Back to Journals » International Journal of Nanomedicine » Volume 16
Anti-Cancer Activity Based on the High Docetaxel Loaded Poly(2-Oxazoline)s Micelles
Authors Xu M, Yao C, Zhang W, Gao S ![]() , Zou H, Gao J
, Zou H, Gao J ![]()
Received 18 December 2020
Accepted for publication 18 March 2021
Published 9 April 2021 Volume 2021:16 Pages 2735—2749
DOI https://doi.org/10.2147/IJN.S298093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Min Xu, 1, 2,* Chong Yao, 3,* Wei Zhang, 1 Shen Gao, 1 Hao Zou, 1 Jing Gao 1, 4
1School of Pharmacy, Second Military Medical University, Shanghai, 200433, People’s Republic of China; 2Department of Pharmacy, Changzheng Hospital, Shanghai, 200003, People’s Republic of China; 3Clinical Pharmacy Center, Department of Pharmacy, Chinese PLA General Hospital, Beijing, 100850, People’s Republic of China; 4State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing, 100850, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Gao; Hao Zou
School of Pharmacy, Second Military Medical University, No. 325 Guohe Road, Yangpu District, Shanghai, People’s Republic of China
Tel +86-10-6687-4665
Email [email protected]; [email protected]
Purpose: Nanocarriers, with a high drug loading content and good safety, to achieve desirable therapeutic effect are always the goals for industry and research.
Methods and Results: In the present study, we developed a docetaxel loaded poly-2-oxazoline polymer micellar system which employed poly-2-butyl-2 oxazoline and poly-2-methyl-2 oxazoline as the hydrophobic chain and hydrophilic chain, respectively. This micellar system achieves a high load up to 25% against the docetaxel, and further demonstrates an IC50 as low as 40% of the commercialized docetaxel injection in vitro and a double maximum tolerated dose in MCF-7 cells in vivo.
Conclusion: The high drug loading content, superior safety, and considerable anti-cancer activity make this newly developed docetaxel loaded poly(2-oxazoline) micelle go further in future clinical research.
Keywords: poly(2-oxazoline)s, micelle, docetaxel, high loading, anti-cancer activity
Corrigendum for this paper has been published
Introduction
Cancer is a major public health problem worldwide, with a rapidly growing incidence and mortality. It was estimated that by 2018 there would be 18.1 million new cancer cases and 9.6 million cancer-related deaths reported.1 Taxanes are a class of anti-cancer drugs extracted or semi-synthesized from the bark of Taxus plants. Taxanes promote the assembly of microtubules and inhibit the depolymerization of microtubules by binding to the β-protein and blocking cells in the G2/M phase, resulting in the inhibition of mitosis and proliferation of cancer cells.2,3 Its active monomers, including paclitaxel (PTX), docetaxel (DTX), and cabazitaxel (CBZ), have been successfully developed and used clinically in advanced ovarian, breast, and non-small cell lung cancers.4,5
In 2014, ASCO reviewed taxol-therapy in a sixty-year study of breast cancer as a landmark finding that taxanes significantly reduced breast cancer recurrence and mortality rate.6 The toxic side effects and low drug loading of the taxane formulations remain the main problems in clinical applications. The active drug content in Taxol® is only 1% and the remaining 99% is polyoxyethylene castor oil and ethanol. The main excipients in docetaxel and cabazitaxel are Tween 80 and ethanol. These excipients can cause serious allergic reactions, hemolysis, and cholestasis.7,8 Although the drug loading in albumin-bound paclitaxel nano-form, Abraxane®, is higher than that of Taxol®, it is still less than 10%. Moreover, compared to Taxol®, the main excipient albumin disables radiotherapy during administration and causes peripheral neuropathy.9–11 Improving tumor accumulation is a critical mission of nanomedicine-based drug delivery.12 In recent years, some polymeric nanocarriers have spang up to focus on the improvement of antitumor efficacy through high drug loading content.13–15
Polymeric micelles based on amphiphilic block polymers represent a class of nanocarriers with well-defined core-shell nanostructures that facilitate the loading of hydrophobic chemotherapeutic drugs. In addition, their nanosize enables passive accumulation in tumor tissues, resulting in increased antitumor effects owing to the enhanced permeation and retention (EPR) effect.16–18 As a type of polymeric micelles, poly(2-oxazoline)s (POx) have recently attracted considerable attention in biomedical applications due to their tunable properties and structure, which compare favorably to polyethers such as PEG.19–21 Furthermore, poly(2-ethyl-2-oxazoline) (PEtOx) has been approved by the FDA as an indirect food additive.22 Poly(2-methyl-2-oxazoline)- block-poly(2-butyl-2-oxazoline)- block-poly(2-methyl-2-oxazoline) (PMeOx-bPBuOx-b-PMeOx) is a poly(2-oxazoline)s micelle (POx) based triblock copolymer. Kabanov and co-workers reportedly loaded POx with almost 50 wt.% of paclitaxel (PTX) in stable and injectable formulations,23 which exhibited a significantly increased therapeutic efficacy in vivo against ovarian cancer.15 The high loading and high stability of the poly(2-oxazoline)s micelles may be due to the block copolymers comprising poly(2-butyl-2-oxazoline) as hydrophobic core.24
In the present study, we developed a docetaxel loaded poly-2-oxazoline polymer micellar system which employed poly-2-butyl-2 oxazoline and poly-2-methyl-2 oxazoline as the hydrophobic chain and hydrophilic chain, respectively. Followed by self-assembly in an aqueous solution and achieves a high load up to 25% against the docetaxel. The anti-cancer activity was investigated in MCF-7 and A549 cells and compared with the commercially available docetaxel injection. The newly docetaxel loaded poly-2-oxazoline micelles demonstrated a promising therapeutic effect in the breast cancer MCF-7 cells in vitro and in vivo, with good safety, stability, and a considerably higher maximum tolerated dose compared to the clinically approved docetaxel injection, indicating a potential for the future development.
Materials and Methods
Materials
DTX drug was purchased from Dalian Meilun Biotechnology Co. Ltd. (Dalian, China). Docetaxel injection (DJ) was purchased Qilu Pharmaceutical Co. Ltd. (Jinan, China). Amphiphilic triblock poly(2-oxazoline)s were purchased from Advanced Polymer Materials Inc. (Montreal, Canada). Coumarin-6 was purchased from Sigma-Aldrich (Shanghai, China). MCF-7 and A549 cells were purchased from the Institute of Biochemistry and Cell Biology (Shanghai, China). Dulbecco’s modified eagle medium (DMEM), fetal bovine serum (FBS), and penicillin-streptomycin solution (5 kU/mL) were purchased from Thermo Fisher Scientific (Shanghai, China). 1,1ʹ-Dioctadecyltetramethyl indotricarbocyanine iodide (DiR) was purchased from Biotium (CA, USA). All animal experiments were approved by the ethics committee of Beijing Institute of Pharmacology and Toxicology, and were performed in accordance with the Guidelines on Laboratory Animal Welfare issued by Ministry of Science and Technology of the People’s Republic of China.
Methods
Preparation of DTX Loaded Poly(2-Oxazoline)s Micelles (DPM) and Blank Poly(2-Oxazoline)s Micelles (BPM)
The thin-film hydration method was used to prepare BPM and DPM. Briefly, 10 mg/mL POx ethanol stock solution and 10 mg/mL DTX ethanol stock solution were prepared by ultrasound using 10 mg POx and 10 mg DTX dissolved in 1 mL of ethyl alcohol. Next, precision transferred 100 μL of POx stock solution and 40 μL of DTX stock solution were mixed and heated over 47°C steam to form a thin film. Subsequently, 40 μL of normal saline was added, hydrated at 60°C, and followed by high-speed centrifugation at 10,000 rpm for 5 min to prepare the DPM. The BPM were prepared in the same manner as DPM without the addition of DTX.
Particle Size, Stability and Transmission Electron Microscopy (TEM)
The particle size of DPM (1 mg/mL) and BPM (1 mg/mL) were measured by dynamic light scattering (Zetasizer Nano ZS90, Malvern) at 25°C, and the stability at 1, 3, 6, 9, 12, 15 days was investigated using the same method.
BPM, DPM and the DPM, stored at 4°C for 10 days, eliminated NaCl by dialysis and were examined using a transmission electron microscopy (TEM, Jeol, Japan).
Drug Loading Content (DL) and Encapsulation Efficiency (EE)
DL and EE were measured using the HPLC method using an Ultimate 3000 (Thermo Fisher, US) with a UV detector. All reagents were of HPLC grade and were filtered and degassed prior to use. A C18 reversed-phase column (150 mm × 4.6 mm, 5 μm) was used. The injection volume was 20 µL. The flow rate was set at 1 mL/min. The mobile phase consisted of acetonitrile in water (50:50, v/v). The UV detection was performed at 228 nm and the column temperature was 30°C. DPM solution (1 mg/mL) was diluted 10-fold using the mobile phase. Samples were filtered through a 0.45 µm filter prior to injection. DL and EE values were calculated using the following formula:
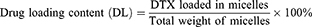
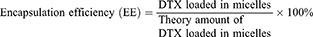
Measurement of Critical Micelle Concentration (CMC) of DPM
The CMC was determined using the standard procedure.25,26 Briefly, a pyrene solution in acetone (5.0 × 10−5 M) was added dropwise to a brown volumetric flask. Next, the acetone was completely evaporated at room temperature. The polymer solutions at appropriate concentrations (10−3~103 μg/mL) in the assay buffer were added to the vials to obtain a final concentration of 5×10−7 M pyrene. The solutions were incubated at 37°C, at 50 rpm for 6 h in the dark. The pyrene fluorescence spectra were recorded using the F-7000 fluorescence spectrometer (Hitachi, Japan) at λex=333 nm, λem=360–390 nm, step width 0.5 nm, and an integration time of 0.1 s. The CMC value is assumed where a steep increase in fluorescence intensity is observed. Furthermore, the fluorescence intensity of the I1 (373nm) band was compared to the intensity of the I3 (384 nm) band, estimating the polarity of the pyrene probe environment.
In vitro Drug Release Study
In order to investigate the in vitro drug release profile of DTX from DPM, 0.9 mL of DPM was transferred into a dialysis bag (MWCO 3500 Da). The dialysis bag was immersed in 45 mL of PBS at pH 7.4 and pH 5.5, with a stirring speed of 100 rpm to achieve sink conditions at 37°C. At selected time intervals, 1 mL of the external solution was withdrawn and replaced with the same volume of fresh PBS. The samples were filtered and measured using HPLC as previously described.
Cellular Uptake Assay
In this assay, coumarin-6 was used as a fluorescent probe and loaded into the micelles (C6-POx). The preparation was as described in section 2.2.1 except the stock concentration of C6 and POx was 50 μg/mL and 1mg/mL, respectively. The micelles were prepared using 10 mL from each of the two stock solutions. The particle size of C6-POx was measured using dynamic light scattering (Zetasizer Nano ZS90, Malvern) at 25°C. The concentration of C6 was measured using the F-7000 fluorescence spectrometer (Hitachi, Japan) at λex=450 nm and λem=508 nm, and subsequently, DL was calculated.
Confocal laser scanning microscopy (CLSM) was used to observe the cellular uptake of C6-POx. MCF-7 and A549 cells were seeded in confocal dishes at a density of 2×105 per dish and grown for 12 h at 37°C in 5% CO2. C6-POx micelles were diluted with saline to 20 μg/mL and with DMEM (10% FBS) to 1 μg/mL. Next, the medium was replaced, C6 and C6-POx micelles were added into a dish and incubated for 2 h and 4 h at 37°C in the dark. After removal of the culture medium, cells were washed with PBS and fixed with fresh 4% paraformaldehyde, followed by treatment with 4ʹ, 6-diamidino-2-phenylindole (DAPI) for nuclear staining. Next, the cells were sealed with the mounting medium and visualized using a confocal laser scanning microscope (Leica, US). The fluorescence intensity was calculated using the Image J software.
Cell Viability Assay
The cytotoxicity of BPM, DPM, and DJ was assessed in MCF-7 and A549 cells. MCF-7 and A549 cells were seeded in 96-well plates at a density of 6×103 cells per well overnight under 5% CO2, at 37°C for 12 h. Next, the depleted medium was replaced with fresh culture medium containing DPM and DJ at various concentrations (0 μg/mL~100 μg/mL DTX) or blank micelles at various concentrations (0 μg/mL~1000 μg/mL), and incubated for 48 h. Cell viability was determined using the CCK-8 assay according to the manufacturer’s protocol (Dojindo, Japan). Briefly, 10 μL of CCK-8 solution was added to each well of the plate and incubated for 2 h in the incubator. A microplate reader (Thermo Fisher Scientific, Waltham, MA) was used for optical density (OD) determination at 450 nm. Each treatment condition was repeated in quintuplicate and all data have been expressed as the means ± SD.
Cell Apoptosis
The rate of apoptosis in the MCF-7 and A549 cells was determined using the Annexin V-FITC/PI double dyeing method. MCF-7 and A549 cells were seeded in 6-well plates at a density of 3×105 cells per well overnight under 5% CO2, at 37°C for 12 h. After treatment with DPM, DJ (both DTX equivalent 20 ng/mL and 350 ng/mL), and BPM (50 ng/mL and 875 ng/mL) for 48 h, the cells were trypsinized, harvested, washed with PBS, and stained with Annexin V-FITC and propidium iodide (PI) according to the manufacturer’s protocol. Each sample was analyzed using an analytical flow cytometer (FACSCalibur; BD Biosciences, US).
In vivo Distribution Assay
DiR, a near-infrared fluorophore, was used to investigate the vivo distribution of BPM and DPM. The DiR loaded micelle preparation is as mentioned in section 2.2.1 except the stock concentrations of DiR and POx were 200 μg/mL and 2 mg/mL, respectively. The micelles were prepared by using 5 mL from the two stock solutions. The particle size of DiR-POx was measured by dynamic light scattering (Zetasizer Nano ZS90, Malvern) at 25°C. The concentration of DiR was measured using the UV-VIS spectrophotometer (TECHCOMP, China) at 749 nm and DL was calculated.
The xenograft tumor model was generated by injecting 0.2 mL of the MCF-7 cell suspension (2×106) into the right flank of male BALB/C nude mice. Once tumors had grown to approximately 400 mm3 in size, the mice were injected with saline, DiR, and DiR-POx (DiR equivalent 15 μg) through the caudal vein and scanned at 2, 4, 6, 8, 12, and 24 h post-inoculation using IVIS imaging system (IVIS Lumina II, Caliper Life Sciences, Hopkinton, MA) with a filter (excitation at 745 nm and emission at 780 nm). The exposure time was set to 1 s per image. The mice were sacrificed 24 h after administration and the heart, liver, spleen, lungs, kidneys, and tumors were isolated, washed in saline, and immediately imaged. Images were analyzed using the Living Imaging software (Caliper Life Sciences, Hopkinton, MA).
In vivo Maximum Tolerated Dose (MTD) and Anti-Tumor Effects
The MTD was investigated by dose escalation in healthy nude mice injected through the caudal vein (q3d×4) with BPM (125 mg/kg POx), DPM (DTX equivalent: 5 mg/kg~50 mg/kg), and DJ (DTX equivalent: 5 mg/kg~20 mg/kg). The body weights of the nude mice were recorded and MTD is defined as the dose at which the weight loss of the highest dose group is less than 15%.27
The xenograft tumor model was generated as described in In vivo Distribution Assay. Once the tumors had grown to approximately 200 mm3 in size, mice with visible MCF-7 tumors were randomly divided into five groups and injected through the caudal vein (q3d × 4) with saline, BPM (50 mg/kg POx), DJ (10 mg/kg DTX), DPM (10mg/kg DTX), and DPM (20 mg/kg DTX). The longest diameter (D) and shortest diameter (d) were measured every 2 days using a Vernier caliper. The body weights of the nude mice were also recorded. Tumor volume (V) is calculated as: V= D × d2/2. Mice were sacrificed on day 24, and tumors were excised from mouse carcass and weighed. Inhibition rate of tumor (IRT) is calculated as: IRT (%) = |(WEG -WCG)|/WEG×100%. “WEG” means the weight of mice in the experimental group and “WCG” means the weight of mice in the control group.
In vivo Cell Apoptosis and Histological Analysis
After the mice were sacrificed, tumors were isolated, fixed in 4% paraformaldehyde, and subsequently embedded in paraffin. Subsequently, the samples were stained using the TdT-mediated dUTP Nick-End Labeling (TUNEL) method or the hematoxylin and eosin (H&E) method to observe cell apoptosis and histological changes.
Results
Preparation and Characterization of DPM and BPM
The DPM and BPM were prepared by the thin-film hydration method (Figure 1). As shown in Table 1, the ratio of drug to carrier was 2:5, the DL was 24.67 ± 0.98%, and the EE was 86.31 ± 3.44%. The DLS analysis indicated that the diameter of BPM was 21.84 ± 0.31 nm, and the average diameter of DPM increased to 48.92 ± 0.33 nm. Within 15 days, the average diameter of DPM was stable around 50 nm (Figure 2A) and the polymer dispersity index (PDI) < 0.2 (Figure 2B). The TEM images (Figure 2A) demonstrated that DPM and BPM were spherical, the blank micelles have a particle size of about 20 nm, and the drug-loaded micelles have a particle size of about 35 nm, consistent with the DLS results. In addition, the drug-loaded POx micelles, placed at 4°C for 10 days, were observed under TEM, and the shape of DPM was still near-spherical (Figure 2A). The CMC of POx was 1.34 ± 0.48 μg/mL.
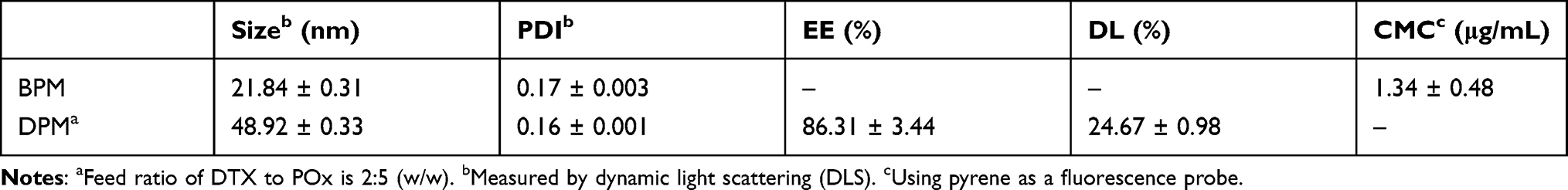 |
Table 1 Particle Size Distribution, CMC, Encapsulation Efficiency and Drug Loading Content of the Prepared Micelles |
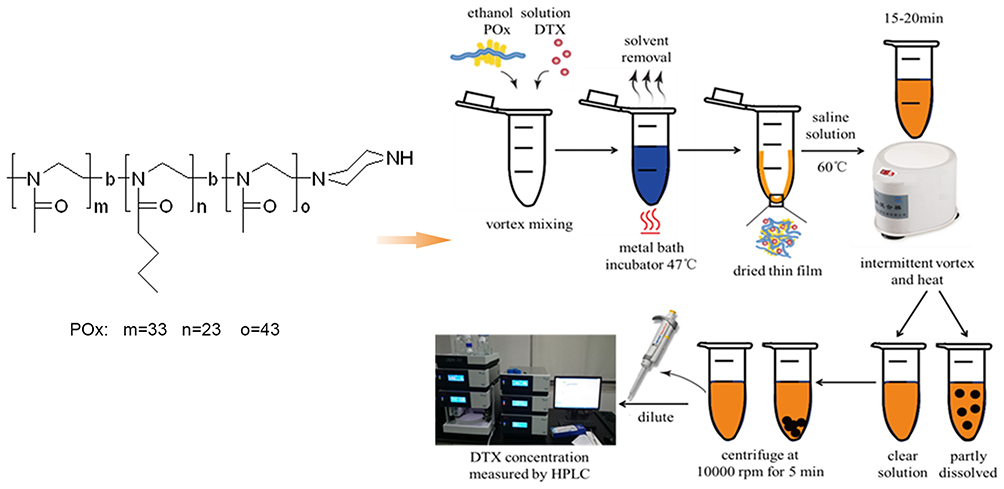 |
Figure 1 Chemical structure of amphiphilic poly(2-oxazoline)s (POx) triblock copolymers and the standard operating procedure for small scale micelle production (1–5mg) (blue: ethanol solution, yellow: DI water solution, black: undissolved drug). |
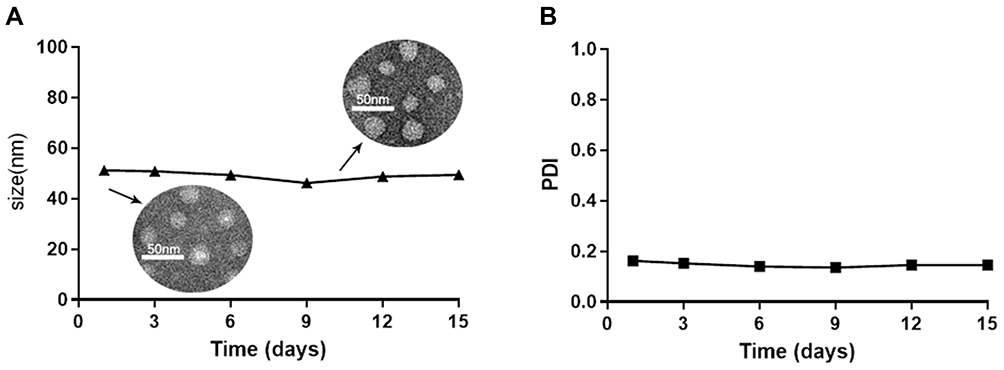 |
Figure 2 Stability of the DTX loaded micelles determined by the size (A) and PDI (B) measurements over time. |
Release Behavior of DPM and DJ in vitro
The DTX release profiles of DPM and DJ were investigated at pH 5.5 and pH 7.4. As shown in Figure 3, the release rate of DPM in the initial stage was higher than that of DJ in both pH environments, followed by a slower release. At pH 5.5, the cumulative release rate of DPM at 24 h was as high as 81.42 ± 1.87% or more, while the cumulative release rate of DPM was approximately 62.6 ± 3.78% at pH 7.4.
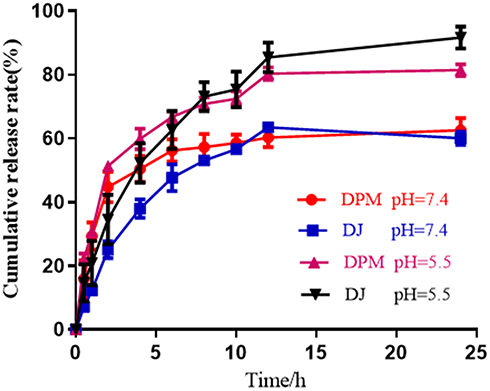 |
Figure 3 Drug release profiles of DTX from different formulations at 37°C in PBS buffer at pH 5.5 and pH 7.4. Data are expressed as mean ± SD (n= 3). |
Cellular Uptake
The DLS analysis indicated that the average diameter of C6-POx was 24.60 ± 1.13 nm and the PDI was 0.222 ± 0.015 nm. The C6 loading content was 3.75 ± 0.38% measured using a fluorescence spectrometer.
CLSM was performed to analyze the cellular uptake of C6-POx in MCF-7 and A549 cells. As shown in Figure 4, the nuclei were stained with DAPI, and the green fluorescence corresponded to the intracellularly released C6. In MCF-7 cells (Figure 4A), after incubation with C6-POx for 2 h, green fluorescence was observed in the perinuclear regions of the cytoplasm and the intensity in the C6-POx treated group was stronger than that in the free C6 treated group (C6-POx: 64.75, free C6: 25.88). After 4 h of incubation (Figure 4B), the green fluorescence intensity (C6) in the free C6 treated group was stronger than that observed at 2 h. However, the green fluorescence intensity in the C6-POx treated group (64.00) was considerably stronger than the free C6 treated group (43.67). In A549 cells, no difference in the green fluorescence intensity between the free C6 and C6-POx group was observed at 2 h (Figure 4C). Furthermore, no obvious change in the green fluorescence intensity developed 4 h after administration (Figure 4D).
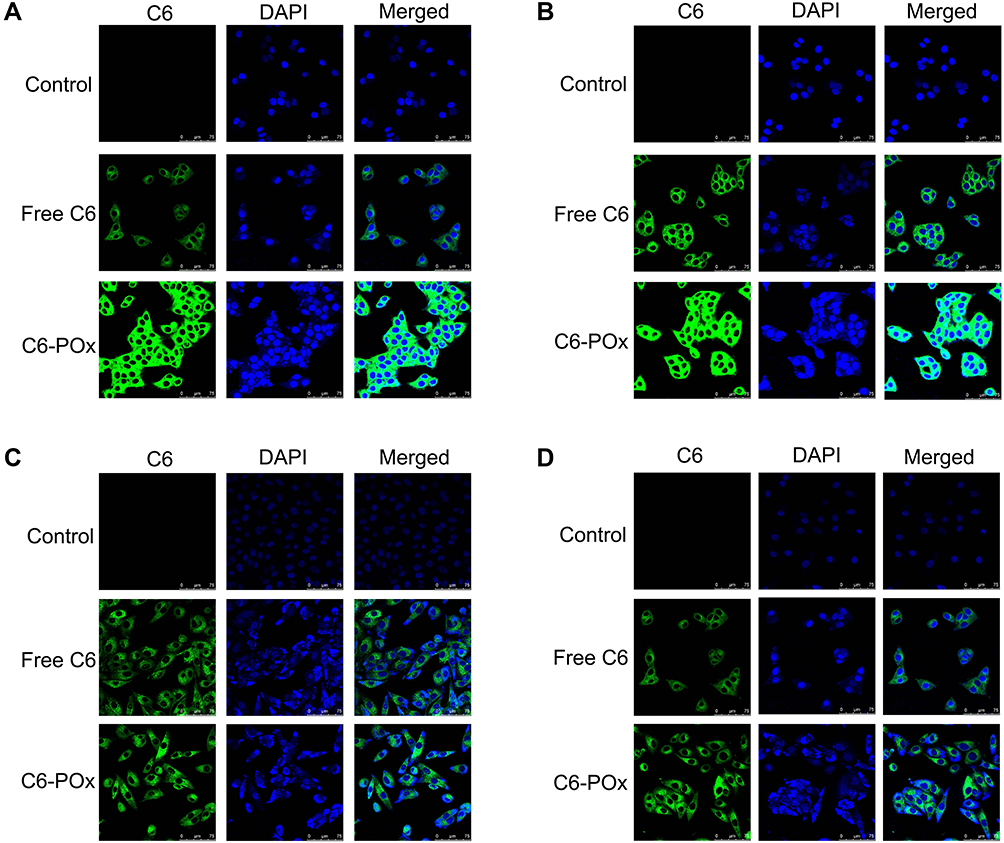 |
Figure 4 The cell uptake of C6-POx towards MCF-7 and A549 cells by laser scanning confocal microscopy. ((A) MCF-7 cells, treated for 2h; (B) MCF-7 cells, treated for 4h; (C) A549 cells, treated for 2h; (D) A549 cells, treated for 4h). |
Cell Viability Assay
The cytotoxicity of DPM and BPM was investigated in MCF-7 and A549 cells. As shown in Figure 5A, the cell viability of MCF-7 and A549 cells treated with 1~500 μg/mL BPM was greater than 95%, and the cell viability was maintained at 80% when the POx concentration was increased to 1000 μg/mL for 48 h.
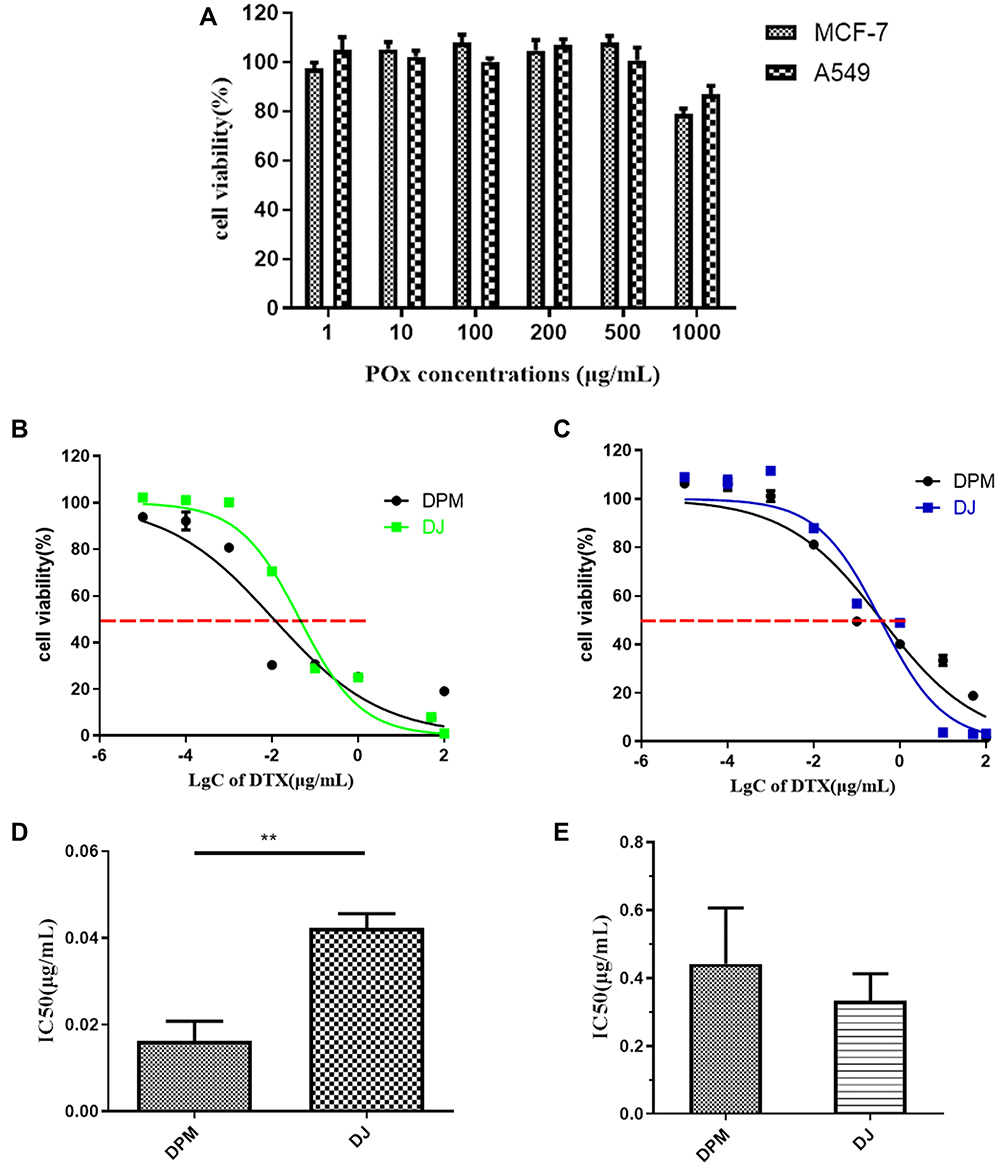 |
Figure 5 In vitro cytotoxicity evaluation. (A) Blank POx micelles, (B) DPM and DJ in MCF-7 cells, (C) DPM and DJ in A549 cells, (D) IC50 in MCF-7 cells, (E) IC50 in A549 cells. Data are represented as mean ± SD (n=3). **p < 0.01. |
Figure 5B and D report the cytotoxicity of DPM in MCF-7 cells 48 h after incubation. The IC50 of DJ is 2.5-fold that of the DPM treated group (DJ: 0.042 ± 0.003 μg/mL vs DPM: 0.016 ± 0.005 μg/mL, p < 0.01). However, no difference was observed between the DPM and DJ treated groups in the A549 cells (Figure 5C and E)
Cell Apoptosis in vitro
Cell apoptosis was detected using the Annexin V-FITC/PI double staining method. The control and BPM treated groups (Figure 6A and B) showed negligible apoptotic and necrotic cells (less than 10% in both MCF-7 and A549 cells). The upper and lower right quadrants indicated the late apoptotic cells and early apoptotic cells, respectively. In the MCF-7 cell line, the DPM treated group demonstrated a higher number of apoptotic cells than the DJ treated group (DPM: 24.52 ± 1.92% vs DJ: 18.11 ± 0.83%, p < 0.01) (Figure 6A and C). In A549 cells (Figure 6B and D), no significant differences were observed between the cell apoptosis rates of DJ and DPM (DJ: 12.50 ± 1.77% vs DPM: 15.41 ± 2.83%, p > 0.05).
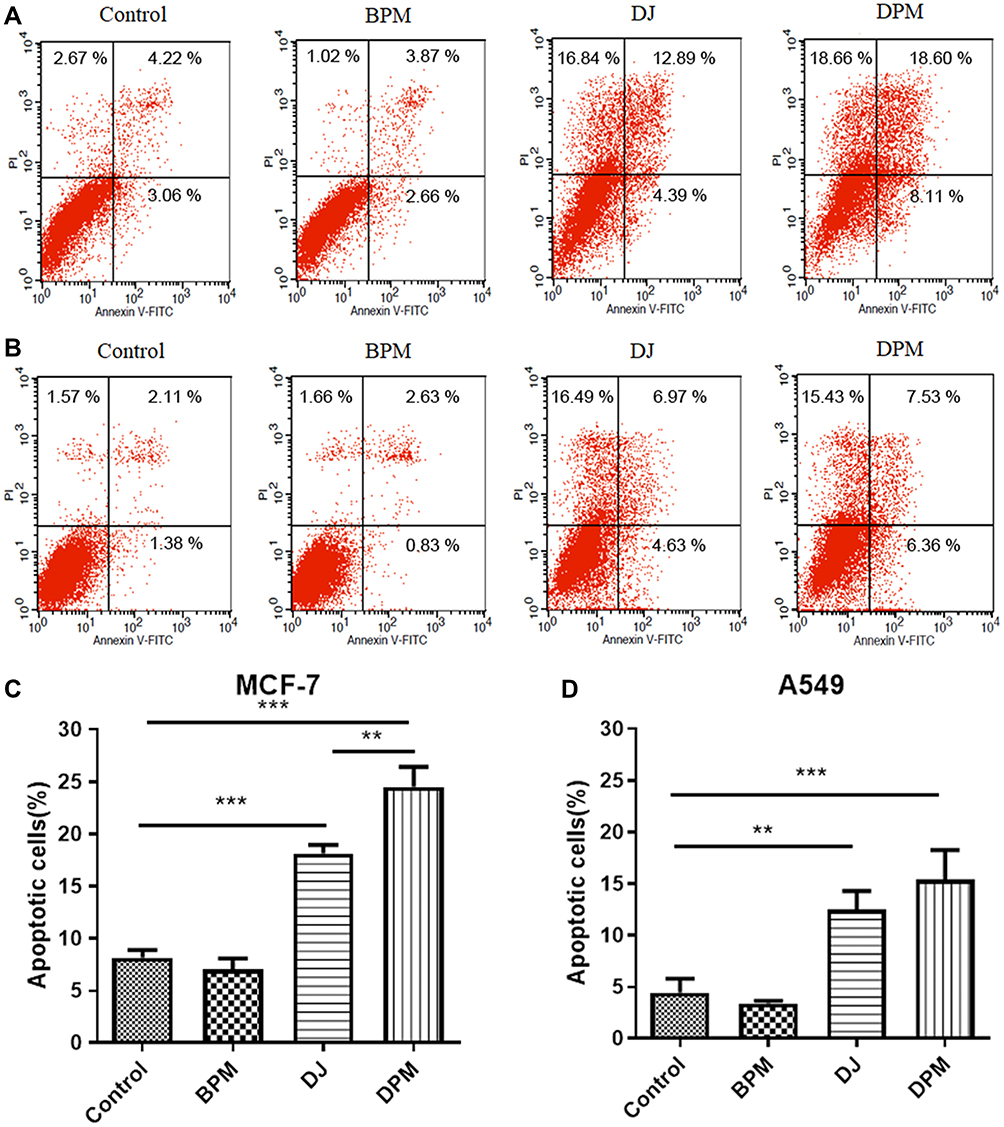 |
Figure 6 Cell apoptotic assay. MCF-7 cells (A and C) and A549 cells (B and D) were treated with different formulations for 48h and measured by flow cytometry using V-APC kit and PI staining. Data are represented as mean ± SD (n=3). **p<0.01, ***p<0.001. |
Biodistribution of DiR-POx in Tumor-Bearing Mice
Following the preparation of DiR-POx, DLS indicated that the average diameter was 28.26 ± 0.61 nm (PDI: 0.275 ± 0.014) and the DL was 8.52 ± 0.69%. The free DiR and DiR micelles (DiR-POx) were administered using a caudal vein injection. As shown in Figure 7A, the nude mice injected with normal saline failed to be detected by the fluorescence signal at any given time point. A detectable fluorescent signal in the liver was present at all time points; however, no signal was detected in the tumor of the free DiR treated group. After 2 h, the nude mice injected with DiR-POx micelles demonstrated significant DiR fluorescence signals differentiated from the surrounding tissues, and the fluorescence intensity increased with the prolongation of injection time. Compared to the observations at 12 h, the fluorescence intensity at 24 h did not decrease significantly and was maintained at a high level.
 |
Figure 7 The evaluation of the distribution of DiR loaded micelles in MCF-7 tumor-bearing nude mice after tail vein injection of the formulations. (A) Images taken at 2, 4, 6, 8, 12, 24h after administration of DiR and DiR loaded micelles, respectively. (B) Ex vivo fluorescence images of organs and tumors were collected at 24h post-injection of formulations. (C) Total fluorescence signals of the ex vivo organs and tumors uptake DiR by Imaging Software. Data are expressed as mean ± SD (n=3). ***p<0.001,**p<0.01, nsIndicates p>0.05. |
After 24 h of administration, the three nude mice groups were sacrificed by cervical dislocation. The heart, liver, spleen, lung, kidney, and tumors of the nude mice were carefully dissected, and the fluorescence signal intensity of each tissue was quantitatively detected using the software of the IVIS imaging system. As shown in Figure 7B, no fluorescence signal was generated in the tissues of nude mice injected with normal saline. The lungs and liver of the nude mice injected with free DiR demonstrated strong fluorescence signals, with almost no fluorescence signal detected at the tumor site. The tumor and liver fluorescence signals in nude mice treated with DPM were significantly stronger than that of other tissues, and the intensity of the tumor fluorescence signal was 40-fold higher than that of the free DiR group (p < 0.001)(Figure 7C).
Maximum Tolerated Dose (MTD) Profiles of DPM
The MTD values for the nude mice were examined by the dose escalation method, and the changes in body weight were recorded daily during the administration period. As shown in Figure 8A, following normal saline and BPM injections, the body weights of nude mice indicated a fluctuating upward trend. Similarly, the weight of nude mice in both the DJ and DPM groups at 5 mg/kg (DTX equivalent) fluctuated and increased. Furthermore, as the DTX equivalent dose increased, the bodyweight of the nude mice decreased. At the 10 mg/kg dose (DTX equivalent), the weight of nude mice in the DJ group decreased to less than 90% of the initial body weight, while the body weight of nude mice in the DPM group remained above 95%. In case of the 20 mg/kg dose (DTX equivalent), the nude mice in the DJ group reported a loss of appetite and the body weight less than 80% of the initial weight, while the DPM group still demonstrated 85% of the initial weight without abnormal behavior. The nude mice in the DPM group expressed a loss of appetite and weight as the DTX equivalent dose increased to 30 mg/kg, with more serious symptoms emerging such as significant weight loss and malformations. As shown in Figure 8B, the MTD for the DPM group was 20 mg/kg, twice that of the DJ group.
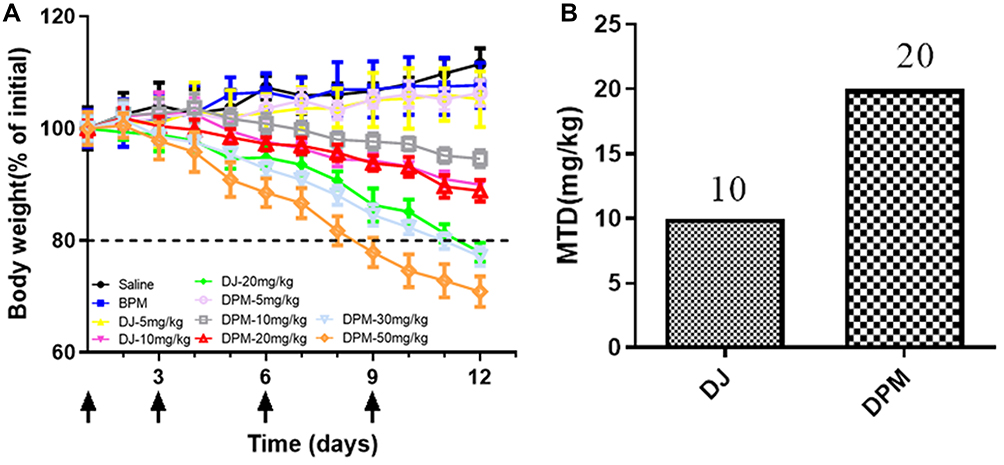 |
Figure 8 MTD in tumor-bearing nude mice. (A) Mice body weight change (% of initial) after repeated administration of different DTX formulation. (B) The maximum tolerated dose of BPM and DJ. Data are expressed as mean ± SD (n=4). |
Antitumor Effects in vivo
As shown in Figure 9A, the tumor growth curve demonstrated no difference between the saline and BPM groups, with almost no inhibitory effect on tumor growth. The DJ (MTD) group, DPM (1/2 MTD) group, and DPM (MTD) group all expressed inhibitory effects on tumor growth. The DPM (MTD) group demonstrated the most significant tumor growth inhibition. On the 24th day, the tumor volume in the DPM (MTD) group was only 3.2% of the saline group (p < 0.001), 13.9% (p < 0.001) of the DJ group, and 21.2% (p < 0.01) of the DPM (1/2 MTD) group. In addition, the tumor volume in the DJ (MTD) group and the DPM (1/2 MTD) group was 23.2% and 15.2% of the saline group, respectively.
 |
Figure 9 Anti-tumor efficacy of DTX formulations in MCF-7 tumor-bearing nude mice. (A) Tumor growth curve of mice administrated every three days for four times via tail vein. (B) The weight of the excised tumors from all groups. **p<0.01 and *p<0.05. (C) The images of excised tumor, scale bar 1cm. (D) Changes in body weight of tumor-bearing nude mice in each group during anti-tumor efficacy study. Data are expressed as mean ± SD (n= 6). ***p<0.001, vs saline; Δp<0.001, vs DJ(MTD); □p<0.01, vs DPM (1/2MTD). |
At the end of the administration, the mice were sacrificed, the tumors were isolated, and the tumor inhibition rate (IRT) was calculated based on the tumor weight. As shown in Figure 9B and Figure 9C, the tumor masses in the saline group and the BPM group were the largest, 2.01 ± 0.36 g and 1.91 ± 0.29 g, respectively. The mean tumor weight in the DPM (MTD) group was the least (0.05 ± 0.02 g), which significantly differed from the DJ and DPM (1/2 MTD) group (p < 0.01). The three groups demonstrated different tumor inhibition levels. The DPM group demonstrated the strongest antitumor effect at the MTD dose, and the IRT was higher than that at 1/2 MTD dose (97.76 ± 1.03% vs 88.47 ± 6.84%, p < 0.01). In addition, DJ (MTD) and DPM (1/2 MTD) were administered at the same DTX equivalent dose, however, the IRT of the DPM group was higher than that of the DJ group (88.47 ± 6.84% vs 79.10 ± 3.52%, p < 0.05).
The safety profiles of DTX loaded micelles were evaluated in terms of changes in body weight. As shown in Figure 9D, no treatment, except for treatment with DJ (p < 0.01), decreased body weight (> 15% of initial weight), and no difference was observed between the BPM and saline treated groups.
Histological Analysis and Cell Apoptosis in vivo
The in vivo antitumor effect of drug-loaded micelles was examined by HE staining. As shown in Figure 10A, the tumor tissues of the saline and BPM groups demonstrated intensive tumor cells, high karyoplasmic ratio, and less cytoplasm. The nucleus shrinkage in the tumor tissue was observed in the DJ and DPM treated group and the nuclear area was further reduced, with more cytoplasm produced in the latter group.
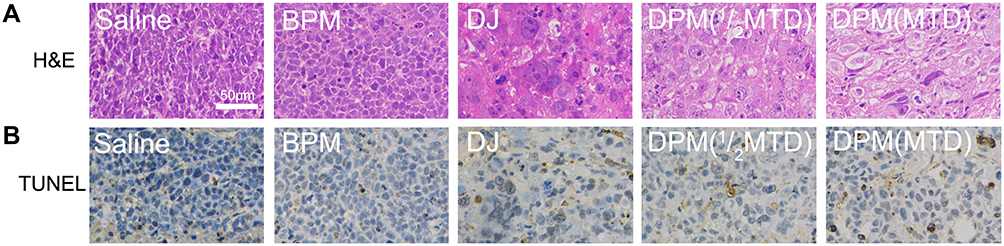 |
Figure 10 H&E and TUNEL assay. (A) Histological examination of tumors by hematoxylin and eosin (H&E) staining. (B) TUNEL staining from mice treated with different formulations. Images were taken with 200 × magnification, scale bar 50 μm. |
Cell apoptosis in the tumor tissues was observed using TUNEL staining. In Figure 10B, the brown-yellow particles refer to the positive expression of apoptotic cells, with no increase in the BPM group compared to the saline group. The brown-yellow particles in the drug-administered group increased significantly, indicating that the apoptotic cells increased in the two doses of the DPM treated group and were higher than the DJ treated group.
Discussion
The nano-polymer delivery system has been considerably developed at the laboratory level, and its advantages can be summarized as follows:28–33 (1) A suitable particle size range (< 200 nm) and a small dispersion index prevents quick elimination by the kidney and permits accumulation at the tumor site by the EPR effect; (2) The hydrophilic outer shell stabilizes and protects the micelles against recognition and phagocytosis by the reticuloendothelial system, prolonging blood circulation; (3) The micelle surface can be modified to precisely target the desired tissue; (4) Good biocompatibility of the micellar material makes it possible to degrade into non-toxic substances, to be absorbed or excreted by the body. However, poor clinical use has been reported due to the low drug loading content and the instability of the micelles in vivo.34,35
In this study, the DTX loaded poly-2-oxazoline micelles were designed to overcome these two flaws. The DL of DTX was observed as high as 24.67 ± 0.98% in the DPM group owing to the hydrogen bond that exists between poly-2-oxazoline micelles and taxanes,36 higher than that reported with the new clinical drug Genexol-PM (17%).37 In addition, the DTX release profiles of DPM and DJ at pH 5.5 reportedly simulate the tumor cell microenvironment,38,39 and DTX can be released from the DPM in large amounts at the tumor site in a sustainable manner.
Coumarin-6 has been widely used as a fluorescent probe to replace the hydrophobic drug and has been loaded in micelles to investigate cellular uptake.40 The MCF-7 cell uptake of POx was not time-dependent, with a greater C6 cell-mediated entry by the POx micelles than the free C6 owing to the change in the cellular uptake patterns. It has been reported that the cellular uptake of the free drug is usually by a passive transfer pattern. Polymer micellar systems alter this cellular uptake to an endocytosis pattern, leading to a higher uptake and facilitate the resolution of the multi-drug resistance problem.34 Interestingly, in the A549 cells, no difference was observed between the cellular uptake of C6 mediated by the polymer micellar system and the uptake of free C6. This may be attributable to the differential protein expression in the MCF-7 and A549 cells.41
The BPM exhibited low cytotoxicity in the cell viability assay, indicating the good biocompatibility of POx micelles. Cell viability inhibition of the DJ and DPM groups was concentration-dependent, with the increasing drug concentration, cell viability decreased. The group treated with DPM demonstrated an IC50 as low as 40% of the DJ treated group in MCF-7 cells, indicating promising clinical use. Cellular apoptosis demonstrated by the BPM, DPM, and DJ was measured by the sum of the early apoptotic and late apoptotic cells.42 Compared to the control group, the effect of BPM on tumor cell apoptosis was negligible, further illustrating the safety of the poly-2-oxazoline micelles. In the MCF-7 cells, an increase in cell apoptosis was detected in the DJ treated and DPM treated groups, which is in agreement with previous reports demonstrating that DTX induces cancer cell apoptosis, resulting in cell death.43 In the MCF-7 cells, the higher amount of cell apoptosis detected in the DPM treated group than the DJ group could be attributed to the high uptake of the DPM.44 Based on the overall cytological screening results, the drug-loaded micelles expressed a more powerful effect in the MCF-7 cells compared to the commercially available DTX injections. Additionally, the MCF-7 cells were used to establish a nude mouse model of breast cancer xenografts for further in vivo evaluations.
The growing research on nano-polymeric micellar delivery systems has demonstrated that antitumor drugs can be delivered to the tumor site by passive targeting, using the tumor EPR effect to achieve accumulation at the tumor site.39 It is important to investigate the in vivo distribution of drug-loaded nanoformulations by fluorescence imaging to assess the effectiveness and safety.45 Notably, the iodide fluorescent dye (DiD or DiR) has become the preferred fluorescent probe in fluorescence imaging owing to its hydrophobicity, and ability to be loaded and avoid interference with inherent fluorescence in small animals.46 In this study, a hydrophobic near-infrared DiR fluorescent probe was used to trace the distribution of docetaxel loaded poly-2-oxazoline micelles. We demonstrated that DiR can be used to simulate the in vivo distribution of the poly-2-oxazoline delivery system. Furthermore, DiR in the micellar delivery system accumulated at the tumor site in large quantities compared to the free DiR. This was attributed to the fluorescent probe micelles loaded with poly-2-oxazoline maintaining small particle size characteristics, escaping rapid elimination by the kidney, and accumulating at the tumor site by the tumor EPR effect.18,34 Hence, this indicates that the micellar delivery system is conducive to the accumulation of drugs at the tumor site. Liver serves important scavenger functions executed by heterogeneous cells (eg, Kupffer and endothelial cells) and related receptors to clear the nanomedicine as foreign materials.12 In this study, the low liver uptake of the DiR-POx micelles, which means the accumulation in tumor, could be beneficial to improve the bioavailability and the therapeutic effect of DPM.
The CMC of POx was at the low level of 10−3 mg/mL, with a spherical shape of about 20 nm for POx at 1 μg/mL diluted 25,000 times from the initial solution. CMC is an important feature for the characterization of micellar systems, especially the stability. When the amphiphilic polymer reaches the CMC or higher in aqueous solution, it can self-assemble to form a micellar system. A low CMC indicates good anti-serum dilution stability,47 which can be used as the polymer micelle delivery system, retaining the micellar morphology during blood circulation. Moreover, the CMC value is still very important for in vivo application. The polymer micelle delivery system is generally administered using the caudal vein for in vivo evaluation, with the carrier concentration diluted by more than 25 times in the blood circulation.48 A micelle may collapse, resulting in drug deposition and side effects.49 The POx micelle system demonstrated a low CMC and it was observed that the concentration of the POx at the lowest dose was still hundreds of times larger than CMC, indicating good stability in vivo. It is well known that most hydrophobic drugs loaded in the micellar delivery system are cytotoxic anti-cancer drugs, and their efficacy is closely related to the dose.34 In this study, the MTD of the poly-2-oxazoline drug-loading system in nude mice was explored and was determined to be twice that of the commercially available docetaxel injection. Undoubtedly, a higher antitumor efficacy can be expected if a greater drug dose is administered and delivered to the tumor. In this regard, DPM may have a clear advantage over the docetaxel injection. Furthermore, the subsequent pharmacodynamic experiments confirmed that the antitumor effect of the drug was improved with an increasing dose. The changes in the nude mice body weight, tumor weight, and the pathological features of HE staining proved the safety of the POx micelles and the effectiveness of DPM compared to commercially available docetaxel injection.
Conclusion
In the present study, we demonstrated the successful development of docetaxel loaded with self-assembling poly(2-oxazoline)s micelles in the breast cancer MCF-7 cells, both in vitro and vivo. Our results indicate that the amphiphilic micellar system possesses high stability and is capable of entrapping docetaxel with a high drug loading content. The antitumor effect of docetaxel loaded poly(2-oxazoline) micelles was significantly better than that of the docetaxel injection, with no obvious systemic toxicity. Therefore, this newly developed docetaxel loaded poly(2-oxazoline) micelle presents great potential to improve breast cancer treatment and necessitates further investigations.
Acknowledgment
This study was supported by Military Medical Innovation Project (16CXZ032), National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (No. 2018ZX09J18107-003, 2018ZX09721003-005-009) and NSFC projects (No. 81773278, 81702491).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4(4):253. doi:10.1038/nrc1317
3. Ojima I, Lichtenthal B, Lee S, Wang C, Wang X. Taxane anticancer agents: a patent perspective. Expert Opin Ther Pat. 2016;26(1):1–20. doi:10.1517/13543776.2016.1111872
4. Cragg GM, Kingston DG, Newman DJ. Anticancer Agents from Natural Products. CRC press; 2011.
5. Tan Q, Liu X, Fu X, Li Q, Dou J, Zhai G. Current development in nanoformulations of docetaxel. Expert Opin Drug Deliv. 2012;9(8):975–990. doi:10.1517/17425247.2012.696606
6. Fontanella C, Aprile G, Scartozzi M, Lederer B, Cascinu S, Von Minckwitz G. Perspectives from American Society of Clinical Oncology: translational and clinical research highlights in breast and colorectal cancers. Future Oncol. 2014;10(12):1901–1906. doi:10.2217/fon.14.150
7. Hennenfent K, Govindan R. Novel formulations of taxanes: a review. Old wine in a new bottle? Ann Oncol. 2005;17(5):735–749. doi:10.1093/annonc/mdj100
8. Feng L, Mumper RJ. A critical review of lipid-based nanoparticles for taxane delivery. Cancer Lett. 2013;334(2):157–175. doi:10.1016/j.canlet.2012.07.006
9. Desai NP, Trieu V, Hwang LY, Wu R, Soon-Shiong P, Gradishar WJ. Improved effectiveness of nanoparticle albumin-bound (nab) paclitaxel versus polysorbate-based docetaxel in multiple xenografts as a function of HER2 and SPARC status. Anticancer Drugs. 2008;19(9):899–909. doi:10.1097/CAD.0b013e32830f9046
10. Sparreboom A, Scripture CD, Trieu V, et al. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin Cancer Res. 2005;11(11):4136–4143. doi:10.1158/1078-0432.CCR-04-2291
11. He Z, Wan X, Schulz A, et al. A high capacity polymeric micelle of paclitaxel: implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials. 2016;101:296–309. doi:10.1016/j.biomaterials.2016.06.002
12. Li JJ, Kataoka K. Chemo-physical strategies to advance the in vivo functionality of targeted nanomedicine: the next generation. J Am Chem Soc. 2021;143(2):538–559. doi:10.1021/jacs.0c09029
13. Li JJ, Li YF, Wang YH, et al. Polymer prodrug-based nanoreactors activated by tumor acidity for orchestrated oxidation/chemotherapy. Nano Lett. 2017;17(11):6983–6990. doi:10.1021/acs.nanolett.7b03531
14. Li JJ, Ke WD, Wang L, et al. Self-sufficing H2O2-responsive nanocarriers through tumor-specific H2O2 production for synergistic oxidation-chemotherapy. J Control Release. 2016;225:64–74. doi:10.1016/j.jconrel.2016.01.029
15. Li JJ, Dirisala A, Ge ZS, et al. Therapeutic vesicular nanoreactors with tumor-specific activation and self-destruction for synergistic tumor ablation. Angewandte Chemie. 2017;56(45):14025–14030. doi:10.1002/anie.201706964
16. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. doi:10.1016/j.addr.2010.04.009
17. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392.
18. Nishiyama N, Matsumura Y, Kataoka K. Development of polymeric micelles for targeting intractable cancers. Cancer Sci. 2016;107(7):867–874. doi:10.1111/cas.12960
19. Barz M, Luxenhofer R, Zentel R, Vicent MJ. Overcoming the PEG-addiction: well-defined alternatives to PEG, from structure–property relationships to better defined therapeutics. Polym Chem. 2011;2(9):1900–1918. doi:10.1039/c0py00406e
20. Wang C-H, Wang W-T, Hsiue G-H. Development of polyion complex micelles for encapsulating and delivering amphotericin B. Biomaterials. 2009;30(19):3352–3358. doi:10.1016/j.biomaterials.2009.02.041
21. Schlaad H, Diehl C, Gress A, et al. Poly (2‐oxazoline) s as smart bioinspired polymers. Macromol Rapid Commun. 2010;31(6):511–525. doi:10.1002/marc.200900683
22. Luxenhofer R, Schulz A, Roques C, et al. Doubly amphiphilic poly (2-oxazoline) s as high-capacity delivery systems for hydrophobic drugs. Biomaterials. 2010;31(18):4972–4979. doi:10.1016/j.biomaterials.2010.02.057
23. Schulz A, Jaksch S, Schubel R, et al. Drug-induced morphology switch in drug delivery systems based on poly (2-oxazoline) s. ACS Nano. 2014;8(3):2686–2696. doi:10.1021/nn406388t
24. Seo Y, Schulz A, Han Y, et al. Poly (2‐oxazoline) block copolymer based formulations of taxanes: effect of copolymer and drug structure, concentration, and environmental factors. Polym Adv Technol. 2015;26(7):837–850. doi:10.1002/pat.3556
25. Colombani O, Ruppel M, Schubert F, Zettl H, Pergushov DV, Müller AH. Synthesis of Poly(n-butyl acrylate)-block-poly(acrylic acid) diblock copolymers by ATRP and Their micellization in water. Macromolecules. 2007;40(12):4338–4350. doi:10.1021/ma0609578
26. Kabanov AV, Nazarova IR, Astafieva IV, et al. Micelle formation and solubilization of fluorescent probes in poly (oxyethylene-b-oxypropylene-b-oxyethylene) solutions. Macromolecules. 1995;28(7):2303–2314. doi:10.1021/ma00111a026
27. He Z, Schulz A, Wan X, et al. Poly (2-oxazoline) based micelles with high capacity for 3rd generation taxoids: preparation, in vitro and in vivo evaluation. J Control Release. 2015;208:67–75. doi:10.1016/j.jconrel.2015.02.024
28. Liu J, Lee H, Allen C. Formulation of drugs in block copolymer micelles: drug loading and release. Curr Pharm Des. 2006;12(36):4685–4701. doi:10.2174/138161206779026263
29. Ke X, Ng VWL, Ono RJ, et al. Role of non-covalent and covalent interactions in cargo loading capacity and stability of polymeric micelles. J Control Release. 2014;193:9–26. doi:10.1016/j.jconrel.2014.06.061
30. Ding J, Chen L, Xiao C, Chen L, Zhuang X, Chen X. Noncovalent interaction-assisted polymeric micelles for controlled drug delivery. Chem Commun. 2014;50(77):11274–11290. doi:10.1039/C4CC03153A
31. Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71(3):227–234. doi:10.1016/j.reactfunctpolym.2010.10.009
32. Deng C, Jiang Y, Cheng R, Meng F, Zhong Z. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: promises, progress and prospects. Nano Today. 2012;7(5):467–480. doi:10.1016/j.nantod.2012.08.005
33. Attia ABE, Ong ZY, Hedrick JL, et al. Mixed micelles self-assembled from block copolymers for drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):182–194. doi:10.1016/j.cocis.2010.10.003
34. Zhang Y, Ren T, Gou J, et al. Strategies for improving the payload of small molecular drugs in polymeric micelles. J Control Release. 2017;261:352–366. doi:10.1016/j.jconrel.2017.01.047
35. Yu H, Li J, Shi K, Huang Q. Structure of modified ε-polylysine micelles and their application in improving cellular antioxidant activity of curcuminoids. Food Funct. 2011;2(7):373–380. doi:10.1039/c1fo10053j
36. Jaksch S, Schulz A, Di Z, Luxenhofer R, Jordan R, Papadakis CM. Amphiphilic triblock copolymers from poly (2‐oxazoline) with different hydrophobic blocks: changes of the micellar structures upon addition of a strongly hydrophobic cancer drug. Macromol Chem Phys. 2016;217(13):1448–1456. doi:10.1002/macp.201500465
37. Kim T-Y, Kim D-W, Chung J-Y, et al. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. doi:10.1158/1078-0432.CCR-03-0655
38. Guo M, Yan Y, Liu X, et al. Multilayer nanoparticles with a magnetite core and a polycation inner shell as pH-responsive carriers for drug delivery. Nanoscale. 2010;2(3):434–441. doi:10.1039/B9NR00244H
39. Bazban-Shotorbani S, Hasani-Sadrabadi MM, Karkhaneh A, et al. Revisiting structure-property relationship of pH-responsive polymers for drug delivery applications. J Control Release. 2017;253:46–63. doi:10.1016/j.jconrel.2017.02.021
40. Tang B, Zaro JL, Shen Y, et al. Acid-sensitive hybrid polymeric micelles containing a reversibly activatable cell-penetrating peptide for tumor-specific cytoplasm targeting. J Control Release. 2018;279:147–156. doi:10.1016/j.jconrel.2018.04.016
41. Meschini S, Marra M, Calcabrini A, et al. Role of the lung resistance-related protein (LRP) in the drug sensitivity of cultured tumor cells. Toxicol in Vitro. 2002;16(4):389–398. doi:10.1016/S0887-2333(02)00035-8
42. Qian Y, Lu S, Shi Y, et al. Celastrus orbiculatus extracts induce apoptosis and inhibit invasion by targeting the maspin gene in human gastric adenocarcinoma cells. Oncol Lett. 2018;15(1):243–249. doi:10.3892/ol.2017.7341
43. Kordezangeneh M, Irani S, Mirfakhraie R, Esfandyari-Manesh M, Atyabi F, Dinarvand R. Regulation of BAX/BCL2 gene expression in breast cancer cells by docetaxel-loaded human serum albumin nanoparticles. Med Oncol. 2015;32(7):208. doi:10.1007/s12032-015-0652-5
44. Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577. doi:10.2147/IJN.S36111
45. Feng Q, Yu M-Z, Wang J-C, et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core–shell nanoparticles. Biomaterials. 2014;35(18):5028–5038. doi:10.1016/j.biomaterials.2014.03.012
46. Wang D, Fu J, Shi Y, et al. The modulation of tumor vessel permeability by thalidomide and its impacts on different types of targeted drug delivery systems in a sarcoma mouse model. J Control Release. 2016;238:186–196. doi:10.1016/j.jconrel.2016.07.014
47. Munch MR, Gast AP. Block copolymers at interfaces. 1. Micelle formation. Macromolecules. 1988;21(5):1360–1366. doi:10.1021/ma00183a030
48. Rapoport N. Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery. Prog Polym Sci. 2007;32(8–9):962–990. doi:10.1016/j.progpolymsci.2007.05.009
49. Mikhail AS, Allen C. Block copolymer micelles for delivery of cancer therapy: transport at the whole body, tissue and cellular levels. J Control Release. 2009;138(3):214–223. doi:10.1016/j.jconrel.2009.04.010
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.