.7 Mouse models of renal fibrosis had metabolic changes that were similar to those found in human chronic kidney disease samples: a low fatty acid and carbohydrate metabolism and a high intracellular lipid accumulation.7 In the present study, the concentrations of 44 free fatty acids in mice kidneys were determined using GC-MS. Based on our findings, the main free fatty acids in the kidneys are long chain fatty acids, including docosahexaenoic acid, arachidonic acid, linoleic acid, erucic acid, oleic acid, palmitic acid, and stearic acid. The concentration of these 7 types of long chain fatty acids and total free fatty acids were higher in MRL/lpr mice than in the MRL/MpJ mice. In addition, a higher triglyceride accumulation in the renal tubules of MRL/lpr mice was also found using oil red O staining. Such findings suggest that there is an abnormal lipid accumulation in the kidneys during the pathogenesis of LN.
An abundant lipid accumulation leads to lipotoxicity, thereby promoting the development of renal fibrosis.5,6 However, a study carried out with a FAT transgenic mouse model presenting lipid (including triglycerides and long chain fatty acids) accumulation in tubular epithelial cells found that a higher lipid accumulation alone in the renal tubules was not sufficient to induce the full spectrum of renal fibrosis, but repressed fatty acid oxidation alone was enough to reprogram renal tubular epithelial cells into a profibrotic phenotype, and was related to intracellular lipid accumulation.7 The improvement of fatty acid oxidation by drugs or gene modification in the renal tubules can protect animals from the development of renal fibrosis.7,28 As an essential regulator of fatty acid uptake, oxidation, and energy metabolism, the expression level of PGC-1α is low in chronic kidney disease animal models. In addition, a decreased PGC-1α expression is observed in human kidneys with fibrosis.28 Transforming growth factor β1 (TGF-β1) is one of the most powerful profibrotic cytokines. In fact, TGF-β1-induced fibrosis was found to be related to decreased fatty acid oxidation caused by the inhibition of PGC-1α and its downstream target gene expression. Moreover, PGC-1α transgenic proximal tubular epithelial cells were found to be protected from TGF-β1-induced functional changes, including the expression of profibrotic and apoptosis-associated genes.7 The transgenic expression of PGC-1α improves fatty acid oxidation, reduces apoptosis, and ameliorates kidney fibrosis in Notch1-induced kidney injury animal models.28 In the kidneys of C57BL/6 mice, folic acid treatment led to the accumulation of carnitine-conjugated long chain fatty acids and reduced the levels of key enzymes involved in fatty acid oxidation (ie, CPT 1 and CPT 2), indicating the presence of defects in fatty acid metabolism. In contrast, the transgenic expression of muscle-specific PGC-1α largely corrected these abnormalities and limited fibrosis in the injured kidneys of mice.29 The present study revealed that renal PGC-1α was mainly expressed in tubules, as determined using immunofluorescence staining. Further the expression levels of renal PGC-1α and the downstream target genes (FAT, CPT 1, LACD, and COX II) were much lower in MRL/lpr mice than those in MRL/MpJ mice. FAT mainly mediates the transport of long chain fatty acids into cells, and may also contribute to the oxidation of long chain fatty acids.30 CPT1 is considered to be the rate-limiting enzyme in fatty acid oxidation31 while LACD is involved in fatty acid oxidation32 and COX II is related to oxidative phosphorylation. The reduction in FAT gene expression indicates that the uptake of long chain fatty acids is decreased; however, long-chain fatty acids were still observed to accumulate in the kidneys of mice with lupus. These seemingly contradictory results indicate that there are more obvious defects in the utilization of long-chain fatty acids, such as fatty acid oxidation and oxidative phosphorylation, as confirmed by the decreased expression of CPT1, LACD, and COX II genes. Therefore, we speculate that the decrease in PGC-1α expression levels leads to a fatty acid oxidation defect and oxidative phosphorylation in LN, which contributes to the occurrence of lipid accumulation.
As a proinflammatory factor, TWEAK has been demonstrated to be involved in the metabolic process of glucose and lipids, regulating the production of energy. TWEAK also plays an important role in metabolic diseases, such as obesity, type 2 diabetes mellitus, and atherosclerosis.33–36 TWEAK can destroy the normal energy metabolism of skeletal muscle by inhibiting the expression of PGC-1α and decreasing the content of the mitochondria, eventually leading to muscle atrophy.19,37 In dilated cardiomyopathy, TWEAK, which is highly expressed in the cardiomyocytes of mice, alters the energy supply of the myocardium by reducing the gene expression of PGC-1α, resulting in the remodeling of the myocardial structure and a decline in myocardial contractility.18 In human umbilical vein endothelial cells, TWEAK inhibits the expression of active AMP-activated protein kinase and its downstream protein, PGC-1α, and manganese superoxide dismutase, resulting in endothelial dysfunction.38 TWEAK was also found to negatively affect mitochondrial function by downregulating PGC-1α in an established mouse model of acute kidney injury induced by a folic acid overdose.39 This study showed that treatment with the neutralizing anti-TWEAK antibody reduced triglyceride accumulation and the concentration of free fatty acids, and promoted the expression of renal tubular PGC-1α and the downstream target genes in mice with lupus. Moreover, TWEAK decreased the expression of PGC-1α in renal tubular epithelial cells in vitro. Thus, TWEAK negatively regulates PGC-1α expression in renal tubules, resulting in a deficiency in lipid metabolism in LN.
In vitro, TWEAK activates and differentiates pericytes into cytokine-producing myofibroblasts, and further stimulates established myofibroblasts.40 In mice without prior underlying kidney disease, systemic overexpression of TWEAK induces renal inflammation and fibrosis.24,41 TWEAK deficiency decreases renal fibrosis in a chronic model of experimental unilateral ureteral obstruction.24 Our previous research showed that targeting the TWEAK gene significantly alleviates the progression of renal interstitial fibrosis in MRL/lpr mice.42 The neutralizing anti-TWEAK antibody reduces tubular chemokine expression and interstitial inflammation in a model of acute kidney injury induced by a folic acid overdose.41 Further, this neutralizing antibody eases tubulointerstitial fibrosis in nephrotoxic serum nephritis.17 This study confirmed that the anti-TWEAK antibody is effective at treating renal interstitial fibrosis in LN, which is manifested as a reduction in the deposition of collagen fibers, inhibiting the expression of COL-I and fibronectin, and decreasing the levels of serum creatinine and blood urea nitrogen. However, the therapeutic effect of the anti-TWEAK antibody was abolished by the inhibition of PGC-1α expression, suggesting that PGC-1α mediates the role of the anti-TWEAK antibody in renal interstitial fibrosis. Based on the above results, we concluded that TWEAK induces a deficiency in lipid metabolism by inhibiting renal tubular PGC-1α expression, accelerating the progress of renal interstitial fibrosis.
TWEAK activates multiple signaling pathways, including the canonical NF-κB pathway.24,41 In the current study, the inhibitor of IκBα phosphorylation was used to downregulate the phosphorylation of IκBα and p65, and proved to reverse the decreased PGC-1α protein levels induced by treatment with TWEAK, indicating that TWEAK downregulates PGC-1α expression by activating the canonical NF-κB pathway in renal proximal tubular epithelial cells. Another study also verified that parthenolide (a canonical NF-κB inhibitor) completely prevented PGC-1α downregulation induced by TWEAK at the mRNA and protein levels in renal tubular cells.39 In cardiomyocytes, SC-514, a selective IκB kinase-β inhibitor, completely abolished TWEAK regulation of PGC-1α expression,18 suggesting that NF-κB activation is required for the downregulation of PGC-1α expression.
Conclusion
These findings of the present study suggest that TWEAK prevents renal tubular PGC-1α expression by promoting NF-κB activation, resulting in a deficiency in lipid metabolism and the progress of renal interstitial fibrosis. The upregulation of renal tubular PGC-1α expression to improve lipid metabolism is one of the mechanisms employed by the anti-TWEAK antibody to treat renal interstitial fibrosis. The current work shows that increasing the expression of PGC-1α is an effective method for treating LN.
Abbreviations
AAV, adeno-associated virus; COL-I, type I collagen; COX II, cytochrome oxidase II; CPT1, carnitine palmitoyl-transferase 1; ECM, extracellular matrix; FAT, fatty acid translocase; GC-MS, gas chromatograph system coupled with a mass spectrometer; IP-10, interferon-γ inducible protein 10; LCAD, long-chain acyl dehydrogenase; LN, lupus nephritis; MCP-1, monocyte chemoattractant protein-1; PDTC, ammonium pyrrolidine dithiocarbamate; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator-1α; RANTES, regulated upon activation normal T cell expressed and secreted factor; rhTWEAK, recombinant human TWEAK; SLE, systemic lupus erythematosus; shRNA, short hairpin RNA; TNF, Tumor necrosis factor; TWEAK, tumor necrosis factor-like weak inducer of apoptosis; VCAM-1, vascular endothelial cell adhesion molecule-1.
Ethics Approval
All animal study protocols were approved by the Ethics Committee of Soochow University (Approval No. ECSU-2019000213), Suzhou, China.
Acknowledgments
We acknowledge the support from Central Laboratory at the Second Affiliated Hospital of Soochow University.
Funding
This research was funded by the National Nature Science Foundation of China (81800622); the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJB320020); Suzhou Health and Key Talent Project (GSWS2019011); and Jiangsu social development project (BE2019663).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Fornoni A, Merscher S. Lipid metabolism gets in a JAML during kidney disease. Cell Metab. 2020;32(6):903–905. doi:10.1016/j.cmet.2020.11.002
2. Fu Y, Sun Y, Wang M, et al. Elevation of JAML Promotes diabetic kidney disease by modulating podocyte lipid metabolism. Cell Metab. 2020;32(6):1052–1062.e8. doi:10.1016/j.cmet.2020.10.019
3. Pei K, Gui T, Li C, et al. Recent progress on lipid intake and chronic kidney disease. Biomed Res Int. 2020;2020:3680397. doi:10.1155/2020/3680397
4. Ke Q, Yuan Q, Qin N, et al. UCP2-induced hypoxia promotes lipid accumulation and tubulointerstitial fibrosis during ischemic kidney injury. Cell Death Dis. 2020;11(1):26. doi:10.1038/s41419-019-2219-4
5. Declèves AE, Zolkipli Z, Satriano J, et al. Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 2014;85(3):611–623. doi:10.1038/ki.2013.462
6. Du XG, Ruan XZ. Lipid metabolism disorder and renal fibrosis. Adv Exp Med Biol. 2019;1165:525–541. doi:10.1007/978-981-13-8871-2_26
7. Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21(1):37–46. doi:10.1038/nm.3762
8. Calvo JA, Daniels TG, Wang X, et al. Muscle-specific expression of PPARgamma coactivator-1alpha improves exercise performance and increases peak oxygen uptake. J Appl Physiol (1985). 2008;104(5):1304–1312. doi:10.1152/japplphysiol.01231.2007
9. Chambers JM, Wingert RA. PGC-1α in disease: recent renal insights into a versatile metabolic regulator. Cells. 2020;9:10. doi:10.3390/cells9102234
10. Tadaishi M, Miura S, Kai Y, Kano Y, Oishi Y, Ezaki O. Skeletal muscle-specific expression of PGC-1α-b, an exercise-responsive isoform, increases exercise capacity and peak oxygen uptake. PLoS One. 2011;6(12):e28290. doi:10.1371/journal.pone.0028290
11. Feng X, Pan W, Liu L, et al. Prognosis for hospitalized patients with systemic lupus erythematosus in China: 5-year update of the jiangsu cohort. PLoS One. 2016;11(12):e0168619. doi:10.1371/journal.pone.0168619
12. Liu Z, Xue L, Liu Z, et al. Tumor Necrosis factor-like weak inducer of apoptosis accelerates the progression of renal fibrosis in lupus nephritis by activating SMAD and p38 MAPK in TGF-β1 signaling pathway. Mediators Inflamm. 2016;2016:8986451. doi:10.1155/2016/8986451
13. Michaelson JS, Wisniacki N, Burkly LC, Putterman C. Role of TWEAK in lupus nephritis: a bench-to-bedside review. J Autoimmun. 2012;39(3):130–142. doi:10.1016/j.jaut.2012.05.003
14. Zhao Z, Burkly LC, Campbell S, et al. TWEAK/Fn14 interactions are instrumental in the pathogenesis of nephritis in the chronic graft-versus-host model of systemic lupus erythematosus. J Immunol. 2007;179(11):7949–7958. doi:10.4049/jimmunol.179.11.7949
15. Sanz AB, Izquierdo MC, Sanchez-Niño MD, et al. TWEAK and the progression of renal disease: clinical translation. Nephrol Dial Transplant. 2014;29(Suppl 1):i54–54i62. doi:10.1093/ndt/gft342
16. Xia Y, Herlitz LC, Gindea S, et al. Deficiency of fibroblast growth factor-inducible 14 (Fn14) preserves the filtration barrier and ameliorates lupus nephritis. J Am Soc Nephrol. 2015;26(5):1053–1070. doi:10.1681/ASN.2014030233
17. Xia Y, Campbell SR, Broder A, et al. Inhibition of the TWEAK/Fn14 pathway attenuates renal disease in nephrotoxic serum nephritis. Clin Immunol. 2012;145(2):108–121. doi:10.1016/j.clim.2012.08.008
18. Shi J, Jiang B, Qiu Y, et al. PGC1α plays a critical role in TWEAK-induced cardiac dysfunction. PLoS One. 2013;8(1):e54054. doi:10.1371/journal.pone.0054054
19. Hindi SM, Mishra V, Bhatnagar S, et al. Regulatory circuitry of TWEAK-Fn14 system and PGC-1α in skeletal muscle atrophy program. FASEB J. 2014;28(3):1398–1411. doi:10.1096/fj.13-242123
20. Couto M, Cates C. Laboratory guidelines for animal care. Methods Mol Biol. 2019;1920:407–430. doi:10.1007/978-1-4939-9009-2_25
21. Corna D, Morigi M, Facchinetti D, Bertani T, Zoja C, Remuzzi G. Mycophenolate mofetil limits renal damage and prolongs life in murine lupus autoimmune disease. Kidney Int. 1997;51(5):1583–1589. doi:10.1038/ki.1997.217
22. Tan X, Li Y, Liu Y. Paricalcitol attenuates renal interstitial fibrosis in obstructive nephropathy. J Am Soc Nephrol. 2006;17(12):3382–3393. doi:10.1681/ASN.2006050520
23. Gu L, Weinreb A, Wang XP, et al. Genetic determinants of autoimmune disease and coronary vasculitis in the MRL-lpr/lpr mouse model of systemic lupus erythematosus. J Immunol. 1998;161(12):6999–7006.
24. Ucero AC, Benito-Martin A, Fuentes-Calvo I, et al. TNF-related weak inducer of apoptosis (TWEAK) promotes kidney fibrosis and Ras-dependent proliferation of cultured renal fibroblast. Biochim Biophys Acta. 2013;1832(10):1744–1755. doi:10.1016/j.bbadis.2013.05.032
25. Gao HX, Campbell SR, Burkly LC, et al. TNF-like weak inducer of apoptosis (TWEAK) induces inflammatory and proliferative effects in human kidney cells. Cytokine. 2009;46(1):24–35. doi:10.1016/j.cyto.2008.12.001
26. Tian LX, Tang X, Ma W, et al. Knockout of cytochrome P450 1A1 enhances lipopolysaccharide-induced acute lung injury in mice by targeting NF-κB activation. FEBS Open Biol. 2020. doi:10.1002/2211-5463.12977
27. Martínez-Miguel P, Medrano-Andrés D, Griera-Merino M, et al. Tweak up-regulates endothelin-1 system in mouse and human endothelial cells. Cardiovasc Res. 2017;113(2):207–221. doi:10.1093/cvr/cvw239
28. Han SH, Wu MY, Nam BY, et al. PGC-1α protects from notch-induced kidney fibrosis development. J Am Soc Nephrol. 2017;28(11):3312–3322. doi:10.1681/ASN.2017020130
29. Peng H, Wang Q, Lou T, et al. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun. 2017;8(1):1493. doi:10.1038/s41467-017-01646-6
30. Rey E, Meléndez-Rodríguez F, Marañón P, et al. Hypoxia-inducible factor 2α drives hepatosteatosis through the fatty acid translocase CD36. Liver Int. 2020;40(10):2553–2567. doi:10.1111/liv.14519
31. Mørkholt AS, Oklinski MK, Larsen A, et al. Pharmacological inhibition of carnitine palmitoyl transferase 1 inhibits and reverses experimental autoimmune encephalitis in rodents. PLoS One. 2020;15(6):e0234493. doi:10.1371/journal.pone.0234493
32. Beck ME, Zhang Y, Bharathi SS, et al. The common K333Q polymorphism in long-chain acyl-CoA dehydrogenase (LCAD) reduces enzyme stability and function. Mol Genet Metab. 2020;131(1–2):83–89. doi:10.1016/j.ymgme.2020.04.005
33. Hernández EÁ, Kahl S, Seelig A, et al. Acute dietary fat intake initiates alterations in energy metabolism and insulin resistance. J Clin Invest. 2017;127(2):695–708. doi:10.1172/JCI89444
34. Sastre C, Fernández-Laso V, Madrigal-Matute J, et al. Genetic deletion or TWEAK blocking antibody administration reduce atherosclerosis and enhance plaque stability in mice. J Cell Mol Med. 2014;18(4):721–734. doi:10.1111/jcmm.12221
35. Tiller G, Fischer-Posovszky P, Laumen H, et al. Effects of TWEAK (TNF superfamily member 12) on differentiation, metabolism, and secretory function of human primary preadipocytes and adipocytes. Endocrinology. 2009;150(12):5373–5383. doi:10.1210/en.2009-0488
36. Escoté X, Gómez-Zorita S, López-Yoldi M, et al. Role of omentin, vaspin, Cardiotrophin-1, TWEAK and NOV/CCN3 in obesity and diabetes development. Int J Mol Sci. 2017;18:8. doi:10.3390/ijms18081770
37. Sato S, Ogura Y, Mishra V, et al. TWEAK promotes exercise intolerance by decreasing skeletal muscle oxidative phosphorylation capacity. Skelet Muscle. 2013;3(1):18. doi:10.1186/2044-5040-3-18
38. Liu H, Peng H, Xiang H, et al. TWEAK/Fn14 promotes oxidative stress through AMPK/PGC‑1α/MnSOD signaling pathway in endothelial cells. Mol Med Rep. 2018;17(1):1998–2004. doi:10.3892/mmr.2017.8090
39. Ruiz-Andres O, Suarez-Alvarez B, Sánchez-Ramos C, et al. The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 2016;89(2):399–410. doi:10.1038/ki.2015.332
40. Gomez IG, Roach AM, Nakagawa N, et al. TWEAK-Fn14 signaling activates myofibroblasts to drive progression of fibrotic kidney disease. J Am Soc Nephrol. 2016;27(12):3639–3652. doi:10.1681/ASN.2015111227
41. Sanz AB, Justo P, Sanchez-Niño MD, et al. The cytokine TWEAK modulates renal tubulointerstitial inflammation. J Am Soc Nephrol. 2008;19(4):695–703. doi:10.1681/ASN.2007050577
42. Xue L, Liu Z, Hu J, Huang J, Wen J, Liu Z. Estrogen-induced expression of tumor necrosis factor-like weak inducer of apoptosis through ERα accelerates the progression of lupus nephritis. Rheumatology (Oxford). 2016;55(10):1880–1888. doi:10.1093/rheumatology/kew248
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.