Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Annexin A1 is elevated in patients with COPD and affects lung fibroblast function
Authors Lai TW ![]() , Li YY, Mai ZJ, Wen XX, Lv YY, Xie ZQ, Lv QC, Chen M, Wu D, Wu B
, Li YY, Mai ZJ, Wen XX, Lv YY, Xie ZQ, Lv QC, Chen M, Wu D, Wu B
Received 23 August 2017
Accepted for publication 25 November 2017
Published 5 February 2018 Volume 2018:13 Pages 473—486
DOI https://doi.org/10.2147/COPD.S149766
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Tianwen Lai,1,* Yanyu Li,1,* Zongjiong Mai,2 Xiaoxia Wen,1 Yingying Lv,1 Zhanqing Xie,3 Quanchao Lv,1 Min Chen,1 Dong Wu,1 Bin Wu1
1Department of Respiratory and Critical Care Medicine, 2Department of Oncology, 3Department of Thoracic Surgery, The Affiliated Hospital of Guangdong Medical University, Zhanjiang, People’s Republic of China
*These authors contributed equally to this work
Purpose: Fibrosis in peripheral airways is responsible for airflow limitation in chronic obstructive pulmonary disease (COPD). Annexin A1 modulates several key biological events during inflammation. However, little is known about its role in airway fibrosis in COPD. We investigated whether levels of Annexin A1 were upregulated in patients with COPD, and whether it promoted airway fibrosis.
Methods: We quantified serum Annexin A1 levels in never-smokers (n=12), smokers without COPD (n=11), and smokers with COPD (n=22). Correlations between Annexin A1 expression and clinical indicators (eg, lung function) were assessed. In vitro, human bronchial epithelial (HBE) cells were exposed to cigarette smoke extract (CSE) and Annexin A1 expression was assessed. Primary human lung fibroblasts were isolated from patients with COPD and effects of Annexin A1 on fibrotic deposition of lung fibroblasts were evaluated.
Results: Serum Annexin A1 was significantly higher in patients with Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines stage III or IV than in those with GOLD stages I or II (12.8±0.8 ng/mL versus 9.8±0.7 ng/mL; p=0.016). Annexin A1 expression was negatively associated with airflow obstruction (forced expiratory volume in one second % predicted; r=−0.72, p<0.001). In vitro, Annexin A1 was significantly increased in CSE-exposed HBE cells in a time- and concentration-dependent manner. Annexin A1 promoted lung fibroblasts proliferation, migration, differentiation, and collagen deposition via the ERK1/2 and p38 mitogen-activated protein kinase pathways.
Conclusion: Annexin A1 expression is upregulated in patients with COPD and affects lung fibroblast function. However, more studies are needed to clarify the role of Annexin A1 in airway fibrosis of COPD.
Keywords: COPD, Annexin A1, tissue fibrosis, disease severity
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent reduction of airflow that is usually progressive and associated with an enhanced chronic inflammatory response in the airways and the lung to noxious particles or gases.1 Airway remodeling is one of the main pathologies of COPD airflow limitation. However, the mechanisms of airway remodeling are complex and remain largely unknown. Remodeling involves epithelial abnormalities, airway wall fibrosis, and smooth muscle hypertrophy and hyperplasia.2 As one of these pathologic changes, peribronchial and subepithelial fibrosis in peripheral airways is observed and is responsible for airflow limitation.3,4
Annexin A1 – also known as lipocortin 1, lipomodulin, maerocortin or renocortin – is a 37-kD protein that is encoded by the ANNA1 gene in humans.5,6 It is a member of the superfamily of the Ca+-dependent phospholipid-binding structural-related protein.7–11 Annexin A1 is involved in mediating several host physiological and pathological processes such as endocytosis and exocytosis, signal transduction, proliferation, differentiation, apoptosis, invasion, migration, inflammation, tumor, and immune function.12–19 However, whether the expression of Annexin A1 is upregulated in patients with COPD and contributes to disease severity remain unknown.
Airway fibrosis is characterized by activation of lung fibroblasts occurring under pathological conditions, collagen deposition, and shift of fibroblasts and other cell types such as airway smooth muscle (ASM) cells toward more synthetic cells known as myofibroblasts.2 Fibroblasts are the primary source of extracellular matrix (ECM) proteins. Fibroblast-mediated airway fibrosis and ECM protein deposition are believed to be important in the airway remodeling in COPD. Previous studies have shown that Annexin A1 mainly has effects on anti-inflammatory and anti-fibrosis processes.20,21 Neymeyer et al found that Annexin A1 inhibited alpha-smooth muscle actin (α-SMA) and collagen I gene expression in tumor growth factor-beta (TGF-β) induced renal cortical fibroblasts.22 Damazo et al found that Annexin A1 counter-regulates bleomycin-induced lung fibrosis.23 Although fibroblasts are believed to be critical in the airway fibrosis of COPD, the role of Annexin A1 in the fibroblast-mediated profibrotic responses has not yet been elucidated.
In the present study, we designed to determine the following: 1) whether Annexin A1 is upregulated in the serum of patients with COPD; 2) whether levels of Annexin A1 are correlated with clinical parameters such as lung function; and 3) whether Annexin A1 has profibrotic effects, including proliferation, myofibroblast differentiation, or collagen production, and what signal transduction modulates the profibrotic responses in vitro.
Methods
Human study subjects
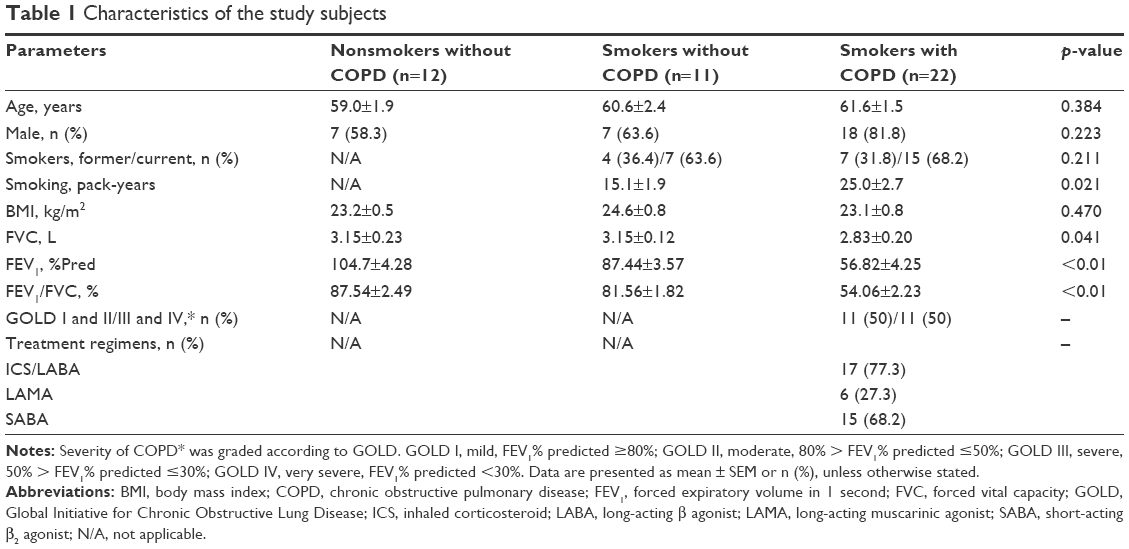
We enrolled both healthy subjects (12 nonsmokers and 11 smokers) and 22 smokers with COPD; COPD was defined according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria (post-bronchodilator forced expiratory volume in one second (FEV1)/forced vital capacity (FVC) <0.7).1 Patients with COPD were clinically stable, with no evidence of respiratory infection or an acute exacerbation for at least 4 weeks prior to screening. Exclusion criteria were as follows: chronic lung disease other than COPD (eg, asthma, bronchiectasis) and received oral or intravenous corticosteroids or anti-inflammatory drugs in the preceding 4 weeks. Clinical characteristics are described in Table 1.
 | Table 1 Characteristics of the study subjects |
To isolate primary human lung fibroblasts, we enrolled control subjects (two nonsmokers and two smokers) and three smokers with COPD. All subjects had undergone a surgical operation for lung cancer after pulmonary function testing. Exclusion criteria were as follows: respiratory infection in the preceding 4 weeks; had undergone radiotherapy or chemotherapy; or had a previous history of other cancer. Specimens were dissected at a distance of ≥5 cm away from the tumor (avoiding areas involving tumors).
Study protocols were approved by the Ethics of Research Committee of the Affiliated Hospital of Guangdong Medical University. Written informed consent was obtained from all participants.
Cigarette smoke extract preparation
Preparation of cigarette smoke extracts (CSE) Commercial cigarettes (Marlboro; Philip Morris USA, Richmond, VA, USA) were used in this study. CSE was prepared as previously described.24 Briefly, smoke from one cigarette was bubbled slowly through 10 mL RPMI 1640, which was considered as a 100% CSE solution, and was then sterilized and stored at −80°C as previously described.24
Isolation of human lung fibroblasts
Primary lung fibroblasts were isolated from lung tissue obtained from donors undergoing resection for localized lung carcinoma who gave informed consent, as described previously.25 Briefly, tissue was minced in 1–2 mm pieces into sterile Hanks Buffered Saline Solution (Hanks) and centrifuged for 5 min at 1,000 rpm. The supernatant was aspirated and the tissue pellet was resuspended and plated onto tissue-culture-grade plastic flasks in 10% (vol/vol) fetal bovine serum (FBS)/2% antibiotics/Dulbecco’s Modified Eagle Medium. Identification of fibroblasts was based on the expression of vimentin.
Human bronchial epithelial (HBE) cells culture
HBE cells were purchased from the American Type Culture Collection (ATCC, CRL-2741), and were maintained in RPMI 1640 with 10% FBS at 37°C in a humidified 5% CO2 atmosphere.
Enzyme-linked immunosorbent assay (ELISA) assays
Human Annexin A1 was measured using an ELISA kit (LOT#SEE787Hu, CLOUD-CLONE CORP) with detection limits of 0.113 ng/mL.
Real-time polymerase chain reaction analysis
cDNA was prepared by reverse transcription of RNA using random hexamer primers and Superscript II (Invitrogen). Each polymerase chain reaction used the following thermal cycling conditions: 37°C for 10 min, 95°C for 5 min, followed by 50 cycles of 95°C for 15 s, 60°C for 20 s, and 68°C for 20 s. For each sample, ΔCt was used to calculate the differences between target cycle threshold (Ct) values and the normalizer (housekeeping) gene: ΔCt = [(target) − Ct (normalizer)]. The comparative ΔΔCt was used to calculate the differences between each sample’s ΔCt value and the baseline ΔCt. The comparative expression level was obtained by transforming the logarithmic values to absolute values using 2−ΔΔCt. Sequences of the primers used for amplification are listed in Table S1.
Western blotting assays
Western blot analysis was used to detect changes in Annexin A1 (LOT# sc-53158; Santa Cruz Biotechnology, Santa Cruz, CA, USA), collagen type I (LOT#14695-1-AP, Proteintech™), collagen type III (LOT#13548-1-AP, Proteintech™), α-SMA (LOT#14968, Cell Signaling Technology), p-p38 antibody (LOT#4511, Cell Signaling Technology), p38 antibody (LOT#8690, Cell Signaling Technology), p-ERK1/2 antibody (LOT#4370, Cell Signaling Technology), and ERK1/2 antibody (LOT#4695, Cell Signaling Technology) as previously described.26 Images were subsequently analyzed using Quantity One to quantify the protein expression (BioRad, CA, USA).
Cell proliferation assays
Fibroblasts were seeded into 96-well plates at a density of 3,000 cells/well. Cells were grown for 24 h prior to treatment with Hyclone RPMI 1640 medium supplemented with 10% FBS. Fibroblasts were treated with various concentrations of recombinant human Annexin A1 (LOT#3770-AN, R&D Systems) for 48 h. Cell proliferation was detected using Japan Dojindo Molecular Technologies Cell counting kit-8 (LOT#JE603) according to manufacturer’s instructions.
Scratch wound-healing assays
A scratch wound-healing assay was undertaken in order to detect fibroblast migration as previously described.27 The healing course of the wound was recorded and images were photographed at 400× magnification using Life Technology EVOS® XL Core inverted microscope.
Immunofluorescent analysis
The localization of collagen type I, collagen type III, and α-SMA was investigated by immunofluorescence as previously described.26 Antibodies used in appropriate combinations were collagen type I (LOT#14695-1-AP, Proteintech™), collagen type III (LOT#13548-1-AP, Proteintech™), and α-SMA (LOT#14968, Cell Signaling Technology).
Statistical analysis
Data were presented as the mean ± SEM or n (%), unless otherwise stated. The normality of distribution was checked with Kolmogorov–Smirnov test. Statistical analyses were conducted using the parametric two-tailed Student’s t-test or non-parametric Mann–Whitney U-test for the comparison of two groups, and ANOVA with post hoc Tukey’s corrections for the comparison of more than two groups. Spearman’s rank correlation was used to test for dependence between two numerical variables. Statistical analysis was undertaken using GraphPad Prism 7.0 (San Diego, CA, USA). p<0.05 was considered statistically significant.
Results
Clinical data
The characteristics of the study subjects are shown in Table 1. There was no significant difference in age, gender, or body mass index (BMI) in study groups. Compared with nonsmokers and smokers, smokers with COPD had lower lung function (p<0.01), but longer smoking history (p=0.021).
Serum Annexin A1 levels were correlated with clinical parameters in COPD
The serum Annexin A1 levels for patients in smokers with COPD were higher than those in smokers without COPD (11.3±0.6 ng/mL versus 7.3±0.5 ng/mL; p<0.01) and those in never-smoker individuals (11.3±0.6 ng/mL versus 3.6±0.2 ng/mL; p<0.01; Figure 1A). The serum Annexin A1 levels of GOLD III/IV patients were higher than those in GOLD I/II patients (12.8±0.8 ng/mL versus 9.8±0.7 ng/mL; p=0.016; Figure 1B). Spearman’s rank correlation analysis showed that serum Annexin A1 levels in patients with COPD correlated negatively with FEV1 predicted % (r=−0.72, p<0.001), but positively associated with pack-years (r=0.47, p=0.006) (Figure 1C and D). However, the concentration of Annexin A1 in former and current smokers was not significantly different – both in smokers without or with COPD (p=0.895, p=0.578, respectively; Figure S1). Moreover, there was no difference in the smoking duration between GOLD I/II and III/IV subjects (p=0.759; Figure 1E).
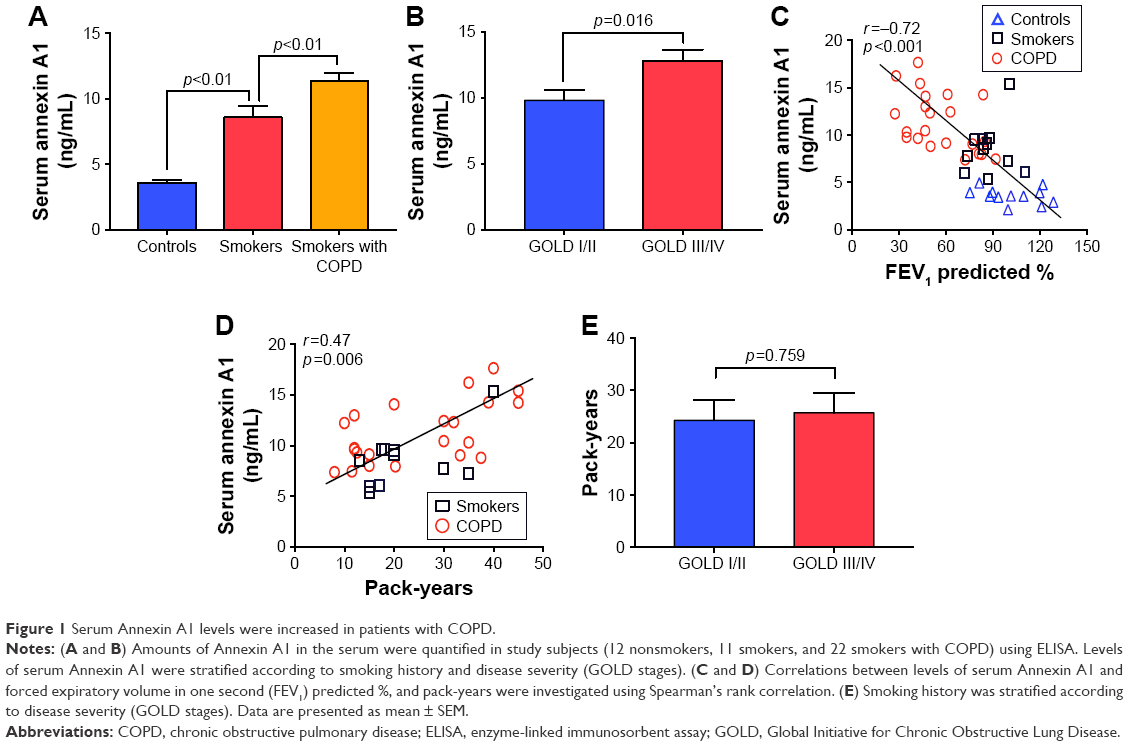 | Figure 1 Serum Annexin A1 levels were increased in patients with COPD. |
CSE-induced Annexin A1 expression in airway epithelial cells
Dysregulation of airway epithelial cell function related to environmental triggers, such as cigarette smoke, may contribute to the pathogenesis of COPD.1 The above data showed that Annexin A1 expression was associated with cigarette smoke in patients with COPD. Thus, we further investigated whether CSE induced Annexin A1 expression in airway epithelial cells (HBE cells). As shown in Figure 2A and B, we found that Annexin A1 was significantly increased in CSE-exposed HBE cells. In addition, CSE promoted Annexin A1 expression in a time-dependent manner. Elevated Annexin A1 expression was confirmed by Western blot (Figure 2C–F). These findings suggested that HBE cells could be a source of Annexin A1 in response to CS and may be involved in the pathogenesis of COPD.
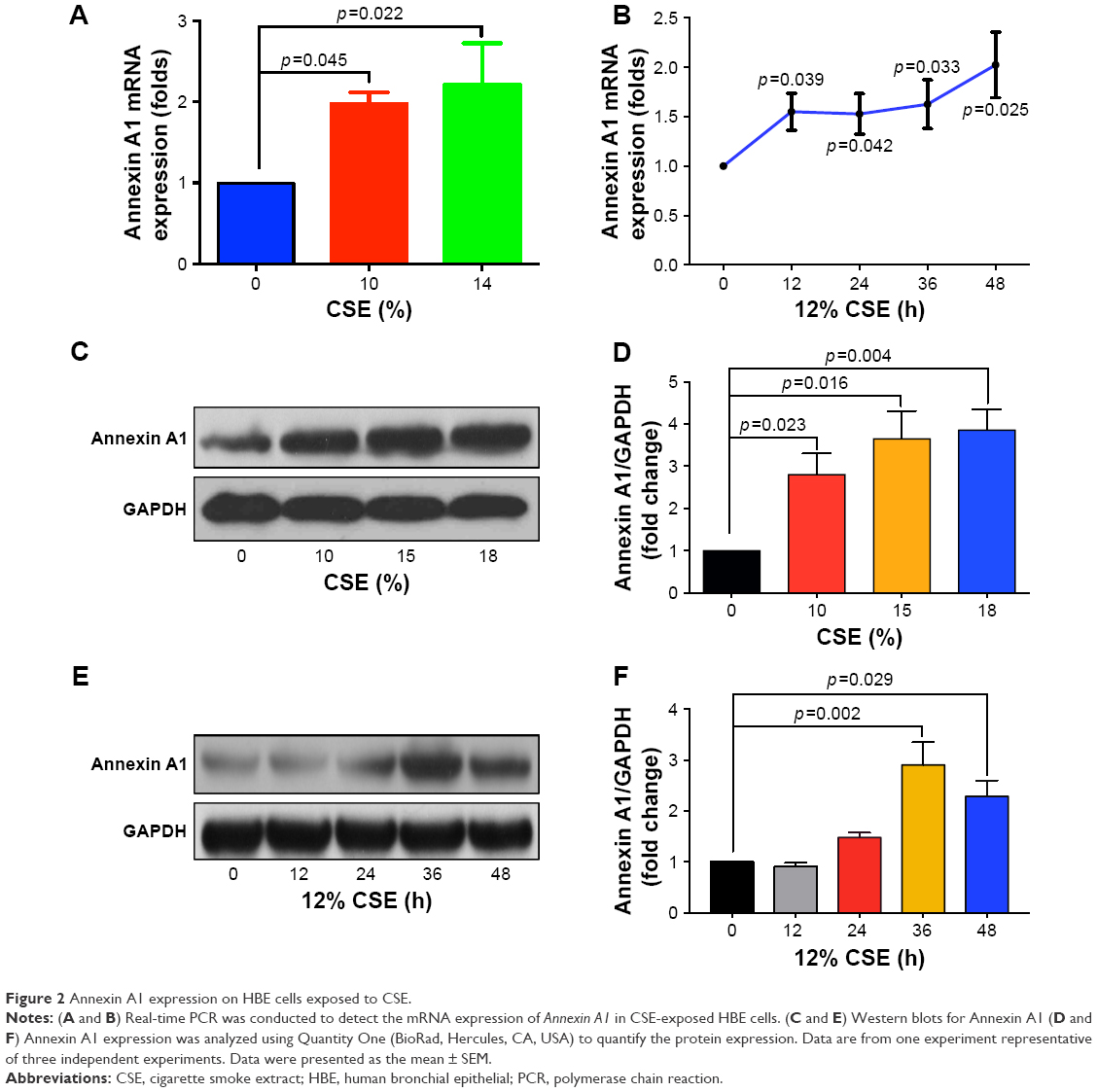 | Figure 2 Annexin A1 expression on HBE cells exposed to CSE. |
Annexin A1 induced lung fibroblasts activation
Fibroblasts play a key role in tissue remodeling and fibrosis. Fibroblast-mediated airway fibrosis has been shown to contribute to airway remodeling by upregulating matrix deposition.4,26 However, the contribution of Annexin A1 to modulating lung fibroblast phenotype and activation is not known. Therefore, we next attempted to investigate whether Annexin A1 exerted effects on primary human lung fibroblast migration and proliferation in vitro. Fibroblast migration was evaluated by the scratch wound-healing assay. As shown in Figure 3A and B, treatment with Annexin A1 increased the migration capacity of human lung fibroblasts, compared with the untreated control, as determined by the percentage of recovered wound area. Moreover, treatment with Annexin A1 significantly increased lung fibroblast proliferation as compared to the untreated control (Figure 3C). Annexin A1 increased the expression of collagen I production compared with untreated control fibroblast cells (Figure 4A–C). Annexin A1 induced collagen III production at the doses of Annexin A1 at 50 and 100 ng/mL. However, when the dose of Annexin A1 increased to 150 ng/mL, Annexin A1 attenuated collagen III production (Figure 4D–F). The upregulation of α-SMA expression is a characteristic marker for differentiation of lung fibroblasts to myofibroblasts.4,26 Using immunofluorescence and Western blotting, we detected the expression of α-SMA in human lung fibroblast cells after 48 h of treatment with Annexin A1 and found that it significantly increased α-SMA protein expression compared with the untreated control cells (Figure 5). We further examined whether the cytokine synthesis of fibroblast was inhibited or augmented by the addition of Annexin A1. We found that levels of IL-6, IL-8, and IL-1β were increased before 6 h in Annexin A1-induced fibroblast cells, but were decreased after 6 h (Figure 6).
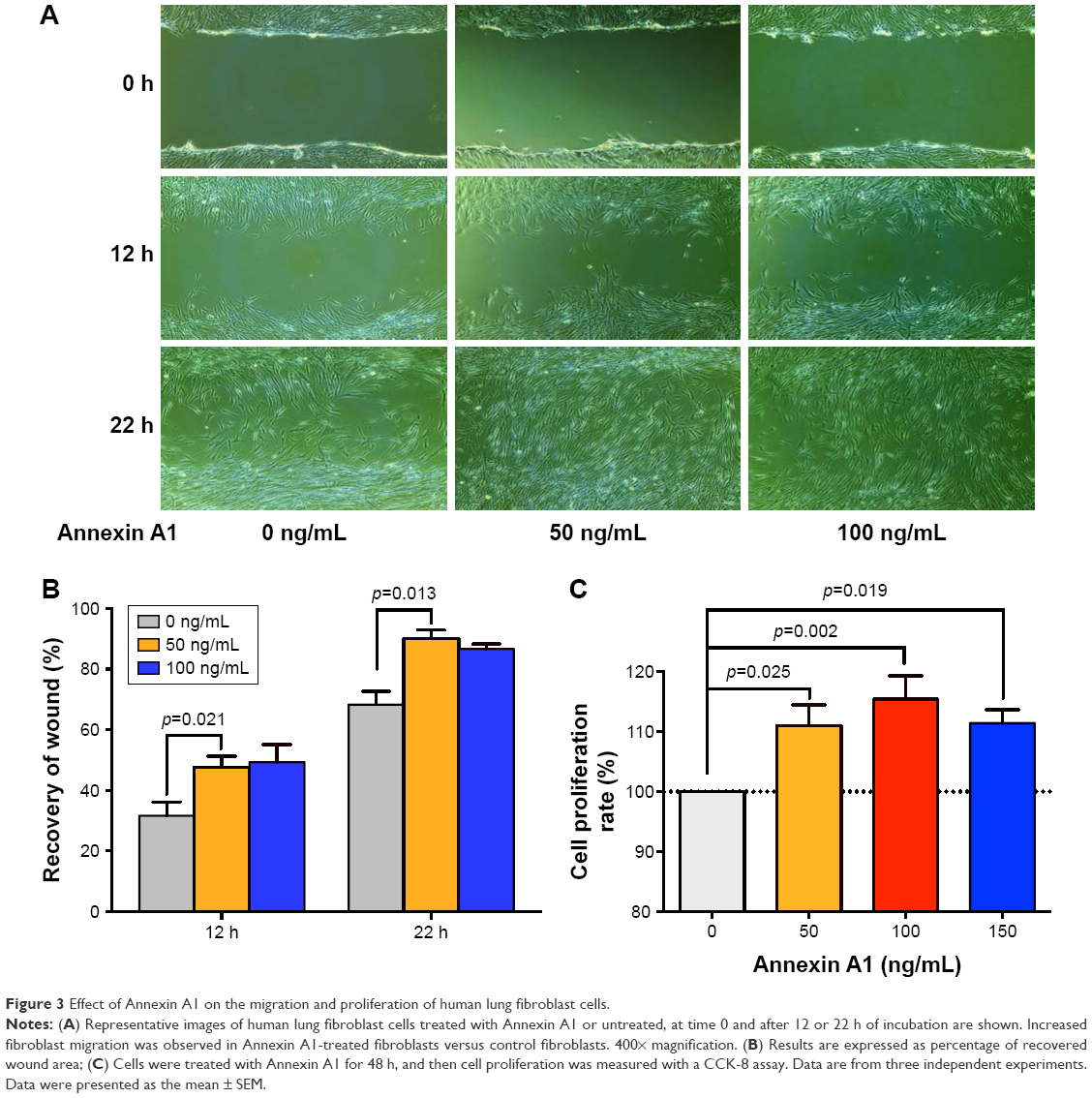 | Figure 3 Effect of Annexin A1 on the migration and proliferation of human lung fibroblast cells. |
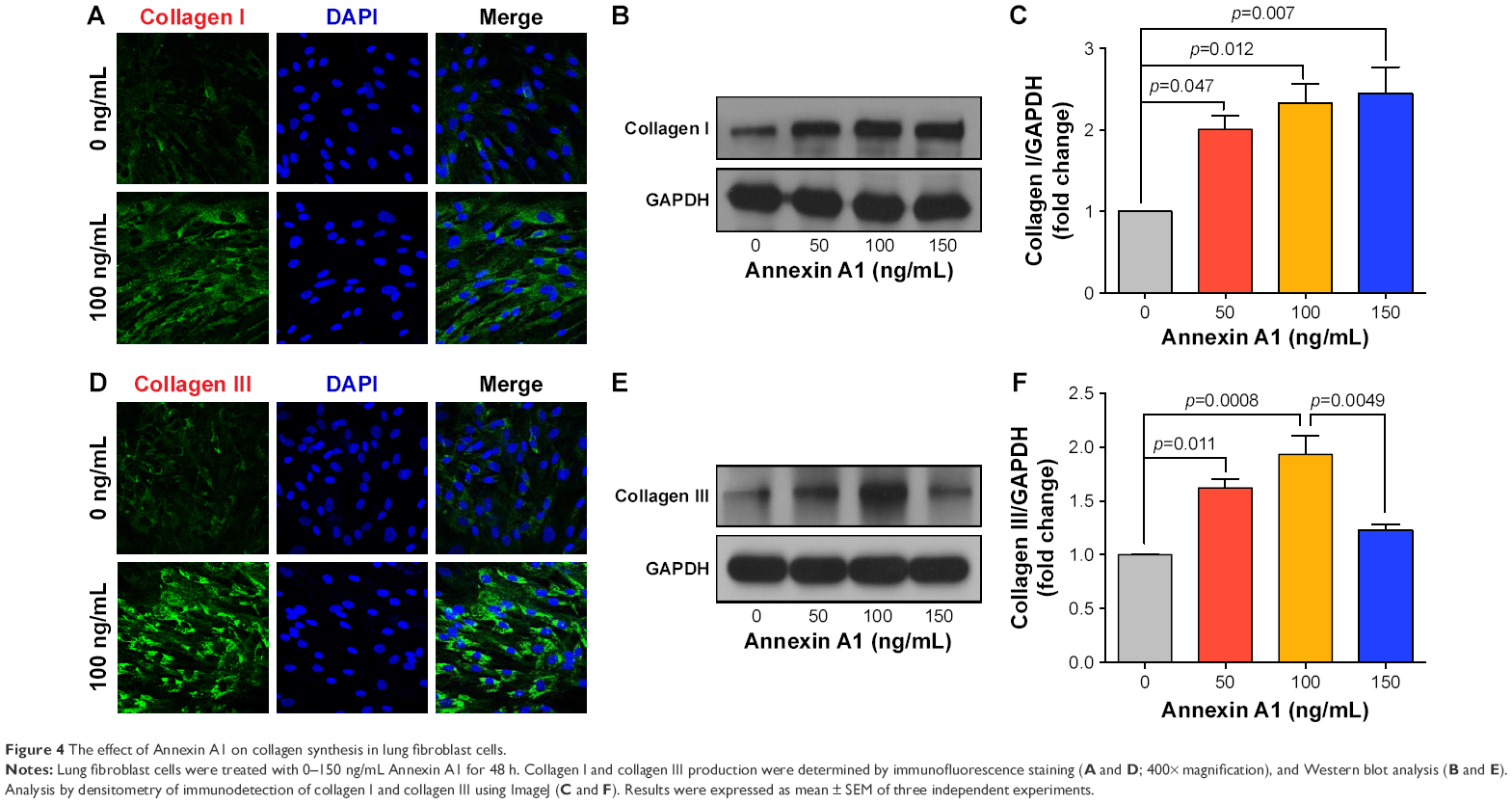 | Figure 4 The effect of Annexin A1 on collagen synthesis in lung fibroblast cells. |
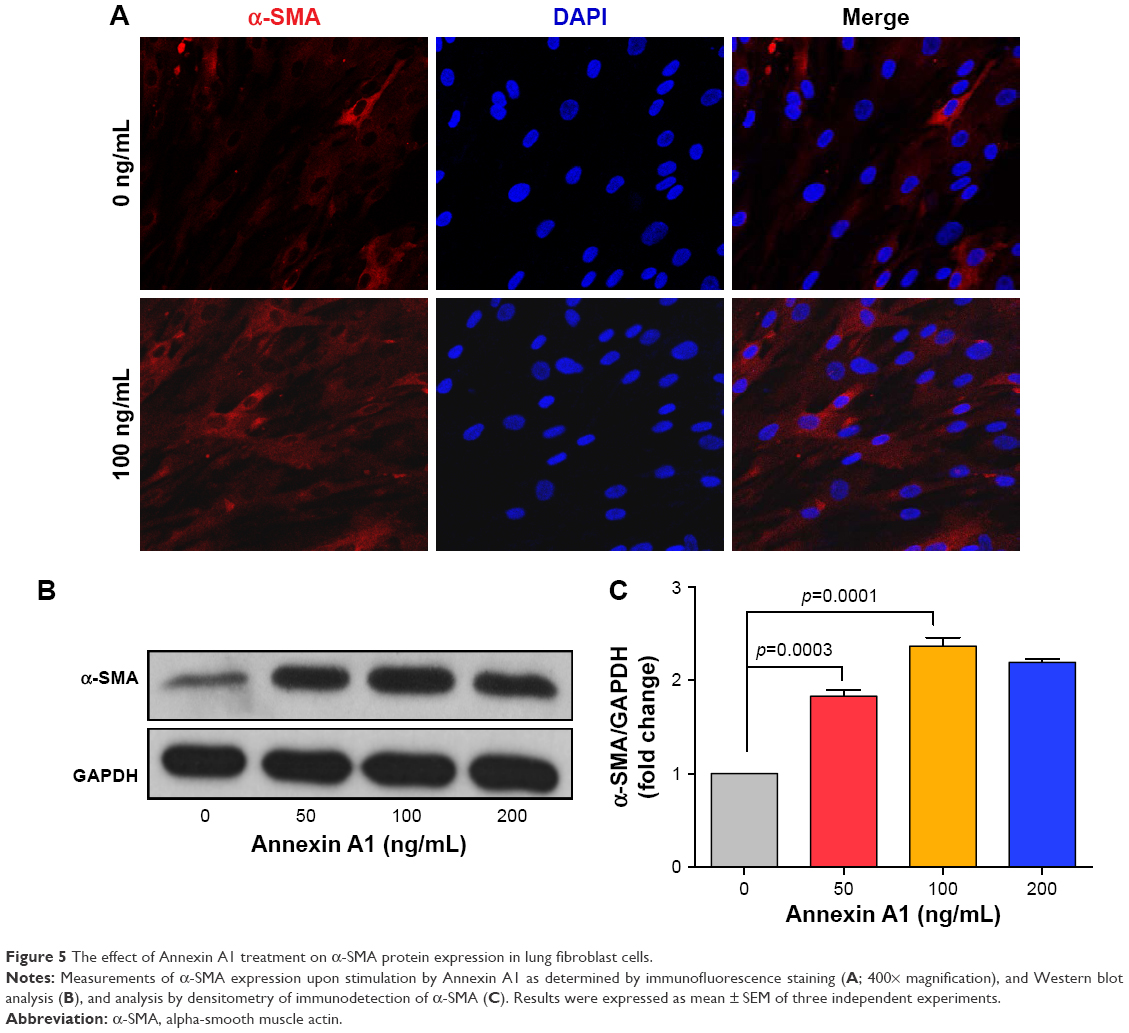 | Figure 5 The effect of Annexin A1 treatment on α-SMA protein expression in lung fibroblast cells. |
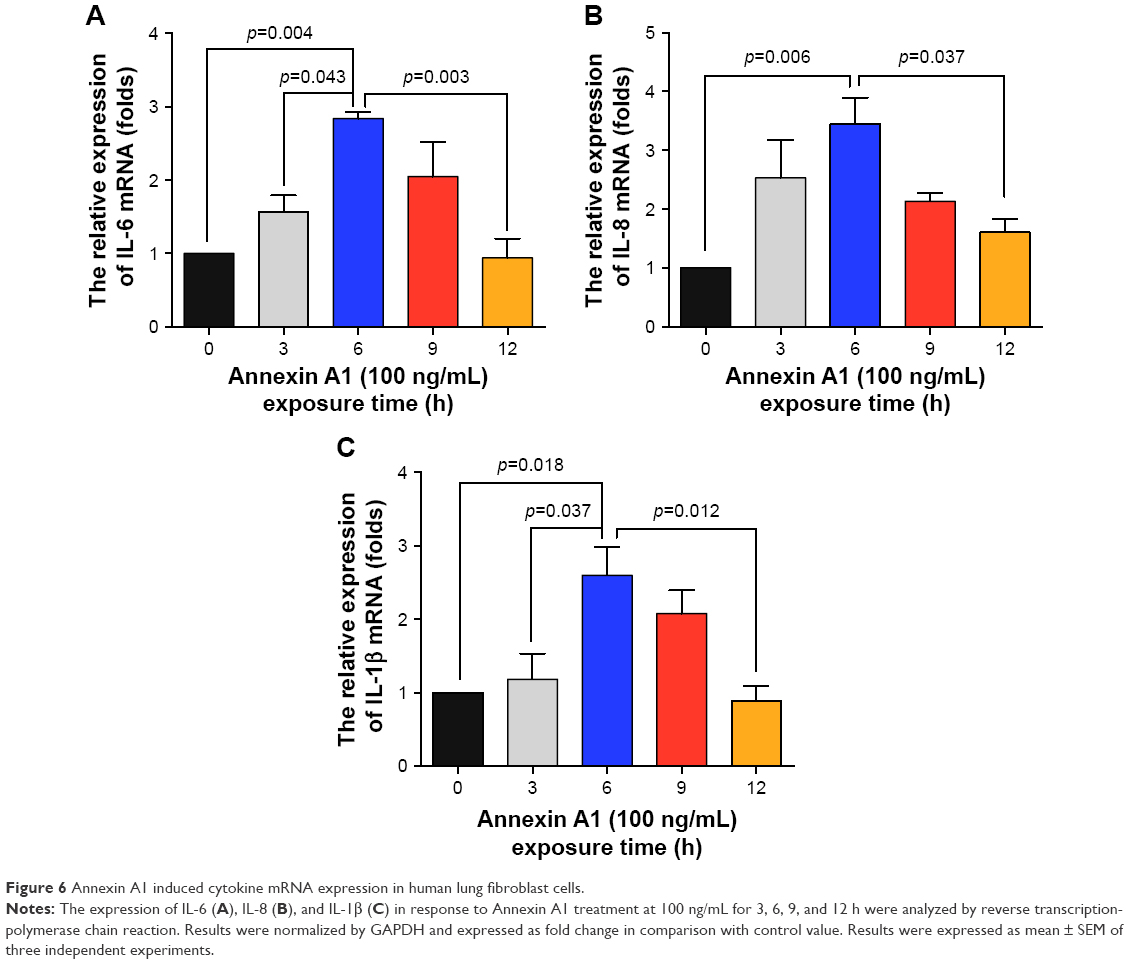 | Figure 6 Annexin A1 induced cytokine mRNA expression in human lung fibroblast cells. |
Annexin A1 induces activation of ERK1/2 and p38 mitogen-activated protein kinase (MAPK) signaling in lung fibroblasts
The MAPK family is well known to play a key role in mediating inflammatory responses.28 To assess whether MAPK pathways are involved in Annexin A1-induced collagen production, the effect of Annexin A1 on activation of ERK1/2 and p38 was evaluated by Western blotting. As shown in Figure 7A, Annexin A1 activated ERK1/2 phosphorylation in a dose-dependent manner. In addition, we cultured fibroblasts with a series of concentrations of Annexin A1 for 60 min and observed p38 MAPK phosphorylation. As shown in Figure 7B, Annexin A1 activated the phosphorylation of p38 MAPK in a dose-dependent manner. Finally, we stimulated fibroblasts for various periods of time with 100 ng/mL recombinant Annexin A1 and observed the phosphorylation of ERK1/2 and p38 MAPK. As shown in Figure 7C and D, Annexin A1 activated ERK1/2 and p38 phosphorylation in a time-dependent manner.
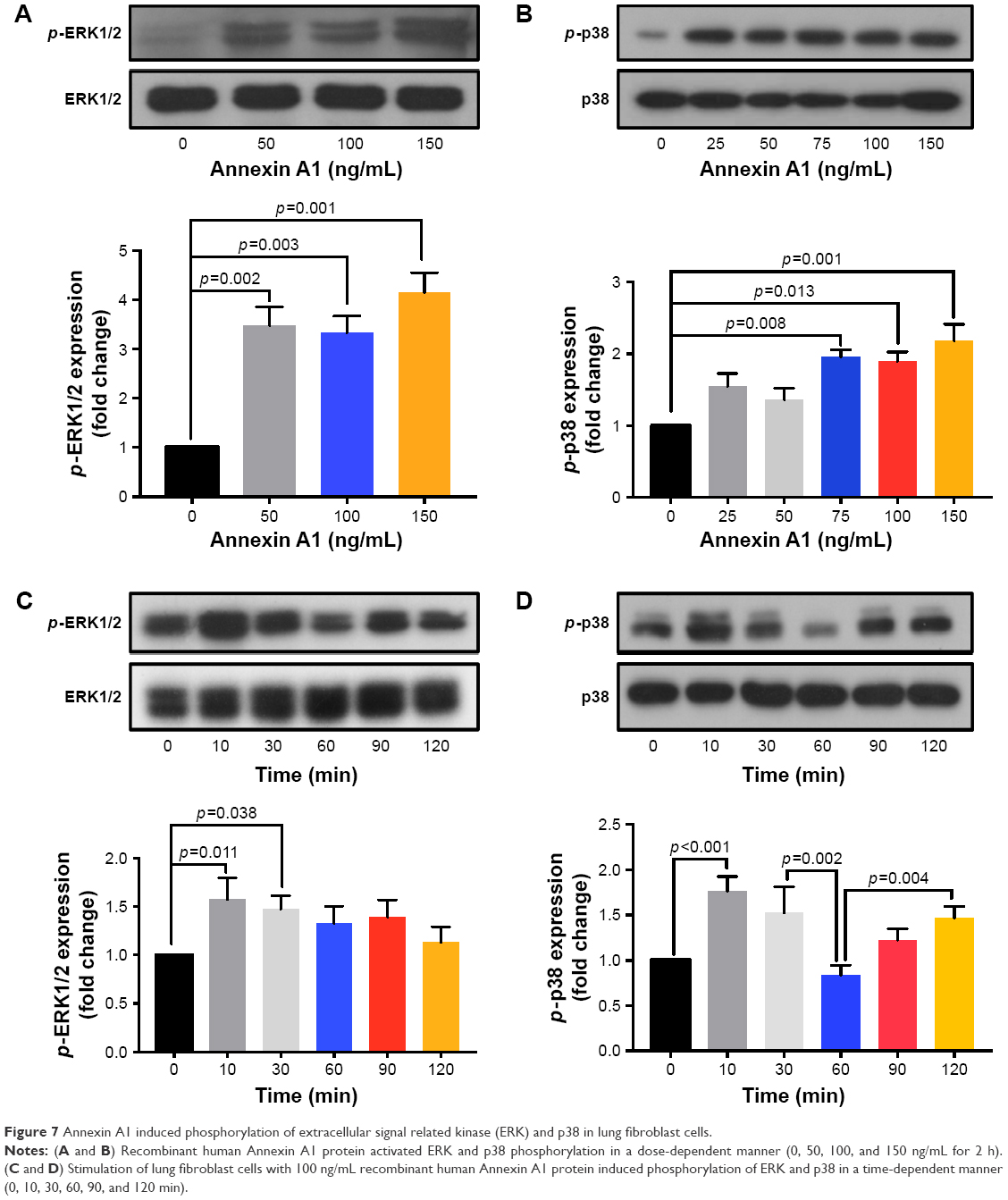 | Figure 7 Annexin A1 induced phosphorylation of extracellular signal related kinase (ERK) and p38 in lung fibroblast cells. |
Discussion
In the present study, we demonstrated that Annexin A1 expression was upregulated in patients with GOLD III/IV stage than in those with GOLD I/II stage, smokers without COPD, and nonsmokers. Levels of Annexin A1 had a negative correlation with lung function (FEV1% predicted). Elevated Annexin A1 induced lung fibroblast activation, suggesting that Annexin A1 may be involved in airway fibrosis of COPD. However, more studies are needed to clarify the role of Annexin A1 in airway fibrosis of COPD.
COPD is accompanied by systemic inflammation that occurs as a result of many mechanisms, particularly airway inflammation and smoking. Annexin A1 is a member of the Annexin superfamily of calcium- and phospholipid-binding proteins. Annexin A1 has been shown to be capable of modulating a number of biological events such as chronic inflammation, tissue growth, and apoptosis. It has been shown that the absence of Annexin A1 is associated with the development of more severe inflammation in different models of inflammatory diseases.15,29,30 However, relatively little is known about the role of Annexin A1 in regulating airway inflammation in COPD. In our study, we found that serum Annexin A1 levels were elevated in patients with COPD. Moreover, serum Annexin A1 levels correlated positively with disease severity (GOLD stages). Epithelial cells of the airway are a major source of inflammatory mediators.31 Bronchial epithelial cells are one of the first cell types contacting the toxins present in inhaled cigarette smoke, and, when exposed to environmental tobacco smoke, bronchial epithelial cells may cause the release of inflammatory factors.32 In this study, we found that CSE can stimulate HBE cells to produce Annexin A1. These findings suggested that HBE cells could be a source of Annexin A1 in response to CSE and may be involved in the pathogenesis of COPD. Cigarette smoking is the most dangerous risk factor for COPD and approximately 90% of COPD patients are smokers or ex-smokers.1 Currently, tobacco smoking is considered the main pathogenic factor for COPD. Thus, the difference of Annexin A1 between smokers and smokers with COPD may be associated with smoking duration. However, COPD is a systemic and chronic inflammatory disease associated with the abnormal inflammatory response of the lungs to tobacco smoke.1 We further examined whether there is any difference in smoking duration between GOLD I/II and III/IV subjects. We found there was no difference in smoking duration between GOLD I/II and III/IV categories. However, serum Annexin A1 levels of patients with GOLD III/IV were higher than those in patients with GOLD I/II. This finding suggests that the concentration of circulatory Annexin A1 in patients with COPD may also be affected by other local or systemic factors in addition to cigarette smoke exposure. Annexin A1 may modulate inflammatory responses via regulation of cytokine production. Previous studies have shown that Annexin A1 and its mimetic peptides inhibit neutrophil tissue accumulation by reducing leukocyte infiltration and activating neutrophil apoptosis.15 Additionally, Annexin A1 promotes monocyte recruitment and clearance of apoptotic leukocytes by macrophages, resulting in reduced production of proinflammatory cytokines and increased release of immunosuppressive and proresolving molecules.15,33 Our data may imply that Annexin A1 is increased in patients with COPD in an attempt to attenuate the exacerbated inflammatory response in these patients. Chronic inflammation in COPD suggests that the resolution of inflammation is dysfunctional. Accordingly, elevated Annexin A1 expression seems to be insufficient to resolve inflammation. Indeed, systemic levels of proresolving mediators are increased in other chronic inflammatory diseases such as inflammatory bowel disease, preeclampsia, and Alzheimer’s disease, suggesting that this resolution pathway is dysfunctional.30,34,35
Collagens are the classical components of the ECM. Peribronchiolar fibrosis is an important feature of COPD and results from the increased ECM deposition. Collagens are synthetized primarily by fibroblasts as precursor molecules, with the propeptides being cleaved during the process of secretion of newly formed collagens.26 Recent studies have shown that Annexin A1 plays an important role in the regulation of differentiation, immigration, proliferation, and secretion of fibroblasts. Jia et al found that silencing constitutive Annexin A1 expression in human normal lung fibroblasts using small-interfering RNA (siRNA) was associated with moderate but significant increases in tumor necrosis factor (TNF)-induced proliferation and interleukin (IL)-6 production.36 Damazo et al suggested that Annexin A1 inhibits bleomycin-induced lung fibrosis.23 Neymeyer et al showed that Annexin A1 exerts antifibrotic effects in human renal fibroblasts.22 Skouteris and Schröder suggested that the expression of Annexin A1 exhibited a positive correlation with cell proliferation in human foreskin fibroblasts.37 However, whether Annexin A1 participates in the onset of fibrosis of the small airways in patients with COPD remains unclear. On the basis of the above findings, we further explored the role of Annexin A1 in collagen production in human lung fibroblasts in vitro. We found that Annexin A1 induced collagen production in lung fibroblast cells, which is inconsistent with previous studies. The reason for the discrepancies of the results is currently unclear. However, it may be due to differences in cell/tissue type, different sources of Annexin A1, as well as in vitro and in vivo experiments. Previous studies conducting in vivo experiments showed that annexin A have antifibrotic effects.23,38,39 In contrast, we isolated primary human fibroblast cells from lung tissues in vitro, and Annexin A1 was an exogenous protein (recombinant human Annexin A1 protein) in our study. The regulation of the body’s internal environment is a complex network, and our in vitro experiment cannot fully reflect the role of Annexin A1 in the body. Another reason may be differences in concentrations of Annexin A1 used and/or duration of stimulation with Annexin A1. We observed that Annexin A1 induced collagen III production at the doses of Annexin A1 50 and 100 ng/mL. However, when the dose of Annexin A1 increased to 150 ng/mL, Annexin A1 attenuated collagen III production. We further examined whether the cytokine synthesis of fibroblasts was inhibited or augmented by the addition of Annexin A1. We found that the levels of IL-6, IL-8, and IL-1β were increased before 6 h in Annexin A1-induced fibroblast cells, but decreased after 6 h. We hypothesized that Annexin A1 induced inflammatory cytokines (eg, IL-6, IL-8), and these cytokines induced collagen deposition at the early stage and in low doses of Annexin A1. During this period, Annexin A1 was insufficient to resolve inflammation and elevated inflammatory cytokines induced collagen deposition. When the concentration and duration of stimulation with Annexin A1 were increased, Annexin A1 was sufficient to resolve inflammation and attenuated inflammatory cytokine-induced collagen deposition. Our finding suggest that Annexin A1 can either stimulate or inhibit inflammatory cytokines and collagen production depending upon the experimental conditions in vitro. However, further studies are needed to investigate whether there is a difference in roles between the exogenous and endogenous Annexin A1 in airway fibrosis of COPD, such as by using Annexin A1-deficient mice.
Given the role of Annexin A1 in collagen production in human lung fibroblasts, we sought to determine the key signaling mechanisms by which this occurs. Some previous studies have shown that ERK or p38 is increased in COPD. Yang et al showed that the level of phospho-p38 MAPK was increased in peripheral blood neutrophils of acute exacerbation of COPD patients.40 Li et al demonstrated that the ERK/p38 MAPK pathway was enhanced in CSE-induced HBE cells.41 Li et al indicated that the phosphorylation of ERK and p38 was increased in mice exposed to cigarette smoke.42 Moreover, p38 MAPK and ERK were increased in ASM cells and bone marrow-derived mesenchymal stem cells (MSCs) in COPD.43,44 We found that MAPK signaling was involved in Annexin A1-induced lung fibroblasts activation. We demonstrated a biphasic effect in p-p38 activation in response to Annexin A1. The mechanism underlying this phenomenon is unknown. A previous study has showed that biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis.45 Annexin A1 is a member of a phospholipid and calcium binding family of proteins; it is involved in the regulation of apoptosis.12,46,47 One of the potential mechanisms that could be set in motion following p-p38 activation is the induction of proapoptotic pathways. The time course of p-p38 activation suggested that activation of apoptosis may be downstream to, and a consequence of, p-p38 activation. The earlier peak of p-p38 activation is part of the mechanism leading to fibroblast activation, whereas the later peak of p-p38 may not be essential for fibroblast activation. However, further studies are needed to determine the exact role of Annexin A1 in the activation of p38MAPK. This is the aim of our further efforts.
There are some limitations to this study that need to be considered. First, Annexin A1 significantly upregulated proliferation in fibroblasts obtained from nonsmokers, smokers without COPD, and smokers with COPD. However, due to the small sample size, we could not precisely analyze whether fibroblasts from these different patient groups behaved differently with regard to their responses to Annexin A1. Future studies with larger sample sizes as well as animal experiments need to be conducted to clarify this issue; second, we only assessed the role of Annexin A1 on collagen production in lung fibroblast cells in vitro, further studies are needed to investigate Annexin A1 in airway fibrosis of COPD in animal models.
Conclusion
Our study demonstrated that elevated Annexin A1 levels are associated with disease severity in patients with COPD. Elevated Annexin A1 induced lung fibroblast activation, indicating that Annexin A1 may be involved in airway fibrosis of COPD. However, more studies are needed to clarify the role of Annexin A1 in airway fibrosis of COPD.
Acknowledgments
This work was supported by the Doctoral Startup Foundation of Affiliated Hospital of Guangdong Medical University (Tianwen Lai) and the National Natural Science Foundation of China (81420108001). The authors thank Dongming Li (Department of Respiratory and Critical Care Medicine, The Affiliated Hospital of Guangdong Medical University) for drafting the article. The authors also thank the participants of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
GOLD Committee. Global Strategy for the Diagnosis, Management, and Prevention of COPD. Available from: http://www.goldcopd.org/guidelines-pocket-guide-to-copd-diagnosis.html. Accessed January 21, 2016. | ||
Salazar LM, Herrera AM. Fibrotic response of tissue remodeling in COPD. Lung. 2011;189(2):101–109. | ||
Lv Y, Lv Q, Lv Q, Lai T. Pulmonary infection control window as a switching point for sequential ventilation in the treatment of COPD patients: a meta-analysis. Int J Chron Obstruct Pulmon Dis. 2017;12:1255–1267. | ||
Kikuchi T, Sugiura H, Koarai A, et al. Increase of 27-hydroxycholesterol in the airways of patients with COPD: possible role of 27-hydroxycholesterol in tissue fibrosis. Chest. 2012;142(2):329–337. | ||
Perretti M. The annexin 1 receptor(s): is the plot unravelling? Trends Pharmacol Sci. 2003;24(11):574–579. | ||
D’Acunto CW, Gbelcova H, Festa M, Ruml T. The complex understanding of Annexin A1 phosphorylation. Cell Signal. 2013;26(1):173–178. | ||
John CD, Christian HC, Morris JF, Flower RJ, Solito E, Buckingham JC. Annexin 1 and the regulation of endocrine function. Trends Endocrinol Metab. 2004;15(3):103–109. | ||
Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82(2):331–371. | ||
Bizzarro V, Belvedere R, Milone MR, et al. Annexin A1 is involved in the acquisition and maintenance of a stem cell-like/aggressive phenotype in prostate cancer cells with acquired resistance to zoledronic acid. Oncotarget. 2015;6(28):25076–25092. | ||
He J, Yi B, Chen Y, et al. The ET-1-mediated carbonylation and degradation of ANXA1 induce inflammatory phenotype and proliferation of pulmonary artery smooth muscle cells in HPS. PLoS One. 2017;12(4):e175443. | ||
Violette SM, King I, Browning JL, Pepinsky RB, Wallner BP, Sartorelli AC. Role of lipocortin I in the glucocorticoid induction of the terminal differentiation of a human squamous carcinoma. J Cell Physiol. 1990;142(1):70–77. | ||
Zhao Y, Li X, Gong J, et al. Annexin A1 nuclear translocation induces retinal ganglion cell apoptosis after ischemia-reperfusion injury through the p65/IL-1β pathway. Biochim Biophys Acta. 2017;1863(6):1350–1358. | ||
Belvedere R, Bizzarro V, Popolo A, et al. Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer. 2014;14:961. | ||
Cheng SX, Tu Y, Zhang S. FoxM1 promotes glioma cells progression by up-regulating Anxa1 expression. PLoS One. 2013;8(8):e72376. | ||
Sugimoto MA, Vago JP, Teixeira MM, Sousa LP. Annexin A1 and the resolution of inflammation: modulation of neutrophil recruitment, apoptosis, and clearance. J Immunol Res. 2016;2016:8239258. | ||
Boudhraa Z, Bouchon B, Viallard C, D’Incan M, Degoul F. Annexin A1 localization and its relevance to cancer. Clin Sci (Lond). 2016;130(4):205–220. | ||
Biaoxue R, Xiguang C, Shuanying Y. Annexin A1 in malignant tumors: current opinions and controversies. Int J Biol Markers. 2014;29(1):e8–e20. | ||
Guo C, Liu S, Sun MZ. Potential role of Anxa1 in cancer. Future Oncol. 2013;9(11):1773–1793. | ||
Weyd H, Abeler-Dörner L, Linke B, et al. Annexin A1 on the surface of early apoptotic cells suppresses CD8+ T cell immunity. PLoS One. 2013;8(4):e62449. | ||
Leoni G, Nusrat A. Annexin A1: shifting the balance towards resolution and repair. Biol Chem. 2016;397(10):971–979. | ||
Bozinovski S, Anthony D, Vlahos R. Targeting pro-resolution pathways to combat chronic inflammation in COPD. J Thorac Dis. 2014;6(11):1548–1556. | ||
Neymeyer H, Labes R, Reverte V, et al. Activation of annexin A1 signalling in renal fibroblasts exerts antifibrotic effects. Acta Physiol (Oxf). 2015;215(3):144–158. | ||
Damazo AS, Sampaio AL, Nakata CM, Flower RJ, Perretti M, Oliani SM. Endogenous annexin A1 counter-regulates bleomycin-induced lung fibrosis. BMC Immunol. 2011;12:59. | ||
Higashi T, Mai Y, Mazaki Y, Horinouchi T, Miwa S. A standardized method for the preparation of a gas phase extract of cigarette smoke. Biol Pharm Bull. 2016;39(6):898–902. | ||
Sandbo N, Ngam C, Torr E, Kregel S, Kach J, Dulin N. Control of myofibroblast differentiation by microtubule dynamics through a regulated localization of mDia2. J Biol Chem. 2013;288(22):15466–15473. | ||
Lai T, Wu D, Chen M, et al. YKL-40 expression in chronic obstructive pulmonary disease: relation to acute exacerbations and airway remodeling. Respir Res. 2016;17:31. | ||
Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2(2):329–333. | ||
Manetsch M, Che W, Seidel P, Chen Y, Ammit AJ. MKP-1: a negative feedback effector that represses MAPK-mediated pro-inflammatory signaling pathways and cytokine secretion in human airway smooth muscle cells. Cell Signal. 2012;24(4):907–913. | ||
Babbin BA, Laukoetter MG, Nava P, et al. Annexin A1 regulates intestinal mucosal injury, inflammation, and repair. J Immunol. 2008;181(7):5035–5044. | ||
Perucci LO, Carneiro FS, Ferreira CN, et al. Annexin A1 is increased in the plasma of preeclamptic women. PLoS One. 2015;10(9):e0138475. | ||
Vijayan VK. Chronic obstructive pulmonary disease. Indian J Med Res. 2013;137(2):251–269. | ||
Mio T, Romberger DJ, Thompson AB, Robbins RA, Heires A, Rennard SI. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am J Respir Crit Care Med. 1997;155(5):1770–1776. | ||
Sugimoto MA, Ribeiro ALC, Costa BRC, et al. Plasmin and plasminogen induce macrophage reprogramming and regulate key steps of inflammation resolution via annexin A1. Blood. 2017;129(21):2896–2907. | ||
Leoni G, Neumann PA, Kamaly N, et al. Annexin A1-containing extracellular vesicles and polymeric nanoparticles promote epithelial wound repair. J Clin Invest. 2015;125(3):1215–1227. | ||
Wang X, Zhu M, Hjorth E, et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015;11(1):40–50.e1–2. | ||
Jia Y, Morand EF, Song W, Cheng Q, Stewart A, Yang YH. Regulation of lung fibroblast activation by annexin A1. J Cell Physiol. 2013;228(2):476–484. | ||
Skouteris GG, Schröder CH. The hepatocyte growth factor receptor kinase-mediated phosphorylation of lipocortin-1 transduces the proliferating signal of the hepatocyte growth factor. J Biol Chem. 1996;271(44):27266–27273. | ||
Trentin PG, Ferreira TP, Arantes AC, et al. Annexin A1 mimetic peptide controls the inflammatory and fibrotic effects of silica particles in mice. Br J Pharmacol. 2015;172(12):3058–3071. | ||
Facio FN Jr, Facio MF, Spessoto LF, et al. Anti-inflammatory and anti-fibrotic effects of annexin1 on erectile function after cavernous nerve injury in rats. Int J Impot Res. 2016;28(6):221–227. | ||
Yang H, Long F, Zhang Y, et al. 1α,25-dihydroxyvitamin D3 induces neutrophil apoptosis through the p38 MAPK signaling pathway in chronic obstructive pulmonary disease patients. PLoS One. 2015;10(4):e0120515. | ||
Li D, Hu J, Wang T, et al. Silymarin attenuates cigarette smoke extract-induced inflammation via simultaneous inhibition of autophagy and ERK/p38 MAPK pathway in human bronchial epithelial cells. Sci Rep. 2016;6:37751. | ||
Li D, Xu D, Wang T, et al. Silymarin attenuates airway inflammation induced by cigarette smoke in mice. Inflammation. 2015;38(2):871–878. | ||
O’Leary L, Sevinç K, Papazoglou IM, et al. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016;590(9):1324–1334. | ||
Gu W, Song L, Li XM, Wang D, Guo XJ, Xu WG. Mesenchymal stem cells alleviate airway inflammation and emphysema in COPD through down-regulation of cyclooxygenase-2 via p38 and ERK MAPK pathways. Sci Rep. 2015;5:8733. | ||
Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS. Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem. 2009;284(18):12524–12532. | ||
Arora S, Lim W, Bist P, et al. Influenza A virus enhances its propagation through the modulation of Annexin-A1 dependent endosomal trafficking and apoptosis. Cell Death Differ. 2016;23(7):1243–1256. | ||
Li X, Zhao Y, Xia Q, et al. Nuclear translocation of annexin 1 following oxygen-glucose deprivation-reperfusion induces apoptosis by regulating Bid expression via p53 binding. Cell Death Dis. 2016;7(9):e2356. |
Supplementary materials
 | Table S1 Primer sets for real-time polymerase chain reaction analysis |
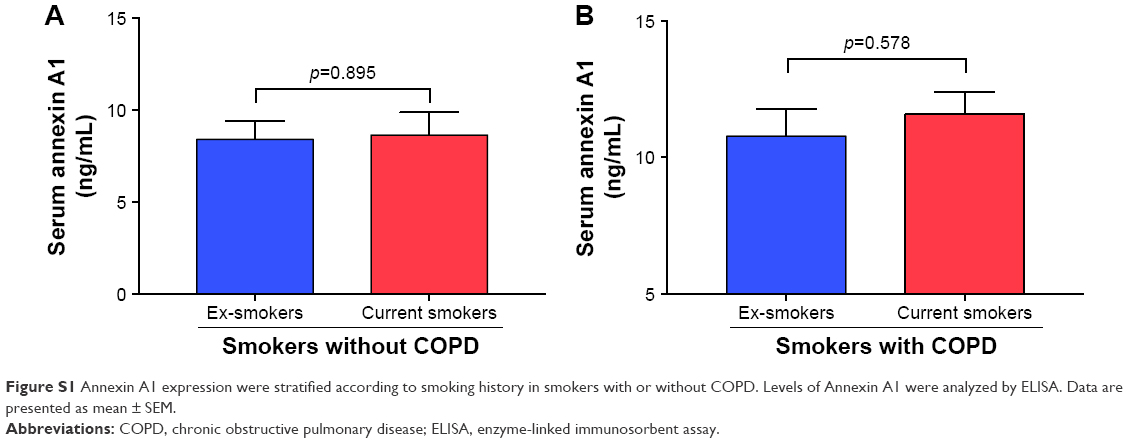 | Figure S1 Annexin A1 expression were stratified according to smoking history in smokers with or without COPD. Levels of Annexin A1 were analyzed by ELISA. Data are presented as mean ± SEM. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.