")
Back to Journals » The Application of Clinical Genetics » Volume 9
Animal models of GM2 gangliosidosis: utility and limitations
Received 3 February 2016
Accepted for publication 24 May 2016
Published 20 July 2016 Volume 2016:9 Pages 111—120
DOI https://doi.org/10.2147/TACG.S85354
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Cheryl A Lawson,1,2 Douglas R Martin2,3
1Department of Pathobiology, 2Scott-Ritchey Research Center, 3Department of Anatomy, Physiology and Pharmacology, Auburn University College of Veterinary Medicine, Auburn, AL, USA
Abstract: GM2 gangliosidosis, a subset of lysosomal storage disorders, is caused by a deficiency of the glycohydrolase, β-N-acetylhexosaminidase, and includes the closely related Tay–Sachs and Sandhoff diseases. The enzyme deficiency prevents the normal, stepwise degradation of ganglioside, which accumulates unchecked within the cellular lysosome, particularly in neurons. As a result, individuals with GM2 gangliosidosis experience progressive neurological diseases including motor deficits, progressive weakness and hypotonia, decreased responsiveness, vision deterioration, and seizures. Mice and cats are well-established animal models for Sandhoff disease, whereas Jacob sheep are the only known laboratory animal model of Tay–Sachs disease to exhibit clinical symptoms. Since the human diseases are relatively rare, animal models are indispensable tools for further study of pathogenesis and for development of potential treatments. Though no effective treatments for gangliosidoses currently exist, animal models have been used to test promising experimental therapies. Herein, the utility and limitations of gangliosidosis animal models and how they have contributed to the development of potential new treatments are described.
Keywords: GM2 gangliosidosis, Tay–Sachs disease, Sandhoff disease, lysosomal storage disorder, sphingolipidosis, brain disease
Introduction
Recent advances ascribe novel and important roles to the lysosome that were previously unappreciated, including nutrient homeostasis and cell signaling in local or even distant tissues.1,2 Still valid, however, is the lysosome’s well-recognized function as a type of intracellular “stomach” or recycling center that contains various enzymes including nucleases, proteases, lipases, phosphatases, and glycosidases at low pH.3–5 Here, complex lipids, oligosaccharides, and other macromolecules are degraded into their building blocks in a stepwise fashion by multiple lysosomal enzymes.4,6 When there are defects in the enzymes or proteins necessary for catabolism, abnormal accumulation of nondegraded products results in a lysosomal storage disorder.4 Lysosomal storage disorders are a group of >70 inherited disorders classified according to the accumulated substrate.7 For example, mucopolysaccharidoses result from the accumulation of mucopolysaccharides (also known as glycosaminoglycans), and gangliosidoses are characterized by the accumulation of gangliosides.
Gangliosides, a class of glycosphingolipids, are essential molecules within the cellular plasma membrane where they often assist in the formation of lipid rafts.8–10 They are involved in adhesion and cytokine signal transduction and cell membrane protein regulation.10 They may also be used as receptors for infectious or normal cellular processes. An example is cholera toxin B, which requires at least one GM1 ganglioside binding site to complete infection.11 Neurons have high levels of gangliosides, with maximum expression in the gray matter of the brain.8,9 As much as 12% of the lipid matter of the brain consists of gangliosides.10 Here, they modulate cell signaling and are essential for normal brain development and function.9,10 However, when ganglioside catabolism is flawed, such as with the inherited gangliosidoses, severe neurological dysfunction ensues.9 Gangliosides consist of a ceramide backbone and oligosaccharide side chain, with or without sialic acid. Gangliosides are classified via the Svennerholm nomenclature in which “G” denotes “ganglioside,” the number of sialic acid residues is identified using the following letters: A=0, M=1, D=2, and T=3, and the number of monosaccharide residues is identified using the following final digits: 1=4, 2=3, and 3=2.12 The structure of GM2 ganglioside is shown in Figure 1.
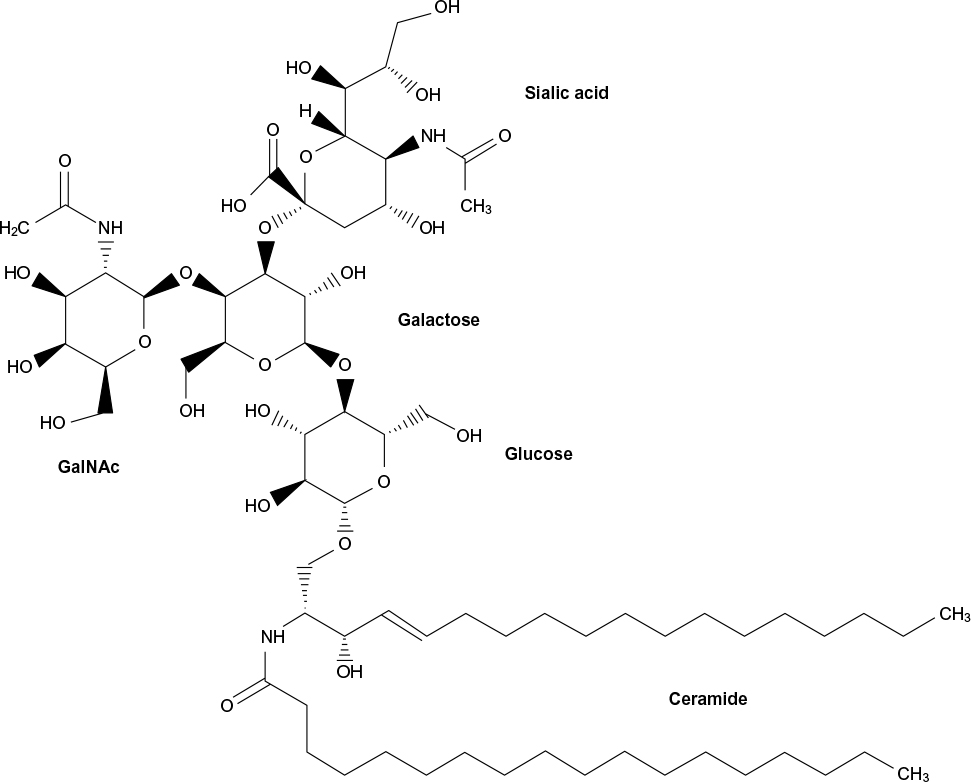 | Figure 1 Structure of GM2 ganglioside. |
Gangliosides are degraded in a stepwise fashion through a series of hydrolytic events mediated by specific lysosomal enzymes. For example, GM2 hydrolysis is initiated by β-N-acetylhexosaminidase (Hex; EC 3.2.1.52). The absence of Hex activity prevents further degradation of the molecule, resulting in GM2 gangliosidosis. Hex consists of three isozymes, each comprised of two subunits. Hexosaminidase A (HexA) is composed of both an α and a β subunit, HexB is composed of two β subunits, and HexS is composed of two α subunits, though it is very unstable and contributes little to GM2 hydrolysis. Each subunit is encoded by specific genes, HEXA for the α subunit and HEXB for the β subunit. Of the three isozymes, only HexA is capable of degrading GM2 ganglioside in humans. The GM2 activator protein, an accessory protein with no hydrolytic activity toward GM2 ganglioside, is nevertheless required for GM2 degradation by Hex.13 Thus, GM2 gangliosidosis may be caused by any of the three protein deficiencies and is classified into the following three variants based on the isozyme(s) that remain functional: variant B (Tay–Sachs disease) is an α subunit deficiency that destroys HexA and HexS; variant 0 (Sandhoff disease) is a β subunit deficiency that destroys both HexA and HexB; and variant AB (GM2 activator deficiency) leaves HexA and HexB intact, but GM2 cannot be degraded.13 Both the diseases along with their associated genes, proteins, isoenzyme subunits, and accumulated substrates are depicted in Figure 2.
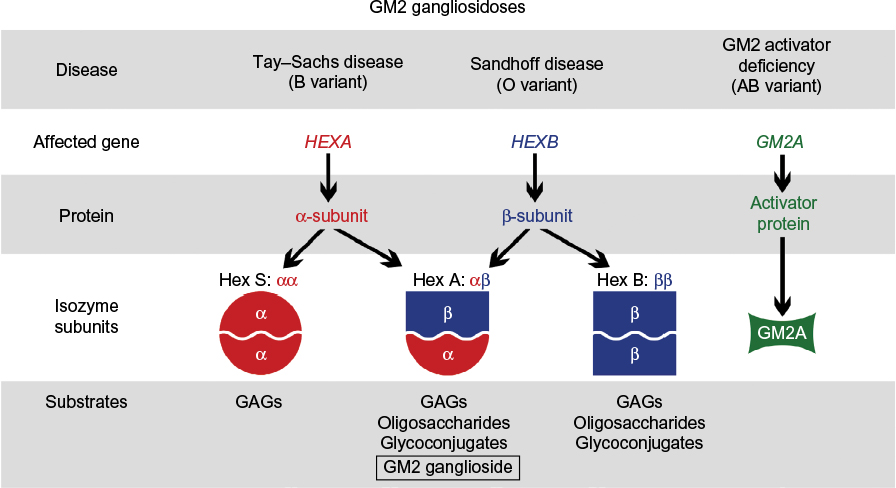 | Figure 2 Hex isozymes, associated substrates, and disorders. |
Human GM2 gangliosidosis
Regardless of the protein deficiency underlying GM2 gangliosidosis, the disease course is quite similar and is segregated into the following clinical phenotypes based on severity and age of onset: infantile (also known as acute), the most severe form with onset at <1 year of age; subacute (which includes late-infantile and juvenile forms), with onset in early childhood; and chronic (also called late- or adult-onset), with initial symptoms in early- to midteens.13 Diagnosis may be made by a number of methods, including sequencing for known mutations, mass spectrometry, and conventional enzyme assays using synthetic substrates. The infantile form has been described most thoroughly. Children are clinically normal at birth, begin showing early clinical signs at ∼3–5 months of age, and experience progressive neurological disease including increased startle reflex, motor deficits, progressive weakness and hypotonia, decreased responsiveness, vision deterioration, and developmental arrest.13–15 Seizures develop in 98% of patients.15 Ophthalmoscopic examination often reveals a “cherry-red spot” due to macular pallor of the fovea centralis. Some infants affected by Sandhoff disease have additional peripheral disease consisting of hepatosplenomegaly and dysostosis multiplex.13 Regardless, children affected with either Tay–Sachs or Sandhoff disease usually die at 3–5 years of age.13,15 The majority of children die due to primary disease (∼46%) and fewer die due to aspiration pneumonia or seizure activity.15 Subacute disease is typically recognized between 2 years and 10 years of age by progressive ataxia and incoordination. These patients do not typically display the “cherry-red spots” and instead experience loss of vision due to optic atrophy and retinitis pigmentosa.13 The majority of patients with this form progress to a vegetative state accompanied by decerebrate rigidity at ∼10–15 years of age and die a few years later.13 The cause of death is most often concurrent infection.13 The chronic GM2 gangliosidoses have similar clinical phenotypes; however, chronic Tay–Sachs disease is more common than chronic Sandhoff disease.13 The age of clinical onset is variable, but symptoms include progressive dystonia, nystagmus, ataxia, tremors, muscle wasting and weakness, and psychosis.13
Normal life span can be expected with residual Hex activity as low as 10%–20% of normal.8 Below that threshold, disease onset generally correlates with the amount of residual enzyme activity produced by a given mutation. That is, severe, early-onset disease is caused by low levels of residual activity, whereas milder, late-onset disease results from relatively high residual activity. Correlation of disease severity with residual enzyme activity suggests that prognosis can be predicted by specific mutations, which is often but not always the case and demonstrates that other unknown factors influence disease progression.
Pathological changes
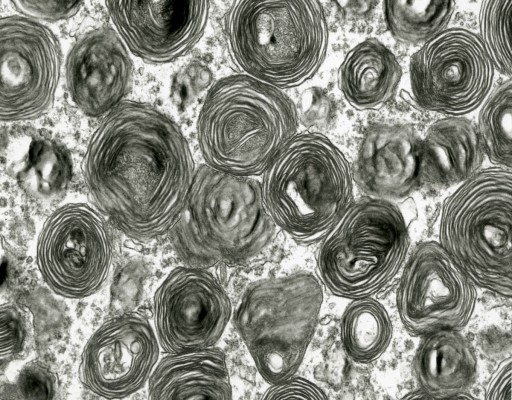
Grossly, the brain of GM2 gangliosidosis patients can vary widely in appearance, but alterations include marked atrophy, thickened and fibrotic leptomeninges, dilated ventricular system, thinned cerebral cortex, cerebellar and brainstem atrophy, and widened gyri.14 Some brain abnormalities, such as diffuse white matter changes, subtle basal ganglia alterations, and hemisphere and cerebellar atrophy, can be detected using advanced imaging modalities such as computed tomography and magnetic resonance imaging (MRI).12 Histopathological changes noted via light microscopy include distention of neurons with storage material, demyelination, atrophic dendrites, and infiltration of macrophages containing abundant storage material.14,16 Central nervous system (CNS) inflammation due to glial cell activation, a hallmark of the gangliosidoses, may be associated with subsequent iron depletion in the brain due to alterations in cellular iron metabolism and homeostasis.17 Ultrastructurally, characteristic cellular changes are termed “membranous cytoplasmic bodies” that appear as concentric ring-like structures in neuronal lysosomes. This term was coined after early electron micrographs of affected brain tissue revealed cytoplasmic, membrane-like structures formed by massive accumulation of the amphiphilic GM2 ganglioside molecule within the aqueous environment of the lysosome (Figure 3).13
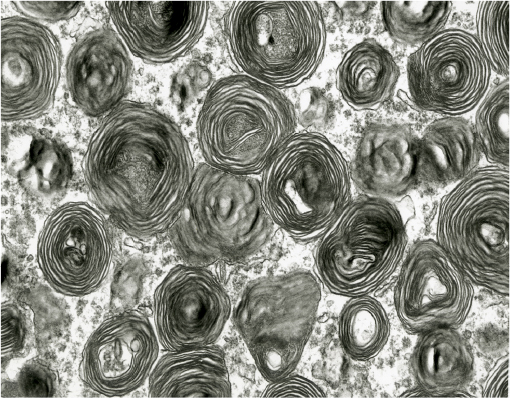 | Figure 3 Transmission electron micrograph of membranous cytoplasmic bodies in the brain of a cat affected with gangliosidosis (Magnification 23,275). |
As previously mentioned, disease is not isolated to the CNS. Besides hepatosplenomegaly and eye abnormalities, alterations in other peripheral tissues such as foamy histiocytes within the bone marrow, cardiac valvular and structural abnormalities, skeletal changes, and iron depletion of the bone marrow, liver, and other organs have been described.8,13,17,18 The presence of vacuolated lymphocytes along with magenta neutrophil inclusions has been well documented in several nonhuman species affected by GM2 gangliosidosis.19 Cytologic cerebrospinal fluid (CSF) analysis of a Korat kitten with GM2 gangliosidosis (Sandhoff disease) revealed large mononuclear cells containing prominent, purple inclusions, whereas CSF from muntjac deer with Tay–Sachs-like disease contained large mononuclear cells with long, needle-like inclusions.20,21 However, little has been described regarding CSF cytologic evaluation in human patients with GM2 gangliosidosis.
Several biomarkers have been investigated recently as minimally invasive monitors of disease progression. Thirteen analytes associated with glial cell activation and subsequent inflammation were markedly increased in the CSF in gangliosidosis patients. Five of these 13 analytes were persistently increased in the infantile-onset form of the disease.22 Blood, CSF, MRI, and electrodiagnostic abnormalities also have been investigated.16
Animal models of GM2 gangliosidosis
No effective treatments exist for GM2 gangliosidosis, and animal modeling is an important tool for therapy development. Subsequently, the species in which GM2 gangliosidosis has been reported and their degree of similarity to the human condition are described.
Mice
Unlike human GM2 gangliosidosis, Tay–Sachs and Sandhoff diseases in mice have dramatically different phenotypes. In 1995, mouse models of both Tay–Sachs and Sandhoff diseases were described. The Tay–Sachs mouse model (HEXA knockout) displayed biochemical and pathological hallmarks of disease only in a few focal areas of the brain such as the septal area, hippocampus, and cerebral cortex. Clinical signs could only be induced in female mice that had at least four litters before a year of age.23 Otherwise, Tay–Sachs mice showed few, if any, clinical signs and had normal life spans.23,24 However, the mouse model of Sandhoff disease (HEXB knockout) showed marked neurological disease, widespread neuropathology, and a short lifespan of ∼4 months.24 The mouse model of GM2 activator deficiency had an intermediate phenotype, with cerebellar pathology and motor deficiencies, but without a reduced life span.25
Discordance of disease severity between the models was explained by the presence of an alternative pathway for the degradation of GM2 ganglioside in mice. In humans, GM2 is almost exclusively degraded by HexA so that Tay–Sachs disease (HexA deficiency) leads to profound ganglioside storage. However, in the mouse model of Tay–Sachs disease, sialidases are able to convert GM2 to GA2, which is then degraded through a minimal yet sufficient activity of HexB. Elegant work from the laboratory of Li suggests that GA2 degradation by HexB results from a mouse GM2 activator protein that is much less restrictive than its human counterpart. Thus, an alternative catabolic pathway for GM2 ganglioside in mice is responsible for a Tay–Sachs model that does not faithfully recapitulate the human condition.26,27
Dogs
Although dogs with GM2 gangliosidosis are not known to exist in a research colony setting, affected dogs of several breeds have been described. The following cases of canine GM2 gangliosidosis have been reported:
- A castrated male golden retriever with reduced HexA and HexB activity has been reported. This patient succumbed to disease at 15 months of age, and the genetic mutation was not identified.28 The approximate average life span of normal dogs is 10–12 years.
- A family of toy poodles affected by Sandhoff disease have also been described. The clinical, pathological, biochemical, and MRI findings were attributed to a frameshift mutation in HEXB.29,30
- A mixed breed dog presented at 10 months of age with hind limb weakness. The weakness progressed and by the age of 13 months, the dog was unable to stand or ambulate normally. Enzyme assays from whole blood revealed a marked decrease in HexA and HexB activity, and GM2 accumulation was detected by immunohistochemistry. DNA genotyping from this dog was negative for the mutation documented in the family of toy poodles previously described.31
- German short-haired pointer and Japanese spaniels were reported with profound storage of GM2 ganglioside, but the underlying protein defect was not pinpointed.32,33
- Recently, a likely case of Tay–Sachs disease was reported in Japanese Chin dogs with a Glu323Lys mutation in the Hex α-subunit.34
Cats
No feline models of Tay–Sachs disease have been described. However, numerous mutations in the feline HEXB gene have been reported. Clinical signs of Sandhoff disease in affected kittens begin at ∼4–7 weeks of age and include hypermetria, head tremor, ataxia, and eventual paresis.35–37 Seizures are part of the natural disease course, and death occurs at ∼6 months of age.35,36 For cats that are used as models of therapy development for Sandhoff disease, a clinical rating scale that describes disease progression in more detail has been developed.38,39 Also, humane end point has been established as the inability to stand, which occurs at ∼4.3 months of age.38,39 The approximate life span of normal cats is 12–14 years.
Several feline mutations have been described in a number of breeds including European Burmese, Korat, domestic short-haired cats, and Japanese domestic cats (Table 1). In 1977, a naturally occurring mutation genetically and biochemically analogous to human Sandhoff disease was reported in domestic short-haired cats (Figure 4A).40 Animals with this mutation have <3% of normal Hex activity and ∼15% of normal protein levels. The mutation has been identified as a 25 base pair inversion at the extreme 3’ terminus of the coding sequence.37 This mutation results in the presence of antigenically cross-reactive material, which may diminish the immune response to therapeutic protein.36 Another mutation was described in 1985 in Korat cats derived from Thailand. The gene mutation in this lineage is due to a single base deletion with a resultant premature termination codon in exon 1.40,41 Since no β subunit protein is produced from this mutation, the immune system of affected Korat cats is likely to recognize a therapeutic β subunit protein as foreign.
 | Figure 4 Representative animal models of GM2 gangliosidosis. |
 | Table 1 Overview of documented feline Sandhoff mutations and their respective breeds |
Japanese domestic cats also have a mutant allele that results in low activities of both HexA and HexB.42,43 This mutation is a single nucleotide substitution resulting in premature termination at codon 223, midway through the β subunit.44,45 Also, a 15 base pair deletion at the 3’ end of intron 11 of HEXB, which included the preferred splice acceptor site, has been identified in European Burmese cats.46
Sheep
A consequence of the alternative pathway for GM2 ganglioside catabolism in mice, described earlier, is that mouse models of Tay–Sachs disease have not been terribly helpful for therapy development. In fact, the lack of an established animal model of Tay–Sachs disease with measurable clinical symptoms has been a hindrance in the quest for effective treatments. Although Tay–Sachs disease has been described in various species, many – such as flamingos – are not appropriate research models.47 In 2010, it was described that Jacob sheep (Figure 4B) with Tay–Sachs disease provide a model with a large brain size and obvious clinical signs that can be easily maintained in a research setting.48,49 Affected sheep have a single base substitution in exon 11 of HEXA, resulting in a glycine to arginine substitution in the α subunit and substantial reduction of HexA activity.48
Utilities and limitations of GM2 animal models
Though GM2 gangliosidosis has been reported in numerous species, only mice, cats, and sheep are maintained as research models. Mice continue to be invaluable as models of numerous diseases, including GM2 gangliosidosis. However, as discussed earlier, the alternative catabolic pathway for gangliosides constitutes a prominent disadvantage of mouse models of Tay–Sachs and Sandhoff diseases, confounding interpretation of results from certain experimental therapies and thus their potential for human application. Additional disadvantages of mice are their comparatively short life span that precludes truly long-term studies16 and their small size that limits the ability to obtain sufficient samples of tissues or body fluids (especially CSF). A decided advantage of mouse models for neurodegenerative disease is that standardized behavioral assays are available for consistent evaluation of disease progression.50 Other utilities of mice as models of Sandhoff disease include small space requirements, short life cycle (∼2 years) and gestation (∼21 days), and large litter size. These considerations allow a large number of animals to be readily available in a short period of time,51 and per diem/maintenance costs are usually far below those of larger animals.
The feline models have been advantageous in translational research endeavors due to their strong resemblance phenotypically, biochemically, and genetically to human Sandhoff disease. Compared with mice, the degree of GM2/GA2 accumulation in cats with Sandhoff disease is more comparable to human patients. Additionally, the reduction in myelin-enriched lipids (associated with dysmyelination) is intermediate to the levels in the mouse with Sandhoff disease and human patients.52 Also, cats have long been used as models of human disease and have large brains (∼60× larger than mice) with complexity and organization that is more similar to humans.53 In addition to providing a greater amount of tissue for experimentation and diagnostics, the increased size eases dissection of certain regions of the brain and permits noninvasive imaging with MRI field strengths identical to those used in human medicine (3 T). Furthermore, cats have substantially larger bodies facilitating repeated sampling of fluids such as blood, urine, and CSF for biomarker development to track disease progression in individual animals over time. The gestation length for cats (∼63 days) is longer than that of mice; however, it is significantly shorter than that of humans. Additionally, cats have a significantly longer life span compared with mice, allowing for long-term studies. However, a significant limitation of the feline model is the limited availability of feline-specific reagents (such as antibodies).
The primary utility of sheep is that they are the only established research model of Tay–Sachs disease with measurable clinical symptoms. Human late-onset Tay–Sachs disease closely resembles the clinical and pathological features of affected sheep, such as ataxia, proprioceptive deficits, and similar histological and ultrastructural appearance.49 Furthermore, the level of residual enzyme activity is also similar.48 They are easily maintained as research animals, often with per diem costs lower than cats, and their brain size is only five to ten times smaller than that of a child. However, it is important to note that sheep have disproportionately larger body versus brain sizes compared with humans, meaning that intravenous or other systemic therapies for people may not be effectively modeled in sheep. Additional disadvantages include the possession of horns (up to six), which creates challenges with imaging modalities such as MRI and computed tomography. Similar to cats, procuring ovine-specific antibodies or other markers is difficult.
Therapy development
As previously mentioned, there is no effective treatment for GM2 gangliosidosis. However, several experimental therapies are being investigated.
Enzyme replacement therapy
Since the GM2 gangliosidoses are the result of absent or defective Hex enzymes with no or little functional activity, replacement of these enzymes with functional molecules has been investigated. There has been some success in treating humans with systemic or peripheral disease associated with other lysosomal storage diseases such as Gaucher disease and mucopolysaccharidoses; however, penetration of infused enzymes into the CNS remains challenging.54 A major obstacle is the delivery of the exogenous enzymes across biochemical or anatomical barriers such as the blood–brain barrier. Additionally, hepatic mechanisms may rapidly clear any circulating enzyme before it can benefit target tissues.55 No attempts to treat GM2 gangliosidosis through systemic enzyme replacement therapy have been successful. However, intracerebroventricular injections of a recombinant human HexA in Sandhoff mice led to a slightly prolonged life span compared with the untreated control animals.56
Substrate reduction therapy
Substrate reduction therapy (SRT, also called substrate deprivation therapy) utilizes small molecules that partially inhibit the synthesis of ganglioside.57,58 N-Butyldeoxynojirimycin (miglustat or Zavesca®), which was initially developed as a treatment for patients with acquired immunodeficiency syndrome, reduces the rate of neuronal ganglioside storage in murine models of Tay–Sachs and Sandhoff diseases.59,60 In treated Sandhoff mice, no increase in life expectancy could be achieved if therapy began during the late presymptomatic period; however, in mice treated earlier (at 3 weeks or 6 weeks of age), the life expectancy was extended by ∼40%.60 In two infantile Tay–Sachs patients treated after the onset of clinical signs, miglustat did not halt disease progression but did slow the advancement in one patient. A decrease in the rate of CNS inflammation was also noted.61 Another study in a patient with chronic Sandhoff disease appreciated minor effects on neurological progression.62 Thus, miglustat has shown only mild efficacy in humans, and it has an associated gastrointestinal toxicity that leads to discontinuance in many patients.
Chaperone therapy
Often, mutated lysosomal enzymes retain a reasonable degree of activity toward native substrates, but are prevented from reaching the lysosome by the quality control machinery of the endoplasmic reticulum. Chaperones are small molecules that stabilize (or “rescue”) the conformation of mutated lysosomal proteins that would otherwise be cleared by endoplasmic reticulum-associated degradation. After “rescued” proteins reach the low-pH environment of the lysosome, they dissociate from the chaperone molecule and are able to restore some level of enzyme activity.63 One study in GM2 fibroblasts found that chaperone therapy is effective in increasing enzyme activity, but concluded that it should be combined with SRT for synergistic effects.62 To date, efficacy of chaperone therapy in GM2 patients has not been confirmed, though fine-tuning of dose and delivery routes may prove beneficial. For example, one clinical trial found a close correlation in HexA activity in leukocytes and plasma levels of pyrimethamine, a pharmacological chaperone drug. However, the plasma level varied due to both the dose and metabolism of each patient.63
Bone marrow/cord blood transplantation
Bone marrow transplants have been investigated in various lysosomal storage diseases including leukodystrophies, mucopolysaccharidosis, Gaucher disease, and Tay–Sachs disease; however, mortality is high (∼25%). Similar to the other potential therapies discussed, studies in animal models indicate that treatment before the onset of neurological symptoms is pivotal to maximize the effects of therapy.64 Results of umbilical cord blood transplantation in Tay–Sachs children have not been promising. Though disease in some children stabilized after the transplant, they remained severely debilitated.65 A synergistic effect of SRT along with bone marrow transplantation has been reported in murine Sandhoff disease models.57 In a human Tay–Sachs patient, bone marrow transplantation with miglustat treatment did not improve outcome, though miglustat was added only after it became apparent that standard bone marrow transplant was not beneficial.66
Neural stem cell therapy
Neural stem cell transplantation (via intracranial injection) has been noted to delay disease onset, preserve motor function, reduce pathological changes, and enhance survival in mice with Sandhoff disease, by migrating extensively throughout the brain, disseminating cross-correcting enzyme, and reducing ganglioside storage and inflammation.67,68 If treatment occurred presymptomatically, there was a 40% increase in life expectancy compared with the 19% improvement in animals treated after onset of clinical disease. Combination treatment with stem cell therapy and SRT synergistically doubled the life span in some of the mice in this study.68 However, another study with Sandhoff mice found no such synergistic relationship with neural stem cell therapy and the substrate reduction molecule N-butyldeoxygalactonojirimycin (NB-DGJ).69 That is, NB-DGJ alone reduced storage as effectively as the combination of NB-DGJ and neural stem cell therapy.70 Neural stem cell transplantation in human GM2 patients has not been reported.
Iron supplementation
One study appreciated a marked depletion of iron in the brain and other organs such as the bone marrow, liver, spleen, and kidneys of GM2 gangliosidosis mice. Supplementation of iron (both oral and parenteral) delayed disease onset, improved motor function, and prolonged survival. The author concluded that further studies regarding iron supplementation combined with other therapeutic strategies and anti-inflammatory drugs should be investigated.17
Gene therapy
Adenoviral-mediated gene therapy has been tested in the knockout mouse model of Sandhoff disease,71 whereas adeno-associated viral (AAV) gene therapy has been intensively investigated in both feline and murine models.38,39,41,70,72,73 AAV gene therapy delivered intracranially in Sandhoff mice showed dramatically increased survival and delayed disease onset. Supranormal Hex activity was widespread and long-lasting throughout the brain and spinal cord, reducing ganglioside storage and inflammation.72 Some treated mice lived normal life spans.
The life span of Sandhoff cats treated via bilateral thalamic injections was prolonged in spite of strong humoral immunity to AAV1 vectors and human HexA protein.74 When vectors were modified to encode feline Hex in the context of an AAVrh8 capsid, the immune response was reduced and survival increased (>2× longer life span than untreated cats).74 In short-term studies with the same feline-specific vectors, thalamic treatment supplemented by injections of the deep cerebellar nuclei or lateral ventricle restored Hex activity to above normal levels throughout the brain and spinal cord. Although storage material was significantly reduced in the CNS, it was not completely cleared. However, neurological function significantly improved 17 weeks after therapy compared with the control animals.38 Long-term studies are underway to determine maximum survival benefit.
Concerns regarding gene therapy for human treatment include permanent genetic modification of neurons and insertional mutagenesis leading to neoplasia after vector integration into the host genome. Also, direct intracranial injection has inherent risks that may be minimized by less invasive routes such as CSF and peripheral blood. Such alternative strategies are being investigated75 but may be confounded by the dilution of vector in large volumes of body fluid. After injection into the CSF or blood, two separate vectors expressing the α and β subunits are unlikely to transduce any given cell, and coexpression of both subunits is necessary for optimal production of HexA.73,74 Construction of a bicistronic AAV vector expressing both subunits has proven challenging due to size constraints. However, a novel construct termed HexM incorporates critical features of both the α and β subunits into a single protein and has produced promising results after intravenous injection in the mouse model of Sandhoff disease.76
Conclusion
Animal models are a useful tool in advancing the understanding of pathogenesis and developing potential therapies for orphan diseases such as the GM2 gangliosidoses. Animal models of GM2 gangliosidosis vary according to biochemistry, size, species, and other important factors. The most appropriate model or models may be chosen according to specific experimental goals. Although there are a few limitations to the use of animal models, they are more than counterbalanced by the utilities. Much of the information gained from research using animal models has been crucial in the advancement of experimental therapies to potential clinical application. With continued development and testing of promising approaches, long-sought therapies will finally be at hand.
Disclosure
The authors report no conflicts of interest in this work.
References
Mony VK, Benjamin S, O’Rourke EJ. A lysosome-centered view of nutrient homeostasis. Autophagy. 2016;12(4):619–631. | ||
Fraldi A, Klein AD, Medina DL, Settembre C. Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci. 2016;39:277–295. | ||
Schulze H, Sandhoff K. Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol. 2011;3(6):a004804. | ||
Kolter T, Sandhoff K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010;584(9):1700–1712. | ||
Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th ed. Philadelphia, PA: Elsevier Saunders; 2015. | ||
Kolter T, Sandhoff K. Glycosphingolipid degradation and animal models of GM2-gangliosidoses. J Inherit Metab Dis. 1998;21(5):548–563. | ||
Cox TM, Cachón-González MB. The cellular pathology of lysosomal diseases. J Pathol. 2012;226(2):241–254. | ||
Sandhoff K, Harzer K. Gangliosides and gangliosidoses: principles of molecular and metabolic pathogenesis. J Neurosci. 2013;33(25): 10195–10208. | ||
Schengrund C. Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem Sci. 2015; 40(7):397–406. | ||
Palmano K, Rowan A, Guillermo R, Guan J, McJarrow P. The role of ganglioside in neurodevelopment. Nutrients. 2015;7(5):3891–3913. | ||
Jobling MG, Yang Z, Kam WR, Lencer WI, Holmes RK. A single native ganglioside GM1-binding site is sufficient for cholera toxin to bind to cells and complete the intoxication pathway. MBio. 2012;3(6):1–9. | ||
Patterson M. Chapter 174: gangliosidoses. Handb Clin Neurol. 2013; 113(3):1707–1708. | ||
Gravel RA, Clarke JTR, Kaback MM, Mahuran D, Sandhoff K, Suzuki K. Chapter 92: the GM2 gangliosidoses. In: Valle D, editor. Metabolic and Molecular Basis of Inherited Disease. 7th ed. New York City, NY: McGraw-Hill Professional; 1995:2839–2869. | ||
Sandhoff K, Conzelmann E, Neufeld EF, et al. Chapter 72: the GM2 gangliosidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Disease. 6th ed. McGraw-Hill Information Services Company; 1989:1807–1839. | ||
Bley AE, Ourania A, Giannikopoulos BA, et al. Natural history of infantile GM2 gangliosidosis. Pediatrics. 2011;128(5):1233–1241. | ||
Bradbury AM, Gray-Edwards HL, Shirley JL, et al. Biomarkers for disease progression and AAV therapeutic efficacy in feline Sandhoff disease. Exp Neurol. 2015;263:102–112. | ||
Jeyakumar M, Williams I, Smith DA, Cox TM, Platt FM. Critical role of iron in the pathogenesis of the murine gangliosidoses. Neurobiol Dis. 2009;34(3):406–416. | ||
Gray-Edwards HL, Brunson BL, Holland M, et al. Mucopolysaccharidosis-like phenotype in feline Sandhoff disease and partial correction after AAV gene therapy. Mol Genet Metab. 2015;116(1–2):80–87. | ||
Harvey JW. Veterinary Hematology: A Diagnostic Guide and Color Atlas. 1st ed. St. Louis, MO: Elsevier Saunders; 2012. | ||
Johnsrude JD, Alleman AR, Schumacher J, et al. Cytologic findings in cerebrospinal fluid from two animals with GM2-gangliosidosis. Vet Clin Path. 1996;25(3):80–83. | ||
Fox J, Li YT, Dawson G, et al. Naturally occurring GM2 gangliosidosis in two Muntjak deer with pathological and biochemical features of human classical Tay-Sachs disease (type B GM2 gangliosidosis). Acta Neuropathol. 1999;97(1):57–62. | ||
Utz JRJ, Crutcher T, Schneider J, Sorgen P, Whitley CB. Biomarkers of central nervous system inflammation in infantile and juvenile gangliosidoses. Mol Genet Metab. 2015;114(2):274–280. | ||
Phaneuf D, Wakamatsu N, Huang J, et al. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1996;5(1):1–14. | ||
Sango K, Yamanaka S, Hoffman A, et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenocytpe and ganglioside metabolism. Nat Genet. 1995;11(2):170–176. | ||
Liu Y, Hoffman A, Grinberg A, et al. Mouse model of GM2 activator deficiency manifests cerebellar pathology and motor impairment. Proc Natl Acad Sci U S A. 1997;94(15):8138–8143. | ||
Bertoni C, Li YT, Li SC. Catabolism of asialo-GM2 in man and mouse. Specificity of human/mouse chimeric GM2 activator proteins. J Biol Chem. 1999;274(40):28612–28618. | ||
Yuziuk JA, Bertoni C, Beccari T, et al. Specificity of mouse GM2 activator protein and beta-N-acetylhexosaminidases A and B. Similarities and differences with their human counterparts in the catabolism of GM2. J Biol Chem. 1998;273(1):66–72. | ||
Yamato O, Matsuki N, Satoh H, et al. Sandhoff disease in a golden retriever dog. J Inherit Metab Dis. 2002;25(4):319–320. | ||
Tamura S, Tamura Y, Uchida K, et al. GM2 gangliosidosis variant 0 (Sandhoff-like disease) in a family of toy poodles. J Vet Intern Med. 2010;24(5):1013–1019. | ||
Rahman MM, Chang H, Mizukami K, et al. A frameshift mutation in the canine HEXB gene in toy poodles with GM2 gangliosidosis variant 0 (Sandhoff disease). Vet J. 2012;194(3):412–416. | ||
Kohyama M, Yabuki A, Kawasaki Y, et al. GM2 gangliosidosis variant 0 (Sandhoff disease) in a mixed-breed dog. J Am Anim Hosp Assoc. 2015;51(6):396–400. | ||
Singer HS, Cork LC. Canine GM2 gangliosidosis: morphological and biochemical analysis. Vet Pathol. 1989;26(2):114–120. | ||
Cummings JF, Wood PA, Walkley SU, deLahunta A, DeForest ME. GM2 gangliosidosis in a Japanese spaniel. Acta Neuropathol. 1985; 67(3–4):247–253. | ||
Sanders DN, Zeng R, Wenger DA, et al. GM2 gangliosidosis associated with a HEXA missense mutation in Japanese Chin dogs: a potential model for Tay Sachs disease. Mol Genet Metab. 2013;108(1):70–75. | ||
Cork LC, Munnell JF, Lorenz MD. The pathology of feline GM2 gangliosidosis. Am J Path. 1978;90(3):723–730. | ||
Neuwelt EA, Johnson WG, Blank NK, et al. Characterization of a new model of GM2-gangliosidosis (Sandhoff’s disease) in Korat cats. J Clin Invest. 1985;76(2):482–490. | ||
Martin DR, Krum BK, Varadarajan GS, Hathock TL, Smith BF, Baker HJ. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant 0. Exp Neurol. 2004;187(1):30–37. | ||
McCurdy VJ, Rockwell HE, Arthur JR, et al. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 2015;22(2): 181–189. | ||
Rockwell HE, McCurdy VJ, Eaton SC, et al. AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro. 2015;7(2):1–13. | ||
Cork LC, Munnell JF, Lorenz MD, et al. GM2 ganglioside lysosomal storage disease in cats with β-hexosaminidase deficiency. Science. 1977;196(4293):1014–1017. | ||
Bradbury AM, Gurda BL, Casal ML, et al. A review of gene therapy in canine and feline models of lysosomal storage disorders. Hum Gene Ther Clin Dev. 2015;26(1):27–37. | ||
Yamato O, Matsunaga S, Takata K, et al. GM2-gangliosidosis variant 0 (Sandhoff-like disease) in a family of Japanese domestic cats. Vet Rec. 2004;155(23):739–744. | ||
Hasegawa D, Yamato O, Kobayashi M, et al. Clinical and molecular analysis of GM2 gangliosidosis in two apparent littermate kittens of the Japanese domestic cat. J Feline Med Surg. 2007;9(3):232–237. | ||
Yamato O, Hayashi D, Satoh H, et al. Retrospective diagnosis of feline GM2 gangliosidosis variant 0 (Sandhoff-like disease) in Japan: possible spread of the mutant allele in the Japanese domestic cat population. J Vet Med Sci. 2008;70(8):813–818. | ||
Kanae Y, Endoh D, Yamato O, et al. Nonsense mutation of feline β-hexosaminidase β-subunit (HEXB) gene causing Sandhoff disease in a family of Japanese domestic cats. Res Vet Sci. 2007;82(1):54–60. | ||
Bradbury AM, Morrison NE, Hwang M, et al. Neurodegenerative lysosomal storage disease in European Burmese cats with hexosaminidase β-subunit deficiency. Mol Gene Metab. 2009;97(1):53–59. | ||
Zeng BJ, Torres PA, Viner TC, et al. Spontaneous appearance of Tay-Sachs disease in an animal model. Mol Genet Metab. 2008;95(1–2):59–65. | ||
Torres PA, Zeng BJ, Porter BF, et al. Tay-Sachs disease in Jacob sheep. Mol Genet Metab. 2010;101(4):357–363. | ||
Porter BF, Lewis BC, Edwards JF, et al. Pathology of GM2 gangliosidosis in Jacob sheep. Vet Pathol. 2011;48(4):807–813. | ||
Chung SK, Lee AYW, Chung SSM. Mouse models for human diseases. Hong Kong Med J. 1997;3(2):201–209. | ||
Bailey KR, Rustay NR, Crawley JN. Behavioral phenotyping of transgenic and knockout mice: practical concerns and potential pitfalls. ILAR J. 2006;47(2):124–131. | ||
Baek RC, Martin DR, Cox NR, Seyfried TN. Comparative analysis of brain lipids in mice, cats, and humans with Sandhoff disease. Lipids. 2009;44(3):197–205. | ||
Griffin B, Baker HJ. Domestic cats as laboratory animals. In: Fox JG, Anderson LC, Loew FM, Quimby FW, editors. Laboratory Animal Medicine. New York, NY: Academic Press; 2002:462. | ||
Valayannopoulos V. Chapter 190: enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. Handb Clin Neurol. 2013;113(3):1851–1857. | ||
Rattazzi MC, Appel AM, Baker HJ. Enzyme replacement in feline GM2 gangliosidosis: catabolic effects of human β-hexosaminidase A. Prog Clin Biol Res. 1982;94:213–220. | ||
Tsuji D, Akeboshi H, Matsuoka K, et al. Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis. Ann Neurol. 2011;69(4):691–701. | ||
Jeyakumar M, Norflus F, Tifft CJ, et al. Enhanced survival in Sandhoff disease mice receiving a combination of substrate deprivation therapy and bone marrow transplantation. Blood. 2001;97(1):327–329. | ||
Jakóbkiewicz-Banecka J, Węgrzyn A, Węgrzyn G. Substrate deprivation therapy: a new hope for patients suffering from neuronopathic forms of inherited lysosomal storage diseases. J Appl Genet. 2007;48(4):383–388. | ||
Maegawa GHB, Banwell BL, Blaser S, et al. Substrate reduction therapy in juvenile GM2 gangliosidosis. Mol Genet Metab. 2009;98(1-2): 215–224. | ||
Jeyakumar M, Butters TD, Cortina-Borja M, et al. Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc Natl Acad Sci U S A. 1999;96(11): 6388–6393. | ||
Chiricozzi E, Niemir N, Aureli M, et al. Chaperone therapy for GM2 gangliosidosis: effects of pyrimethamine on β-hexosaminidase activity in Sandhoff fibroblasts. Mol Neurobiol. 2014;50(1):159–167. | ||
Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D. Pharmacological enhancements of β-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff patients. J Biol Chem. 2004; 279(14):13478–13487. | ||
Tropak MB, Zhang J, Yonekawa S, et al. Pyrimethamine derivatives: insight into binding mechanism and improved enhancement of mutant β-N-acetylhexosaminidase activity. J Med Chem. 2015;58(11): 4483–4493. | ||
Hoogerbrugge PM, Brouwer OF. Allogenic bone marrow transplantation for lysosomal storage diseases. Lancet. 1995; 345: 1398-19402. Blood. 2008;112(7):2979–2989. | ||
Prasad VK, Mendizabal A, Parikh SH, et al. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112(7): 2979–2989. | ||
Jacobs JF, Willemsen MA, Groot-Loonen JJ, Wevers RA, Hoogerbrugge PM. Allogenic BMT followed by substrate reduction therapy in a child with subacute Tay-Sachs disease. Bone Marrow Transplant. 2005; 36(10):925–926. | ||
Lee JP, Jeyakumar M, Gonzalez R, et al. Stem cells act through multiple mechanisms to benefit mice with neurodegenerative metabolic disease. Nat Med. 2007;13(4):439–447. | ||
Jeyakumar M, Lee JP, Sibson NR, et al. Neural stem cell transplantation benefits a monogenic neurometabolic disorder during the symptomatic phase of disease. Stem Cells. 2009;27(9):2362–2370. | ||
Arthur JR, Lee JP, Snyder EY, Seyfried TN. Therapeutic effects of stem cells and substrate reduction in juvenile Sandhoff mice. Neurochem Res. 2012;37(6):1335–1343. | ||
Sargeant TJ, Wang S, Bradley J, et al. Adeno-associated virus-mediated expression of β-hexosaminidase prevents neuronal loss in the Sandhoff mouse brain. Hum Mol Genet. 2011;20(22):4371–4380. | ||
Bourgoin C, Emiliani C, Kremer EJ, et al. Widespread distribution of β-hexosaminidase activity in the brain of a Sandhoff mouse model after coinjection of adenoviral vector and mannitol. Gene Ther. 2003; 10(21):1841–1849. | ||
Cachón-González MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci U S A. 2006;103(27):10373–10378. | ||
Cachón-González MB, Wang SZ, McNair R, et al. Gene transfer corrects acute GM2 gangliosidosis – potential therapeutic contribution of perivascular enzyme flow. Mol Ther. 2012;20(8):1489–1500. | ||
Bradbury AM, Cochran JN, McCurdy VJ, et al. Therapeutic response in feline Sandhoff disease despite immunity to intracranial gene therapy. Mol Ther. 2013;21(7):1306–1315. | ||
Walias JS, Altaleb N, Bello A, et al. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol Ther. 2015;23(3):414–422. | ||
Tropak MB, Yonekawa S, Karumuthil-Melethil S, et al. Construction of a hybrid β-hexosaminidase subunit capable of forming stable homodimers that hydrolyze GM2 ganglioside in vivo. Mol Ther Methods Clin Dev. 2016;3:15057. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.