")
Back to Journals » Vascular Health and Risk Management » Volume 11
Angiotensin receptor-neprilysin inhibitors: clinical potential in heart failure and beyond
Received 31 January 2015
Accepted for publication 10 March 2015
Published 1 June 2015 Volume 2015:11 Pages 283—295
DOI https://doi.org/10.2147/VHRM.S55630
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Daniel Duprez
Jagdeep SS Singh, Chim C Lang
Division of Cardiovascular and Diabetes Medicine, Ninewells Hospital and Medical School, University of Dundee, Dundee, UK
Abstract: Heart failure remains a major concern across the globe as life expectancies and delivery of health care continue to improve. There has been a dearth of new developments in heart failure therapies in the last decade until last year, with the release of the results from the PARADIGM-HF Trial heralding the arrival of a promising new class of drug, ie, the angiotensin receptor-neprilysin inhibitor. In this review, we discuss the evolution of our incremental understanding of the neurohormonal mechanisms involved in the pathophysiology of heart failure, which has led to our success in modulating its various pathways. We start by examining the renin-angiotensin-aldosterone system, followed by the challenges of modulating the natriuretic peptide system. We then delve deeper into the pharmacology and mechanisms by which angiotensin receptor-neprilysin inhibitors achieve their significant cardiovascular benefits. Finally, we also consider the potential application of this new class of drug in other areas, such as heart failure with preserved ejection fraction, hypertension, patients with renal impairment, and following myocardial infarction.
Keywords: heart failure, angiotensin receptor-neprilysin inhibitor, heart failure with preserved ejection fraction, nesiritide, candoxatril, omapatrilat, hypertension, renal impairment, myocardial infarction
Introduction
Advances in acute cardiac care and the ever increasing efficacy of treatment for coronary artery disease mean more and more people are surviving acute coronary events. Although a boon for cardiologists, this has also resulted in a steadily increasing number of patients with chronic heart failure (HF). Together with an ageing population, HF is now a public health problem, with 22 million patients worldwide and costing 38 billion USD a year in the USA alone.1 Defined as a clinical syndrome where the heart is unable to maintain cardiac output to meet metabolic demands and accommodate venous return,1 it takes an indolent, progressively worsening course that results in significant morbidity and eventually a premature death. There has been a substantial increase in our understanding of the pathophysiological pathways involved in HF which has helped to define new therapeutic strategies for combating it. In this review, we journey beyond the drugs that affect the renin-angiotensin-aldosterone system (RAAS) to also look at modulators of the natriuretic peptide (NP) system. Firstly, we will look into the pathophysiology of HF with preserved ejection fraction (HFpEF) and reduced ejection fraction (HFrEF). We then consider our approach toward the development of drugs to treat these conditions, starting with the lessons learnt from the early failures of NP manipulation, to the development of simultaneous NP + RAAS modulation therapy that eventually led to the discovery of the angiotensin receptor-neprilysin inhibitor (ARNI), LCZ 696 (Table 1). Thirdly, we study the potential benefits of this novel class of drug in conditions other than HFrEF, such as in HFpEF and hypertension. Finally, we explore its use in other areas, such as left ventricular dysfunction following a myocardial infarction and renal impairment.
 | Table 1 Pertinent trials in the development of nesiritide, candoxatril, omapatrilat, and LCZ 696 |
HFpEF: what’s all the fuss about?
We have now started to appreciate that our rather naive view of diagnosing HF solely based on a patient’s left ventricular ejection fraction does not encompass the entire spectrum of HF. There are patients with normal ejection fraction who present with typical symptoms of the disease. The previous label of diastolic dysfunction has been proven to be a misnomer, as detailed echocardiographic studies reveal these patients to also have significant systolic dysfunction in the form of impaired long axis function and systolic twist, torsional dyssynchrony, and reduced myocardial systolic reserve.2 There is also impaired atrial function, especially on exercise.2,3 All of this is in addition to the traditional definition of diastolic HF characterized by increased end diastolic pressure as a result of abnormal active relaxation and increased passive left ventricular stiffness.4,5 Another feature that is frequently seen is stiff and noncompliant chambers, albeit with normal volumes. Westermann et al described chronic interstitial inflammation causing elevated profibroblast growth factor beta which stimulates type 1 and 3 collagen formation, resulting in increased myocardial wall stiffness and translating into higher resting ventricular pressures.6 Fittingly, diastolic dysfunction has now been replaced by the term HF with preserved ejection fraction to describe this cohort of patients.
It is important to note that HFpEF patients have similar survival rates to HFrEF patients,7,8 ranging from 8%9 to 23%,10 depending on the region and cut-offs for the definition of HFpEF. Also, the fact that approximately 50% of the patients with symptomatic HF actually have HFpEF11 underscores the importance of correctly identifying and treating these patients. The European Society of Cardiology guidelines for the diagnosis and treatment of acute and chronic HF identify elderly, obese, and female patients to be at increased risk to develop HFpEF; they also note that patients with HFpEF are more likely to have underlying hypertension, atrial fibrillation, and no previous history of coronary artery disease.12 In fact, hypertension and the resultant chronic pressure overload is the single most important predictor for HFpEF across registry, epidemiological, and controlled trials.13 The diagnosis of HFpEF is one of exclusion, and is considered only when cardiac and non-cardiac factors have been ruled out. Non-cardiac considerations include differentials like obstructive sleep apnea, chronic obstructive airway disease, obesity, and liver disease. On the other hand, when investigating cardiac causes for symptoms of HF in the setting of preserved ejection fraction, one has to rule out valvular and pericardial pathologies, as well as high-output conditions (anemia, thyrotoxicosis, arterio-venous fistula) and left ventricular hypertrophy before a final diagnosis of HFpEF is reached.14 Despite the progress we have made in understanding the pathophysiology of HFpEF, we have yet to identify treatment that can confer a significant mortality benefit. Trials studying various agents like angiotensin-converting enzyme (ACE) inhibitors,15 angiotensin receptor blockers (ARBs),16,17 and spironolactone18,19 have all failed to demonstrate a definite mortality benefit, although some did indicate a signal toward improved morbidity.17,19
Neurohormonal mechanisms of HFrEF: RAAS and NP system
There are a myriad causes of HFrEF, the commonest being hypertension and ischemic heart disease,20,21 as well as a host of less common etiologies like cardiomyopathy, valvular heart disease, myocarditis, and cardiotoxic agents.1,21 There is also a small group of patients with no discernible cause, ie, the so-called idiopathic HF.21 Whatever the cause, the resultant symptom complex of systemic and pulmonary congestion remains the same, and is a key element in diagnosing HF.
The pathophysiology of HFrEF has long been elucidated. The first neurohormonal compensatory mechanism to be studied,22 and arguably the most influential, is the RAAS. The RAAS, with its cascade of hormones, generates a multifaceted response affecting the heart, kidneys, and systemic vasculature, in an attempt to reverse the adverse effects of HF. However, a vigorous and sustained activation of this mechanism often results in permanent and deleterious alterations in the patient’s cardiovascular physiology. For instance, the left ventricular hypertrophy triggered by angiotensin (Ang) II23,24 and aldosterone to achieve higher stroke volumes will also increase myocardial oxygen demand, predisposing to or worsening any underlying coronary artery disease.25 Besides that, the cellular proliferative effect of these hormones on cardiac myocytes also increases cell apoptosis and fibroblast activity, thus replacing healthy myocardium with non-contractile fibrotic tissue,26,27 resulting in adverse ventricular remodeling. Pulmonary and systemic congestion, on the other hand, is exacerbated by increased intravascular volume due to the enhanced sodium and water retention, also mediated by both Ang II and aldosterone.28,29 The increased peripheral vascular resistance generated by Ang II and sympathetic hyperactivity will not only diminish peripheral blood supply, but also contribute to elevated blood pressure (BP).30 All of this increases the strain on the heart and sets up a vicious cycle that steadily diminishes left ventricular function.
Remarkably, the heart has yet another mechanism to counteract this chronically activated compensatory system, in the form of NPs. The NP system plays a crucial role in maintaining cardiorenal homeostasis using an arsenal of structurally similar but genetically and physiologically distinct peptides.31 Of the four known NPs, atrial natriuretic peptide (ANP) is released by the atria in response to myocardial stretching, signifying increased intravascular volume,32 whereas brain natriuretic peptide (BNP) is released by the ventricles in response to increased ventricular volume and pressures.33,34 Both ANP and BNP have similar physiological effects, inducing diuresis and natriuresis and vasodilatation as well as inhibiting the sympathetic nervous system (by reduced catecholamine secretion) and RAAS (by reducing renin section).35 C-type natriuretic peptide (CNP), on the other hand, is released by the endothelium of the heart, lungs, kidneys, and vasculature in response to shearing stressors.36,37 CNP possesses minimal diuretic capabilities but is a potent vasodilator and prevents vascular cell growth.37 Finally, D-type NP is also associated with chronic HF, but not much of its physiology and significance is known.38 These peptides exert their effects via transmembrane receptors from the guanylyl cyclase family. There are three types of natriuretic peptide receptors (NPRs), namely NPR-A, NPR-B, and NPR-C; however, their nomenclature is not consistent with that of their ligands.39 NPR-A and NPR-B have similar effects and bind to all three NPs, but NPR-A has a stronger affinity for ANP and BNP than for CNP. Conversely, NPR-B is more selective toward CNP.40 When the extracellular binding domain of both these receptors is activated by an NP, the receptor catalyzes the intracellular formation of cyclic guanosine monophosphate from guanosine triphosphate.41 The cyclic guanosine monophosphate then acts as a second messenger to mediate the effects of the NP. NPR-C, on the other hand, has a very different effect, in that its function is to remove NPs from the circulation. It internalizes the bound NP for lysosomal degradation within the cell. This is a minor catabolic pathway for NPs. NPs are predominantly catabolized via enzymatic cleavage by a membrane-bound enzyme called neprilysin, also known as neutral endopeptidase or membrane metallo-endopeptidase.33 Neprilysin, which is widely expressed but most abundant in the kidneys,42 cleaves the ring structures of the NPs, thus rendering them biologically inactive.43 There has been much interest in the positive physiological effects of the NP system, with various groups studying its possible application in treating acute decompensated HF, for instance with BNP (the VMAC trial)44 (discussed below), and more recently with uraglitide (TRUE-HF,45 ClinicalTrials.gov identifier NCT01661634), also in retarding the progress of chronic HF,46,47 and some are even considering its use in preventing symptomatic HF (stage C) among asymptomatic but structurally compromised stage B patients.31 There are still others who are developing chimeric NPs that are superior to their endogenous counterparts in terms of potency and duration of action.48,49
In spite of the enthusiasm, the counter-regulatory effects of the NP are no match for the far more potent effects of the RAAS and other facets of the neurohormonal system. In an attempt to mitigate these effects, we have developed therapy that block the RAAS cascade at almost every level. Right from its origin with the renin inhibitor, aliskiren and beta-blockers that reduce renin secretion, to ACE inhibitors that prevent the enzymatic conversion of Ang I to Ang II, to ARBs that block the binding of Ang II to the angiotensin II type I (AT1) receptors, and finally mineralocorticoid receptor antagonists that act at the very end of the RAAS cascade, by blocking the binding of aldosterone to its receptors on the distal convoluted tubules. In spite of this multi-level blockade on a key pathogenetic cascade, we realize that we still have not won the battle with this complex disease. Hence, it is only natural that we have now ventured beyond the RAAS to identify new targets in other cascades that may be addressed to treat patients with HF. One therapeutic target that has shown much promise is the NP system itself. Its physiological benefits are clear, but studies have shown reduced biological activity of NPs among patients with chronic HF, indeed some patients even develop resistance to NP activity over prolonged periods of exposure.50 Researchers have long tried to augment the NP system, initially by adding recombinant NPs and later by inhibiting its breakdown enzyme, eventually leading to the breakthrough discussed in the following sections (Figure 1).
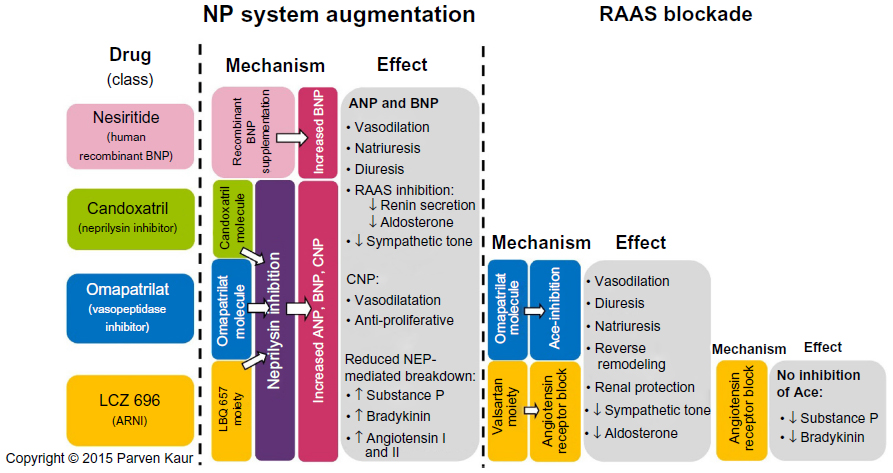 | Figure 1 Overview of the mechanisms and effects of nesiritide, candoxatril, omapatrilat, and LCZ 696. |
Nesiritide: supplementing the NP system
Encouraged by the wide-ranging cardiovascular effects of the NP system, researchers first attempted to harness the system by supplementing it with recombinant human BNP in the form of nesiritide51 (Figure 1). The early VMAC trial demonstrated marked improvements in pulmonary capillary wedge pressures and subjective measurements of dyspnea when compared with other vasodilators during the acute phase of decompensated HF.44,52 They also demonstrated its supposed superiority against dobutamine with regard to ventricular ectopy and average heart rate.53,54 In 2001, it was approved by the US Food and Drug Administration for early relief of dyspnea in acute HF.55 However, concerns followed about the renal toxicity and mortality associated with the use of nesiritide in acute HF. A meta-analysis of three trials revealed a signal for increased 30-day mortality (hazard ratio after adjusting for study, 1.80; 95% confidence interval 0.98–3.31; P=0.057) among patients treated with nesiritide compared with controls.56 In another meta-analysis of five trials pooling 1,269 patients to study the renal effects of nesiritide in acutely decompensated HF patients, Sackner-Bernstein et al demonstrated that treatment with nesiritide increased the risk of worsening renal function (defined as an increase of serum creatinine more than 44 μmol/L) by 1.52 (95% confidence interval 1.16–2.00; P=0.003) and 1.54 (95% confidence interval 1.19–1.98; P=0.001) when compared with a non-inotrope-based control or any (inotrope or non-inotrope) control, respectively.57 An independent panel was convened to evaluate this issue, which prompted the ASCEND-HF58 trial (n=7,141), which showed that nesiritide had no effect on mortality or 30-day rehospitalization rates but a small, non-significant effect on dyspnea when used in combination with other therapies. It was not associated with a worsening of renal function, but was associated with an increase in rates of hypotension. On the basis of those results, nesiritide could not be recommended for routine use in the broad population of patients with acute HF.59 A subgroup analysis was also performed on approximately two thirds of the patients in whom urinary data were available, but failed to show a diuretic effect attributable to nesiritide.60 One could speculate that recombinant BNP is inferior to its endogenous counterpart, or perhaps its effect may become blunted in the acute decompensated phase of HF which itself sees a surge in endogenous BNP levels.
Candoxatril: preventing breakdown of NPs
Following the failure of simply adding NPs, researchers turned to inhibiting their enzymatic breakdown instead. Targeting the enzyme neprilysin with the drug candoxatril, researchers were able to prevent the breakdown of ANP, BNP, and CNP,61,62 thereby prolonging their effects on the cardiovascular system (Figure 1). In addition, neprilysin has a vast array of other substrates including enkephalins, oxytocin, gastrin, Ang I and II, endothelin-1, substance P, and bradykinin,63,64 highlighting the sheer complexity of manipulating such a system. Initial results with this class of drug were encouraging, with significant reductions in pulmonary capillary wedge pressures65 and improved exercise capacity.66 Researchers also found a significant diuretic effect without the concomitant RAAS activation that is usually seen with the use of furosemide.67,68 However, they were unable to demonstrate a significant BP-lowering effect.69 Indeed, one study reported BP elevations among normotensive patients who were given candoxatril.70 This was likely the direct result of increased Ang II71 and endothelin-172 activity as a consequence of neprilysin inhibition. Other groups researching the effect of candoxatril on left ventricular dysfunction were also disappointed when they discovered a dose-dependent increase in peripheral vascular resistance and a reduction of cardiac index in the treatment arm.73 Following a string of negative or equivocal publications, researchers investigating candoxatril conceded that pure neprilysin inhibitors lacked efficacy when used as monotherapy in hypertension and HF.61
Omapatrilat: simultaneously modulating the NP system and RAAS
The next phase of evolution was that of dual inhibition; simultaneously inhibiting neprilysin and ACE to potentiate the beneficial physiological effects of the NP system and undermine the harmful effects of RAAS (Figure 1). This produced a new class of drug, referred to as vasopeptidase inhibitors, of which omapatrilat has been the most developed.74 It was hoped that concurrent ACE inhibition would be able to address the downstream effects of neprilysin inhibition, and early evidence submitted by the authors of the IMPRESS trial were tantalizingly promising. Studying chronic HF patients, they demonstrated improvements in the composite of death and hospitalization, as well as improved New York Heart Association (NYHA) functional class when compared with lisinopril.75 However, the results of larger and more definitive trials, such as OVERTURE and OCTAVE, failed to support this conclusion. OVERTURE, which enrolled nearly 6,000 HF patients to compare omapatrilat and enalapril, did not show any significant difference in death and HF hospitalization rates between groups.76 However, post hoc analysis of a modified definition of its primary end point did reveal a significantly lower risk in the active comparator arm.76 Unfortunately, analysis of its safety revealed a higher incidence of severe angioedema than with any other ACE inhibitor. OCTAVE, which was designed primarily to determine if lower doses of omapatrilat would be safer, enrolled 25,000 hypertensive patients to compare omapatrilat and enalapril. Although it did demonstrate a mean 3/2 mmHg reduction in BP and a larger number of patients achieving goal BP74 at 6 months in the omapatrilat arm, it failed to show a lower incidence of angioedema.77 Investigators discovered that patients on omapatrilat were 3.2 times more likely to develop angioedema than those on enalapril, and the events were also more likely to be severe. This risk of angioedema was more pronounced among blacks and smokers. This included two patients with life-threatening airway compromise, one of which required mechanical ventilation.78 We now believe this is due to a surge in bradykinin and substance P levels as a result of the dual inhibition of their breakdown enzymes, ACE and neprilysin.79,80 The sponsors of the study were quick to point out that the potential benefits of BP reduction by omapatrilat outweighed the risks of angioedema, but failed to convince regulators and were denied approval for the drug.78
ARNI: a new approach to dual inhibition
Recognizing the potential of dual neprilysin and RAAS blockade, researchers persevered in trying to address the concerns of angioedema associated with omapatrilat. The elegant solution to this safety concern came in the form of shifting the RAAS target from ACE inhibition to angiotensin receptor blockade. By doing so, the potentially harmful accumulation of substance P and bradykinin is prevented, as they can now be broken down by the uninhibited ACE (Figure 1). The first in class molecule that was able to achieve this dual angiotensin receptor and neprilysin inhibition was LCZ 696, which is composed of two anionic moieties in a 1:1 molar ratio,80,81 namely AHU 377, which confers potent neprilysin inhibition, and valsartan, a well-studied ARB with a proven track record in treating cardiovascular disease82 (Figure 2).
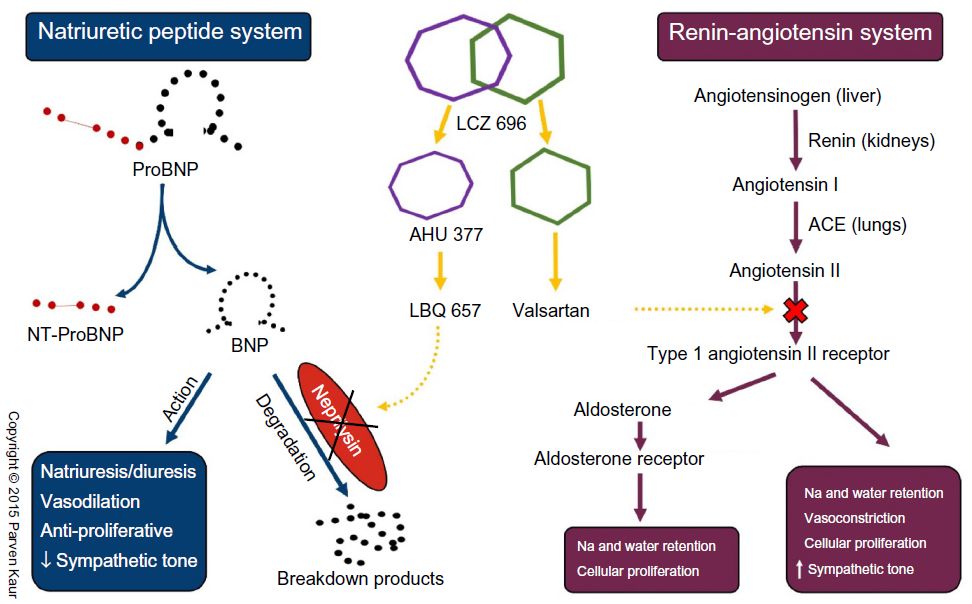 | Figure 2 Schematic representation of mechanism of action of LCZ 696 on the natriuretic peptide and renin angiotensin systems. |
Pharmacology of LCZ 696
As detailed above, LCZ 696 contains equal molar amounts of AHU 377 and valsartan. AHU 377 is a prodrug that is rapidly metabolized into the biologically active neprilysin inhibitor, LBQ 657, by enzymatic cleavage of its ethyl ester,83 reaching its peak concentration in 1.8–2.7 hours. Valsartan, on the other hand, is biologically active in its original form and has a peak concentration at 1.6–4.9 hours, which is comparable with that of LBQ 657.81 With a plasma half-life of approximately 18 hours, LCZ 696 is suitable for once-daily administration,81 although it may also be administered twice daily for indications like HF.84 It is important to note that proBNP, which is triggered by failing ventricles, remains biologically inactive as long as it is attached to its N-terminal polypeptide chain. Only after the N-terminal portion is cleaved from proBNP does it become BNP and start to exert its cardiovascular effects. It should also be remembered that circulating BNP levels do not influence the secretion of proBNP because it is only secreted in response to ventricular volume and pressure overload. This understanding is key to appreciating why proBNP and NT-proBNP levels remain relevant markers of disease severity in patients treated with LCZ 696, simply because they are not substrates of neprilysin80,85 (Figure 2).
ARNI in heart failure
Heart failure with reduced ejection fraction
The recently published PARADIGM-HF trial was the first in nearly two decades to generate excitement in the HF community.86 This Phase III active comparator trial conducted across 47 countries enrolled approximately 8,400 patients with NYHA functional class II–IV HF, an ejection fraction <35% and elevated NT-proBNP levels, who were already on evidence-based treatment for HF.87 The investigators aimed to compare LCZ 696 against enalapril, an ACE inhibitor with proven mortality benefits,88,89 with a primary end point of composite cardiovascular death or hospitalization for HF. They also looked at all-cause mortality, 8-month quality of life score (Kansas City Cardiomyopathy Questionnaire), time to new atrial fibrillation, and renal dysfunction. In an attempt to address the safety concerns with previous attempts at dual RAAS-neprilysin inhibition, the authors elected to have an active run-in phase in which all patients were given a single blind run-in with enalapril for 2 weeks followed by an additional 4–6 weeks of LCZ 696 titrated to a target dose of 200 mg (equivalent to 160 mg of valsartan) twice daily. Only after demonstrating tolerability to both drugs would a patient be randomized to either arm of the trial.87
The baseline characteristics of the cohort included a mean age of 63 years, left ventricular ejection fraction 29%±6%, background therapy of ACE/ARB (99%), beta-blockers (93%), mineralocorticoid receptor antagonists (56%), and predominantly NYHA Class II functional status (70%).90 The outcomes of the trial were so overwhelmingly positive that it was stopped early by its data monitoring committee. With a median follow-up of 27 months, the investigators demonstrated a 20% relative risk reduction in the composite of cardiovascular death or hospitalization for HF and a 16% relative risk improvement in all-cause mortality91 with a number needed to treat of 35.86 They also showed a significant 3 mmHg BP reduction as well as higher quality of life scores in the treatment arm at 8 months. In all, 11% of the recruited patients withdrew due to adverse events to either drug during the run-in period. Following randomization, a further 10.7% from the LCZ 696 arm and 12.3% from the enalapril arm withdrew due to adverse events (P=0.03), leading the investigators to claim that LCZ 696 is at least as safe as enalapril.91
Following the initial enthusiasm about these remarkable results, a number of commentators started to raise some concerns regarding the design and conclusions drawn from the trial. Efficacy concerns revolved around the use of moderate dose enalapril (20 mg/day) instead of its maximal dose (40 mg/day) against 400 mg/day LCZ 696 which delivered an equivalent valsartan dose of 320 mg/day (the maximal daily dose for valsartan). Hence, the supposed benefits may have simply been a result of more effective RAAS blockade conferred by the higher valsartan dose in the intervention arm.92 On the other hand, the safety concerns raised were the use of an active run-in period which would have excluded patients with adverse events from being randomized, thus underestimating the true adverse event risk and possibly even overestimating the efficacy of the study drug. In an edifying paper published in 1998 about the implications of an active run-in design, Pablos-Mendez et al discussed these concerns and added that the applicability of such trials may be diluted because of a trial cohort that cannot be readily defined and reproduced in a clinical setting.93 Apart from that, the community is also concerned that black patients (who are most susceptible to angioedema) and the elderly were under-represented in the trial. In spite of these issues, the general consensus remains positive about the prospects of LCZ 696; it has been granted US Food and Drug Administration fast track status and a rolling submission was completed at the end of 2014. Further post marketing study into the drug will undoubtedly address all the concerns raised.
Heart failure with preserved ejection fraction
The PARAMOUNT trial was the first to evaluate the use of LCZ 696 in patients with HF. It was a Phase II trial studying 300 patients with signs and symptoms of HF, NYHA class II–III with an ejection fraction of ≥45%, and NT-proBNP >400 pg/mL, comparing LCZ 696 200 mg twice daily against a bioequivalent dose of valsartan 160 mg twice daily.94 The primary outcome of a significant reduction in NT-proBNP at 12 weeks was achieved by 4 weeks and sustained up to the 36-week extension phase of the trial. However, the patients in the valsartan arm demonstrated a slow but steady reduction in NT-proBNP levels, resulting in loss of significance between the two groups at 36 weeks (Figure 3). Besides that, the extension phase also revealed improved NYHA scores and left atrial size (an important predictor of outcomes in HFpEF95) in the LCZ 696 arm. The investigators also reported that patients on LCZ 696 had a significantly greater BP reduction than those on valsartan in both the 12-week and 36-week intervals. Just as importantly, they noted no significant difference in side effect profiles between the groups.94
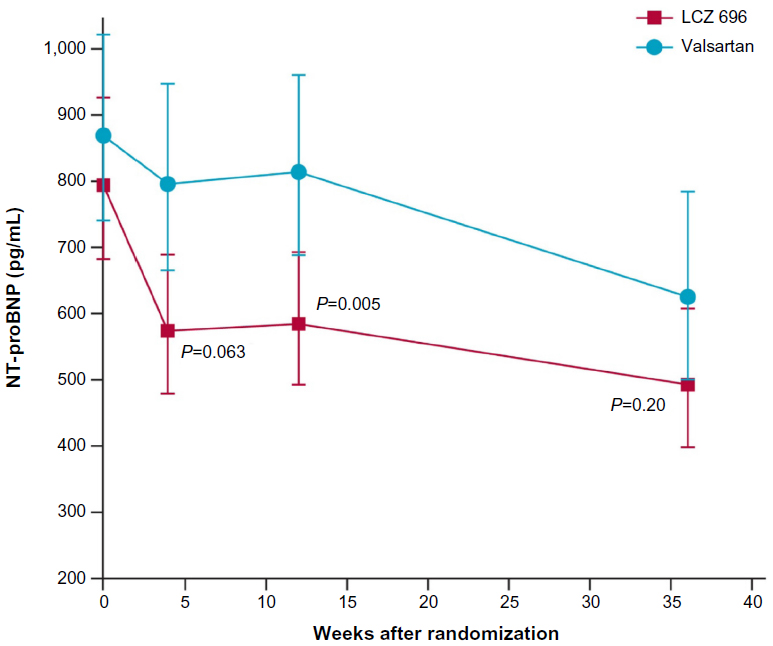 | Figure 3 Results from the PARAMOUNT Trial NT-proBNP at 4, 12, and 36 weeks in the LCZ 696 and valsartan groups. |
The proof of concept PARAMOUNT trial has now lead to the recently initiated PARAGON-HF trial (ClinicalTrials.gov identifier NCT01920711) which aims to compare LCZ 696 against valsartan in approximately 4,000 HFpEF patients to look for hard outcomes like composite cardiovascular death and hospitalization. The authors are also aiming to look for functional improvement as well as other secondary end points like new-onset atrial fibrillation, stroke, myocardial infarction, and all-cause mortality. Patients in NYHA class II–IV, with ejection fraction >45%, structural heart disease (left atrial enlargement/left ventricular hypertrophy), and requiring diuretic therapy will be eligible for the trial. The results for this study are expected in 2019.
ARNI: broadening the horizons
Hypertension
Hypertension was the first indication studied for LCZ 696. In 2010, Ruilope et al96 evaluated 1,300 patients with mild-to-moderate hypertension for 8 weeks to measure the mean difference of three pairwise comparisons of sitting diastolic BP between LCZ 696 and bioequivalent doses of valsartan (ie, LCZ 696 100 mg versus valsartan 80 mg, LCZ 696 200 mg versus valsartan 160 mg, and LCZ 696 400 mg versus valsartan 320 mg) as well as a pairwise comparison between AHU 377 200 mg and placebo. They included secondary outcomes such as systolic BP reduction, pulse pressure, ambulatory BP, and safety parameters, as well as markers for other cardiovascular diseases. Their results demonstrated a significantly greater reduction of BP in the 200 mg and 400 mg LCZ 696 pairs (−5/−3 mmHg and −6/−3 mmHg, respectively) and a greater but non-significant reduction in the 100 mg LCZ 696 versus 80 mg valsartan pair. This effect was also seen in 24-hour ambulatory BP measurements. Interestingly, the researchers pointed out that the BP reduction conferred by LCZ 696 400 mg compared with bioequivalent doses of its components (200 mg AHU 377 and 320 mg valsartan) was fully additive in diastolic BP reduction and more than fully additive in systolic BP reduction, highlighting the complementary effects of dual RAAS and neprilysin inhibition. They also noted positive signals in markers of improvement for HF and pulmonary hypertension.96 Researchers in Japan have also recently confirmed the safety and efficacy of LCZ 696 as an antihypertensive in the Asian population.97
Renal impairment
Patients with chronic kidney disease have an excess risk of developing cardiovascular complications as a result of substantial changes in their internal milieu.98 Numerous studies have demonstrated progressively increasing cardiovascular risk with a worsening glomerular filtration rate. Additionally, patients with albuminuria, even if mild, see a near doubling of cardiovascular mortality risk.99 Treating this cohort of patients with ARNIs offers the exciting prospect of not only improving cardiovascular risk, but also delaying the progression to renal replacement therapy. The evidence for the potential renoprotective effects of ARNIs is currently inferred from animal models using other vasopeptidase inhibitors like omapatrilat and ilepatril, which have shown promise in reversing proteinuria and halting glomerulosclerosis and tubulointerstitial fibrosis.100,101 Reassuringly, secondary outcomes of the two major HF trials conducted so far also do not signal any negative impact of ARNIs on renal function. The PARAMOUNT trial showed no significant difference in new-onset renal dysfunction, hyperkalemia, or a >50% reduction in glomerular filtration rate between the LCZ 696 and valsartan arms.94 In fact, there was a higher degree of change in estimated glomerular filtration rate in the valsartan group (LCZ 696, −1.6 mL/min/1.73 m2 versus valsartan, −5.2 mL/min/1.73 m2; P=0.007). The analysis from PARADIGM-HF was also as expected, with no significant difference in protocol-defined decline in renal function or progression to end-stage renal disease.91 In an effort to explore this potential benefit further, the pilot UK HARP-III Study102 (ISRCTN 11958993), with the primary objective of documenting the change in renal function over 6 months, aims to recruit 360 patients with chronic kidney disease (estimated glomerular filtration rate 20–60 mL/min/1.73m2 or urinary albumin to creatinine ratio ≥30 mg/mol) to compare LCZ 696 with irbesartan.
Post myocardial infarction
Following a myocardial infarction, the risk for further fatal and non-fatal ischemic events continues to increase.103,104 Although factors such as age, sex, and comorbidities play an important role, the degree of left ventricular dysfunction remains the primary determinant of survivability.105 Trials such as EPHESUS106 and SAVE105 have demonstrated the undeniable importance of attenuating left ventricular dysfunction in this group of patients, and LCZ 696 seems poised to support that theory. Von Lueder et al107,108 have been conducting preclinical studies of the possible benefits of ARNI on cardiac remodeling in rats with induced myocardial infarction. In a recently published article, Von Lueder et al demonstrated lower cardiac weight and reduced fibrosis in the peri-infarct and remote myocardium in the LCZ 696 group compared with a placebo group. The LCZ 696 group also had a lower left ventricular end-diastolic diameter, a higher left ventricular ejection fraction, and higher circular and diastolic wall strain, confirming improved left ventricular function 4 weeks after treatment. Using cell cultures, they showed the LBQ 657 component of LCZ 696 inhibiting hypertrophy but not fibrosis, and the valsartan component inhibiting both hypertrophy and fibrosis. Importantly, they also showed that the inhibitory properties of valsartan monotherapy were augmented by the dual inhibition of LCZ 696, which at the highest doses was able to completely cancel the Ang II-mediated effects.108 These are very promising findings but, to the best of our knowledge, there have not been any groups studying this effect clinically.
Conclusion
ARNIs and its first in class molecule, LCZ 696, have shown much potential in becoming the next big thing in the field of cardiovascular therapeutics. Our knowledge and experience with the drug remains very much in flux, but will undoubtedly mature as more research is carried out. However, it is imperative that we remain mindful of our previous eagerness to embrace new therapeutic agents before a thorough and rigorous evaluation of its safety profile. It is important for us to maintain a healthy dose of skepticism and foster open discussions when evaluating the merits of this novel class of drug. If proven, however, ARNIs may well supersede ACE inhibitors and ARBs, heralding a new age of multisystem modulation that will revolutionize the way we understand and treat cardiovascular disease.
Acknowledgment
The authors wish to thank Ms Parven Kaur for designing the original illustrations used in this review.
Disclosure
CCL has received consultancy fees and research support for the conduct of PARADIGM-HF and PARAGON-HF from Novartis. JSSS has received funding from the European Foundation for the Study of Diabetes for a trial on sodium-glucose cotransporter 2 II inhibitors in heart failure.
References
Kemp CD, Conte JV. The pathophysiology of heart failure. Cardiovasc Pathol. 2013;21(5):365–371. | |
Tan YT, Wenzelburger F, Lee E, et al. The pathophysiology of heart failure with normal ejection fraction exercise echocardiography reveals complex abnormalities of both systolic and diastolic ventricular function involving torsion, untwist, and longitudinal motion. J Am Coll Cardiol. 2009;54(1):36–46. | |
Borlaug BA, Olson TP, Lam CS, et al. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2010;56(11):845–854. | |
Zile MR, Baicu CF, Bonnema DD. Diastolic heart failure: definitions and terminology. Prog Cardiovasc Dis. 2005;47(5):307–313. | |
Zile MR, Baicu CF, Gaasch WH. Diastolic heart failure – abnormalities in active relaxation and passive stiffness of the left ventricle. N Engl J Med. 2004;350(19):1953–1959. | |
Westermann D, Lindner D, Kasner M, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. 2011;4(1):44–52. | |
Vasan RS, Larson MG, Benjamin EJ, Evans JC, Reiss CK, Levy D. Congestive heart failure in subjects with normal versus reduced left ventricular ejection fraction prevalence and mortality in a population-based cohort. J Am Coll Cardiol. 1999;33(7):1948–1955. | |
Bhatia RS, Tu JV, Lee DS, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355(3):260–269. | |
Cohn JN, Johnson G. Heart failure with normal ejection fraction. The V-HeFT Study. Veterans Administration Cooperative Study Group. Circulation. 1990;81(2 Suppl):III48–III53. | |
Digitalis Investigation Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med. 1997;336(8):525–533. | |
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62(16):e147–e239. | |
McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012. Eur J Heart Fail. 2012;14(8):803–869. | |
Rose-Jones LJ, Rommel JJ, Chang PP. Heart failure with preserved ejection fraction: an ongoing enigma. Cardiol Clin. 2014;32(1):151-x. | |
Lindenfeld J, Albert NM, Boehmer JP, et al. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010;16(6): 475–539. | |
Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27(19):2338–2345. | |
Massie BM, Carson PE, McMurray JJV, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359(23):2456–2467. | |
Yusuf S, Pfeffer MA, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet. 2003;362(9386): 777–781. | |
Edelmann F, Wachter R, Schmidt AG, et al. Effect of spironolactone on diastolic function and exercise capacity in patients with heart failure with preserved ejection fraction: the ALDO-DHF randomized controlled trial. JAMA. 2013;309(8):781–791. | |
Pitt B, Pfeffer MA, Assmann SF, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370(15): 1383–1392. | |
Go AS, Mozaffarian D, Roger VL, et al. Heart Disease and Stroke Statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. | |
McMurray JJV, Pfeffer MA. Heart failure. Lancet. 2005;365(9474): 1877–1889. | |
Sayer G, Bhat G. The renin-angiotensin-aldosterone system and heart failure. Cardiol Clin. 2014;32(1):21-vii. | |
Crowley SD, Gurley SB, Herrera MJ, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103(47):17985–17990. | |
Gray MO, Long CS, Kalinyak JE, Li H-T, Karliner JS. Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-²1 and endothelin-1 from fibroblasts. Cardiovasc Res. 1998;40(2):352–363. | |
Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102(4):470–479. | |
Weber KT. Extracellular matrix remodeling in heart failure: a role for de novo angiotensin II generation. Circulation. 1997;96(11):4065–4082. | |
Peng J, Gurantz D, Tran V, Cowling RT, Greenberg BH. Tumor necrosis factor-α-induced AT1 receptor upregulation enhances angiotensin II-mediated cardiac fibroblast responses that favor fibrosis. Circ Res. 2002;91(12):1119–1126. | |
Bart BA. Treatment of congestion in congestive heart failure: ultrafiltration is the only rational initial treatment of volume overload in decompensated heart failure. Circ Heart Fail. 2009;2(5):499–504. | |
Schrier RW. Role of diminished renal function in cardiovascular mortality: marker or pathogenetic factor? J Am Coll Cardiol. 2006;47(1):1–8. | |
Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. J Manag Care Pharm. 2007;13(8):S9. | |
McKie P, Burnett J Jr. Rationale and therapeutic opportunities for natriuretic peptide system augmentation in heart failure. Curr Heart Fail Rep. 2015;12(1):7–14. | |
Thibault G, Amiri F, Garcia R. Regulation of natriuretic peptide secretion by the heart. Annu Rev Physiol. 1999;61(1):193–217. | |
Stein BC, Levin RI. Natriuretic peptides: physiology, therapeutic potential, and risk stratification in ischemic heart disease. Am Heart J. 1998;135(5):914–923. | |
Gackowski A, Isnard R, Golmard J-L, et al. Comparison of echocardiography and plasma B-type natriuretic peptide for monitoring the response to treatment in acute heart failure. Eur Heart J. 2004;25(20):1788–1796. | |
Nathisuwan S, Talbert RL. A review of vasopeptidase inhibitors: a new modality in the treatment of hypertension and chronic heart failure. Pharmacotherapy. 2002;22(1):27–42. | |
Moyes AJ, Khambata RS, Villar I, et al. Endothelial C-type natriuretic peptide maintains vascular homeostasis. J Clin Invest. 2014;124(9): 4039–4051. | |
Cheung BY, Kumana CR. Natriuretic peptides – relevance in cardiovascular disease. JAMA. 1998;280(23):1983–1984. | |
Lisy O, Jougasaki M, Heublein DM, et al. Renal actions of synthetic dendroaspis natriuretic peptide. Kidney Int. 1999;56(2):502–508. | |
Suga S, Nakao K, Hosoda K, et al. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinology. 1992;130(1):229–239. | |
Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988; 241(4861):42–52. | |
Chinkers M, Garbers DL. The protein kinase domain of the ANP receptor is required for signaling. Science. 1989;245(4924):1392–1394. | |
Graf K, Koehne P, Gräfe M, Zhang M, Auch-Schwelk W, Fleck E. Regulation and differential expression of neutral endopeptidase 24.11 in human endothelial cells. Hypertension. 1995;26(2):230–235. | |
Vanneste Y, Michel A, Dimaline R, Najdovski T, Deschodt-Lanckman M. Hydrolysis of alpha-human atrial natriuretic peptide in vitro by human kidney membranes and purified endopeptidase-24.11. Evidence for a novel cleavage site. Biochem J. 1988;254(2):531–537. | |
Young JB, Abraham WT, Stevenson LW, Horton DP. Results of the VMAC trial: vasodilation in the management of acute congestive heart failure. Circulation. 2000;102(22):2794–2794. | |
Cardiorentis. TRUE-AHF Phase III clinical trial. Available from: http://www.cardiorentis.com/de/rd/true-ahf.php. Accessed January 26, 2015. | |
Ahmad T, Felker GM. Subcutaneous B-type natriuretic peptide for treatment of heart failure: a dying therapy reborn? J Am Coll Cardiol. 2012;60(22):2313–2315. | |
Chen HH, Glockner JF, Schirger JA, Cataliotti A, Redfield MM, Burnett JC. Novel protein therapeutics for systolic heart failure: chronic subcutaneous B-type natriuretic peptide. J Am Coll Cardiol. 2012;60(22): 2305–2312. | |
McKie PM, Cataliotti A, Boerrigter G, et al. A novel atrial natriuretic peptide based therapeutic in experimental angiotensin II mediated acute hypertension. Hypertension. 2010;56(6):1152–1159. | |
Lisy O, Huntley BK, McCormick DJ, Kurlansky PA, Burnett JC. Design, synthesis, and actions of a novel chimeric natriuretic peptide: CD-NP. J Am Coll Cardiol. 2008;52(1):60–68. | |
Chen HH. Heart failure: a state of brain natriuretic peptide deficiency or resistance or both!*. J Am Coll Cardiol. 2007;49(10): 1089–1091. | |
Natrecor® [Product Information]. Fremont, CA, USA; Scios Inc; 2013. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/20920s033lbl.pdf. | |
Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. Nesiritide Study Group. N Engl J Med. 2000;343(4): 246–253. | |
Silver MA, Horton DP, Ghali JK, Elkayam U. Effect of nesiritide versus dobutamine on short-term outcomes in the treatment of patients with acutely decompensated heart failure. J Am Coll Cardiol. 2002;39(5):798–803. | |
Burger AJ, Horton DP, LeJemtel T, et al. Effect of nesiritide (B-type natriuretic peptide) and dobutamine on ventricular arrhythmias in the treatment of patients with acutely decompensated congestive heart failure: the PRECEDENT study. Am Heart J. 2002;144(6):1102–1108. | |
Kesselheim AS, Fischer MA, Avorn J. The rise and fall of Natrecor for congestive heart failure: implications for drug policy. Health Aff (Millwood). 2006;25(4):1095–1102. | |
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900–1905. | |
Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111(12):1487–1491. | |
Hernandez AF, O’Connor CM, Starling RC, et al. Rationale and design of the Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure Trial (ASCEND-HF). Am Heart J. 2009;157(2): 271–277. | |
O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011; 365(1):32–43. | |
Gottlieb SS, Stebbins A, Voors AA, et al. Effects of nesiritide and predictors of urine output in acute decompensated heart failure: results from ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide and Decompensated Heart Failure). J Am Coll Cardiol. 2013;62(13): 1177–1183. | |
Richards AM. Vasopeptidase inhibitors for heart failure: where do we go from here? Eur J Heart Fail. 2003;5(4):411–413. | |
Lang CC, Motwani J, Coutie WJ, Struthers AD. Influence of candoxatril on plasma brain natriuretic peptide in heart failure. Lancet. 1991; 338(8761):255. | |
Northridge DB, Jardine AG, Alabaster CT, et al. Effects of UK 69 578: a novel atriopeptidase inhibitor. Lancet. 1989;2(8663):591–593. | |
Erdos EG, Skidgel RA. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989;3(2): 145–151. | |
Kahn JC, Patey M, Dubois-Rande JL, et al. Effect of sinorphan on plasma atrial natriuretic factor in congestive heart failure. Lancet. 1990;335(8681):118–119. | |
Northridge DB, Currie PF, Newby DE, et al. Placebo-controlled comparison of candoxatril, an orally active neutral endopeptidase inhibitor, and captopril in patients with chronic heart failure. Eur J Heart Fail. 1999;1(1):67–72. | |
Northridge DB, Newby DE, Rooney E, Norrie J, Dargie HJ. Comparison of the short-term effects of candoxatril, an orally active neutral endopeptidase inhibitor, and frusemide in the treatment of patients with chronic heart failure. Am Heart J. 1999;138(6 Pt 1): 1149–1157. | |
Westheim AS, Bostrom P, Christensen CC, Parikka H, Rykke EO, Toivonen L. Hemodynamic and neuroendocrine effects for candoxatril and frusemide in mild stable chronic heart failure. J Am Coll Cardiol. 1999;34(6):1794–1801. | |
Bevan EG, Connell JM, Doyle J, et al. Candoxatril, a neutral endopeptidase inhibitor: efficacy and tolerability in essential hypertension. J Hypertens. 1992;10(7):607–613. | |
Ando S, Rahman MA, Butler GC, Senn BL, Floras JS. Comparison of candoxatril and atrial natriuretic factor in healthy men. Effects on hemodynamics, sympathetic activity, heart rate variability, and endothelin. Hypertension. 1995;26(6 Pt 2):1160–1166. | |
Richards AM, Wittert GA, Crozier IG, et al. Chronic inhibition of endopeptidase 24.11 in essential hypertension: evidence for enhanced atrial natriuretic peptide and angiotensin II. J Hypertens. 1993;11(4): 407–416. | |
Ferro CJ, Spratt JC, Haynes WG, Webb DJ. Inhibition of neutral endopeptidase causes vasoconstriction of human resistance vessels in vivo. Circulation. 1998;97(23):2323–2330. | |
Kentsch M, Otter W, Drummer C, Notges A, Gerzer R, Muller-Esch G. Neutral endopeptidase 24.11 inhibition may not exhibit beneficial haemodynamic effects in patients with congestive heart failure. Eur J Clin Pharmacol. 1996;51(3–4):269–272. | |
Mangiafico S, Costello-Boerrigter LC, Andersen IA, Cataliotti A, Burnett JC. Neutral endopeptidase inhibition and the natriuretic peptide system: an evolving strategy in cardiovascular therapeutics. Eur Heart J. 2013;34(12):886–893. | |
Rouleau JL, Pfeffer MA, Stewart DJ, et al. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet. 2000;356(9230):615–620. | |
Packer M, Califf RM, Konstam MA, et al. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation. 2002;106(8):920–926. | |
Kostis JB, Packer M, Black HR, Schmieder R, Henry D, Levy E. Omapatrilat and enalapril in patients with hypertension: the Omapatrilat Cardiovascular Treatment vs. Enalapril (OCTAVE) trial. Am J Hypertens. 2004;17(2):103–111. | |
Pickering TG. The rise and fall of omapatrilat. J Clin Hypertens. 2002; 4(5):371–373. | |
Byrd JB, Touzin K, Sile S, et al. Dipeptidyl peptidase IV in angiotensin-converting enzyme inhibitor associated angioedema. Hypertension. 2008;51(1):141–147. | |
Lang CC, Struthers AD. Targeting the renin-angiotensin-aldosterone system in heart failure. Nat Rev Cardiol. 2013;10(3):125–134. | |
Gu J, Noe A, Chandra P, et al. Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual-acting angiotensin receptor-neprilysin inhibitor (ARNi). J Clin Pharmacol. 2010;50(4):401–414. | |
Mistry NB, Westheim AS, Kjeldsen SE. The angiotensin receptor antagonist valsartan: a review of the literature with a focus on clinical trials. Expert Opin Pharmacother. 2006;7(5):575–581. | |
Ksander GM, Ghai RD, deJesus R, et al. Dicarboxylic acid dipeptide neutral endopeptidase inhibitors. J Med Chem. 1995;38(10): 1689–1700. | |
Langenickel TH, Dole WP. Angiotensin receptor-neprilysin inhibition with LCZ696: a novel approach for the treatment of heart failure. Drug Discov Today Ther Strateg. 2012;9(4):e131–e139. | |
Vardeny O, Tacheny T, Solomon SD. First-in-class angiotensin receptor neprilysin inhibitor in heart failure. Clin Pharmacol Ther. 2013;94(4):445–448. | |
Jessup M. Neprilysin inhibition – a novel therapy for heart failure. N Engl J Med. 2014;371(11):1062–1064. | |
McMurray JJV, Packer M, Desai AS, et al. Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin-converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail. 2013;15(9): 1062–1073. | |
Swedberg K, Kjekshus J. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). J Am Coll Cardiol. 1988;62(2):60A–66A. | |
[No authors listed]. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med. 1991;325(5): 293–302. | |
McMurray JJV, Packer M, Desai AS, et al. Baseline characteristics and treatment of patients in Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur J Heart Fail. 2014;16(7): 817–825. | |
McMurray JJV, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371(11): 993–1004. | |
Stiles S. After sinking in, PARADIGM-HF critiqued at HFSA sessions. Medscape. Available from: http://www.medscape.com/viewarticle/832290. September 25, 2014. | |
Pablos-Mendez A, Barr RG, Shea S. Run-in periods in randomized trials: implications for the application of results in clinical practice. JAMA. 1998;279(3):222–225. | |
Solomon SD, Zile M, Pieske B, et al. The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet. 2012;380(9851):1387–1395. | |
Zile MR, Gottdiener JS, Hetzel SJ, et al. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation. 2011;124(23): 2491–2501. | |
Ruilope LM, Dukat A, Bohm M, Lacourciere Y, Gong J, Lefkowitz MP. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: a randomised, double-blind, placebo-controlled, active comparator study. Lancet. 2010; 375(9722):1255–1266. | |
Kario K, Sun N, Chiang F-T, et al. Efficacy and safety of LCZ696, a first-in-class angiotensin receptor neprilysin inhibitor, in Asian patients with hypertension: a randomized, double-blind, placebo-controlled study. Hypertension. 2014;63(4):698–705. | |
Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, et al. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet. 2013;382(9889):339–352. | |
Matsushita K, van der Velde M, Astor BC, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375(9731):2073–2081. | |
Cao Z, Burrell LM, Tikkanen I, Bonnet F, Cooper ME, Gilbert RE. Vasopeptidase inhibition attenuates the progression of renal injury in subtotal nephrectomized rats. Kidney Int. 2001;60(2): 715–721. | |
Benigni A, Zoja C, Zatelli C, et al. Vasopeptidase inhibitor restores the balance of vasoactive hormones in progressive nephropathy. Kidney Int. 2004;66(5):1959–1965. | |
ISRCTN Registry. UK Heart and Renal Protection (UK HARP-III). Available from: http://www.isrctn.com/ISRCTN11958993. Accessed January 26, 2015. | |
Mukharji J, Rude RE, Poole WK, et al. Risk factors for sudden death after acute myocardial infarction: two-year follow-up. J Am Coll Cardiol. 1984;54(1):31–36. | |
[No authors listed]. Risk stratification and survival after myocardial infarction. N Engl J Med. 1983;309(6):331–336. | |
Pfeffer MA, Braunwald E, Moyé LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 1992;327(10): 669–677. | |
Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348(14):1309–1321. | |
Von Lueder TG, Wang B, Kompa A, Webb R, Jordaan P, Krum H. Angiotensin-receptor neprilysin-inhibition (ARNi) attenuates ischemic cardiac failure in vivo and angiotensin-II-induced cellular cardiac hypertrophy and fibrosis. Global Heart. 2014;9(1 Suppl 1):e237. | |
von Lueder TG, Wang BH, Kompa AR, et al. The angiotensin-receptor neprilysin inhibitor LCZ696 attenuates cardiac remodeling and dysfunction after myocardial infarction by reducing cardiac fibrosis and hypertrophy. Circ Heart Fail. 2015;8(1):71–78. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.