")
Back to Journals » The Application of Clinical Genetics » Volume 9
Analysis of the CFTR gene in Venezuelan cystic fibrosis patients, identification of six novel cystic fibrosis-causing genetic variants
Authors Sánchez K , De Mendonca E, Matute X, Chaustre I, Villalon M, Takiff H
Received 27 November 2014
Accepted for publication 8 January 2015
Published 8 March 2016 Volume 2016:9 Pages 33—38
DOI https://doi.org/10.2147/TACG.S78241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Karen Sánchez,1 Elizabeth de Mendonca,1 Xiorama Matute,2 Ismenia Chaustre,2 Marlene Villalón,3 Howard Takiff4
1Unit of Genetic and Forensic Studies, Venezuelan Institute for Scientific Research (IVIC), 2Hospital JM de los Ríos, 3Hospital José Ignacio Baldo, Algodonal, National Reference Unit, 4Laboratory of Molecular Genetics, Venezuelan Institute for Scientific Research (IVIC), Caracas, Venezuela.
Abstract: The mutations in the CFTR gene found in patients with cystic fibrosis (CF) have geographic differences, but there are scant data on their prevalence in Venezuelan patients. This study determined the frequency of common CFTR gene mutations in a group of Venezuelan patients with CF. The 27 exons of the CFTR gene from 110 Venezuelan patients in the National CF Program were amplified and sequenced. A total of 36 different mutations were identified, seven with frequencies greater than 1%: p.Phe508del (27.27%), p.Gly542* (3.18%), c.2988+1G.A (3.18%), p.Arg334Trp (1.36%), p.Arg1162* (1.36%), c.1-8G.C (1.36%), and p.[Gly628Arg;Ser1235Arg](1.36). In 40% of patients, all with a clinical diagnosis of CF, no mutations were found. This report represents the largest cohort of Venezuelan patients with CF ever examined, and includes a wider mutation panel than has been previously studied in this population. Mutations common in Southern European populations predominate, and several new mutations were discovered, but no mutations were found in 40% of the cohort.
Keywords: c.49_50dupTT, c.3963+1G.A, p.Asp373Asn, p.Glu815*, p.Asn900Lys, p.Trp277*
Introduction
Cystic fibrosis (CF; OMIM number: 219700) is a severe autosomal recessive disorder caused by alterations in the CF transmembrane conductance regulator (CFTR; OMIM number: 602421), and more than 1,400 different mutations have been reported in the CFTR gene in the patients with CF.1 The CFTR gene is located at chromosomal region 7q31.2, and is composed of 27 exons2 that span approximately 190 kb of genomic DNA,3,4 encoding a protein of 1,480 amino acids.5 Mutations in the CFTR gene result in elevated sweat chloride concentrations and pathological features of variable severity affecting the lungs and pancreas, among other symptoms.6 The incidence of CF varies across the globe. In the European Union, one in 2,000–3,000 newborns is found to be affected by the CF. Venezuela reports a lower detection rate of 50%.7
Similar to other Latin American countries, the ethnic composition of the Venezuelan population is highly heterogeneous. In our initial screening of the cohort of Venezuelan patients with CF, we examined exons 7, 10, 11, 19, 20, and 21, which contain the most common CFTR mutations in Latin America. In that preliminary study, the CFTR mutations were found in only 46.67% of the Venezuelan individuals studied suggesting that it would be necessary to do a more complete analysis of the CFTR gene in this population.8
In order to characterize the complete spectrum of the CF-causing mutations in Venezuelan patients, we present a molecular analysis of the 27 exons of the CFTR gene in 110 unrelated Venezuelan individuals with the clinical diagnosis of CF who belong to the National CF Program. This information could be used for implementing a diagnostic method that could rapidly identify a large percentage of patients with CF in the Venezuelan population.
Patients and methods
Patients and sampling
From 2011 to 2013, we recruited 110 patients from an equal number of unrelated families registered in the National CF Program of the Ministry of Health of Venezuela. The patients came from different regions of the country and received care at two pediatric reference centers. This group represents approximately one-third of all patients with CF followed by these references centers. The National CF Program incorporates patients who meet the program’s diagnostic criteria of classic signs and symptoms of the disease and have corroborative laboratory results.9 Most of these patients lack molecular confirmation of the disease, due to the lack of routine molecular diagnostic analysis of the CFTR gene. Detailed questionnaires, including clinical and family history, were obtained from each participating family. Bioethics Committee of the Venezuelan Institute for Scientific Research approved the study and written informed consent was obtained from all participating families.
Molecular analysis
A peripheral blood sample was collected from each patient, from which genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN). Amplification of 27 complete CFTR exon and intron junctions, where splicing or frameshift mutations are present, were performed using CFTR-specific primers previously described for CFTR analysis,2,10 based on NCBI Reference Sequence: NM_000492.3. The amplicons were sequenced on a ABI-3130XL Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA). The Devyser CFTR Core (Devyser) kit was used on DNA from 44 patients for whom no mutations were identified by sequencing.
The sequences were analyzed with the Sequencing Analysis V 5.3.1, SeqScape software V 2.5, and GeneMapper V.3.1 programs (Thermo Fisher Scientific). Allele and genotype frequencies were determined by direct counting.
Results
All of the patients in the study were part of the National Program for CF. Seven families had more than one sibling with the CF: six cases had two affected siblings and one case had three affected siblings. We included only one case per family for the statistical analysis, which resulted in 220 alleles derived from 110 patients. The cohort was comprised of 37 females (33.63%) and 73 males (66.36%) with ages ranging from 2 months to 30 years old. Sixty of these patients were at least third-generation Venezuelan (54.54%) and 50 patients (45.45%) were either foreign born or children of parents who were not born Venezuelan.
Mutation analysis
Forty-three different mutant genotypes were found in 110 patients, which are shown in Table 1. In 52/110 patients (47.27%) two mutations were identified, while in 14/110 patients (12.72%) only one mutation was detected. Eighteen patients (16.36%) were homozygotes for p.Phe508del, one patient (0.91%) was a homozygote for p.Glu815*, one patient (0.91%) was a homozygote for the allele complex p.[Gly628Arg;Ser1235Arg], and one patient (0.91%) was a homozygote for p.Gly542*. Forty-six patients were compound heterozygotes: 14 patients (12.72%) had a wild type allele, 20 patients (18.18%) had one p.Phe508del allele and another mutated allele, and eleven patients (10.00%) had two alleles different than p.Phe508del. In the remaining 44 patients (40%), we failed to detect any CFTR mutation in the exon–intron study.
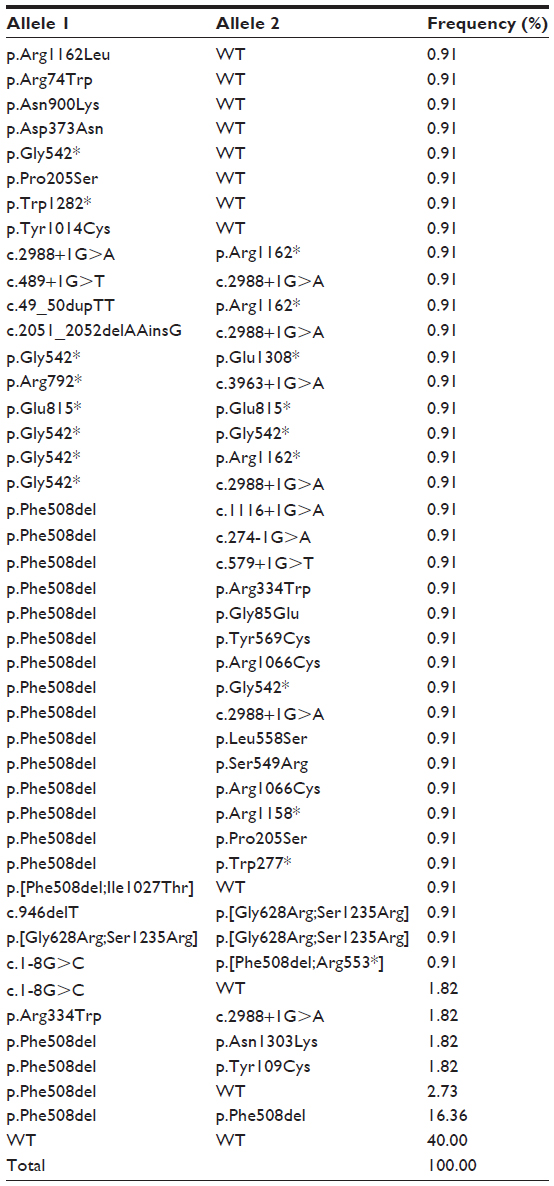 | Table 1 Genotype frequency of mutations found for the CFTR gene studied |
The allelic frequency obtained in this study is shown in Table 2, which also shows the frequencies of the mutations reported for all 27 exons and exon–intron junction examined both in Latin America and the rest of the world. The p.Phe508del mutation had the highest prevalence, and another seven mutations had frequencies greater than 1% (c.1-8G>C, p.Arg334Trp, p.Gly542*, p.[Gly628Arg;Ser1235Arg], p.Arg1162*, and c.2988+1G>A). Twenty-five additional CF-causing mutations and two complex alleles had not been previously reported in the Venezuelan population, they are shaded in the Table 2.
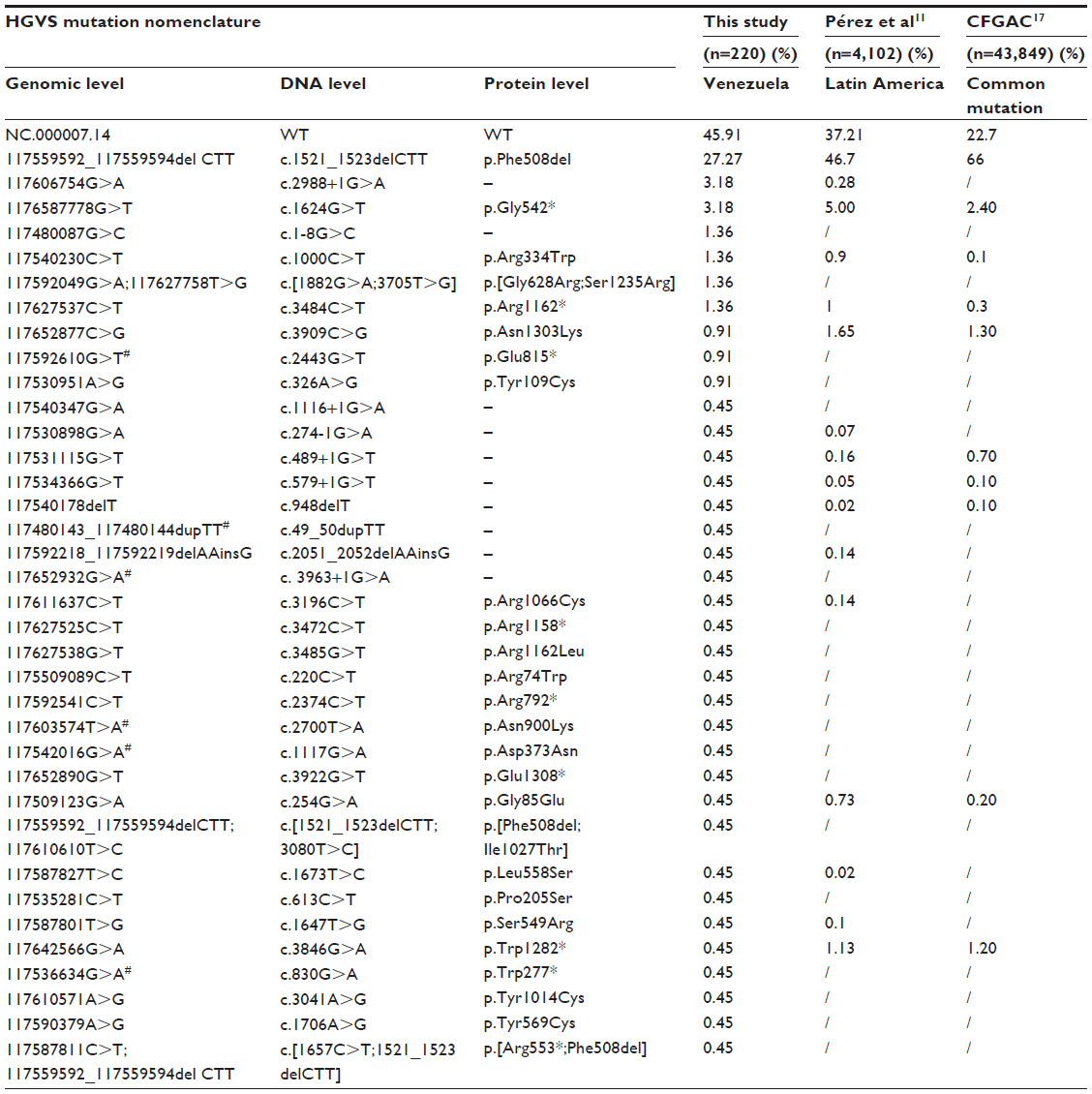 | Table 2 Allelic frequency of mutations found for the CFTR gene studied |
In this study, the frequency of the p.Phe508de variant is lower than has been reported in most populations; seven of the mutations found with lower frequency had not been previously reported in Latin America, and are not present in the databases used as reference.1,2,11–13
The sequence variations found were reported using classical nomenclature; intron 6a: IVS6aGATT(6) 18.6%, IVS6aGATT(7) 45.9%, c.743+40A>G 1.36%; intron 6b: c.869+11G>A 29%; exon 14a: c.2562T>G 0.45%; exon 15: c.2898G>A 0.9%; exon 20: c.3870A>G 3.2%; intron 21: c.3963+27G>A 0.45%, and exon 24: c. 4389G>A 11%.
Novel genetic variants
At present, we have identified a total of eight novel genetic variants: two sequence variations (c.4242+58C>A [46.4]% in intron 23 and c.489+91A>C [4.5%] in intron 4); six CF-causing mutations (four missense mutations – p.Trp277* p.Asp373Asn, p.Glu815*, and p.Asn900Lys); one mRNA splicing defect (c.3963+1G>A); and one frameshift mutation (c.49_50dupTT).
c.49_50dupTT: (record ClinVar accession number: 000047170)
The frameshift mutation c.49_50dupTT in exon 1 is caused by the duplication of two Ts after nucleotide position 49. It was found in a third-generation Venezuelan female patient carrying the p.Arg1162* mutation on the other chromosome. The patient had respiratory and gastric symptomatology from birth, and she had one positive sweat chloride test (124 mmol/L). The patient’s phenotype suggests that she has severe mutations in both CF alleles. Because of the clinical findings, the c.49_50dupTT mutation was classified a CF- causing mutation.
c.3963+1G>A: (record ClinVar accession number: 000149421)
The mRNA splicing defect 3963+1G>A in intron 21 is caused by the transition of G to A at first nucleotide position. It was found in a third-generation Venezuelan female patient carrying the p.Arg792* mutation on the other chromosome. Her clinical diagnosis is CF, and she had two positive sweat chloride tests (122 and 120 mmol/L). Because c.3963+1G>T variant are described causes a mRNA splicing defect,12 we consider the c.3963+1G>A variant potentially cause mRNA splicing defect. The addition of the clinical findings suggest that the c.3963+1G>A mutation can be classified causing a CF mutation.
p.Trp277*: (record ClinVar accession number: 000149425)
The p.Trp277* mutation was caused by the transition of G to A at nucleotide position c.830 in exon 6b. It changes a tryptophan to a stop codon at amino acid 277 of the protein. This mutation was found in a third-generation Venezuelan male patient carrying the p.Phe508del mutation on the other chromosome. The patient has respiratory symptomatology that appeared for the first time at the age of 3 months. He had two positive sweat chloride tests (115 and 100 mmol/L).
p.Asp373Asn: (record ClinVar accession number: 000149423)
The p.Asp373Asn mutation was caused by the transition of G to A at nucleotide position c.1117 in exon 8. It changes an asparagine to an aspartic acid at amino acid position 373 of the protein. This mutation was found in a third-generation Venezuelan male patient carrying no mutation on the other chromosome. The patient has respiratory and gastric symptomatology that appeared for the first time at the age of 2 months. He had two borderline sweat chloride tests (55 and 45 mmol/L).
p.Glu815*: (record ClinVar accession number: 000149424)
The p.Glu815* mutation was caused by the transversion of G to T at nucleotide position c.2443 in exon 13. It changes a glutamic acid to a stop codon at amino acid 815 of the protein. This mutation was found to be homozygotic in a third-generation Venezuelan male patient. The patient has respiratory and gastric symptomatology that appeared for the first time at the age of 6 months, and he had two positive sweat chloride tests (111 and 108 mmol/L). Because of the clinical findings, the p.Glu815* mutation was classified as a CF-causing mutation.
p.Asn900Lys: (record Clinvar accession number: 000149422)
The p.Asn900Lys mutation was caused by the transversion of T to A at nucleotide position c.2700 in exon 15. It changes an asparagine to a lysine at amino acid 900 of the protein. This mutation was found in a third-generation Venezuelan female patient carrying the wild type allele on the other chromosome. The patient has a confused phenotype with late onset of disease (11 years old), but has a low-body weight. She has had two borderline sweat chloride tests (40 and 39 mmol/L).
Analysis of Intron 8
The lengths of the dinucleotide (T: thymidine- G: guanine)n and mononucleotide (T)n repeats, both located at the intron8/exon9 splice acceptor site of the CFTR gene, have been shown to influence the skipping of exon 9 in the CFTR mRNA.14 Seventeen genotypes in the analysis of intron 8 were found in the 110 patients. Of these, 42/110 patients (38.2%) were homozygotic while 68/110 patients (61.8%) were heterozygotic. The intron 8 allelic frequencies obtained in this study are shown in Table 3.
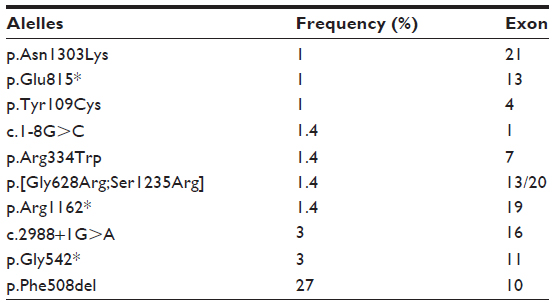 | Table 3 Allelic frequency found for intron 8 |
Discussion
This study examined a larger cohort of Venezuelan patients with CF and included a more comprehensive mutation panel – 27 complete CFTR gene exons and exon–intron junctions – than has been previously reported. As in most countries studied, the allele p.Phe508del was the most common mutation detected in our patients (27.27%). Similar to other studies from Latin America, as reported in the CF Mutation Database,1 we found relatively low detection rates and high allelic heterogeneity compared to studies in Europe. The heterogeneity is likely explained by the complex ethnic composition of the Venezuelan population that has evolved from an unequal admixture of Caucasian–Spanish, Amerindian, and African origins (58.8%, 28.5%, and 12.6%, respectively of the genetic pool).15
This study defined the most common CF mutations in the Venezuelan population, but we wished to determine whether the frequency of particular mutations is different in native Venezuelan compared to recent immigrants. We performed an additional analysis – non-metrics multidimensional scaling – based on the genetic distance between individuals according to their origin, comparing patients from third-generation Venezuelan families to those from families recently immigrated to Venezuela, but this classification analysis showed no differences associated with familial origin (data not shown). The mutation panel shown in Table 3 lists the most common mutations in Venezuelan patients, which we recommend should be included in the design of any detection system for the molecular diagnosis of the CF in Venezuela.
It is important to note that the mutations reported here for the first time were found in third-generation Venezuelan patients, and may be native to the Venezuelan population.
We were unable to detect the CF-causing mutations in 46.6% of the patients analyzed (Table 1). This percentage does not change when the variation IVS8 (T: thymidine- G: guanine)n (T)n is analyzed in these patients, despite the fact that the criteria for clinical diagnosis of all patients were the same. We believe these results to be robust for the following reasons: the cohort included in this study constitutes approximately a third of all patients currently registered in the National CF Program; the results were obtained using an automated sequencing method that allows high sensitivity and resolution in the detection of mutations; and the absence of mutations were confirmed with a commercial primer-specific kit. There are two possibilities that might explain the high percentage of individuals without the CFTR mutations: 1) The patients for whom no mutations were found could have other genetic alterations, such as macro-deletions or chromosomal rearrangements, which are not detectable by the Sanger-sequencing method that we employed. This could be resolved with the use of next-generation sequencing technology in future studies. 2) The patients for whom no mutations were found could be misdiagnosed the patients with CF, who do not really have CF. This possibility suggests that it may be necessary to revise the criteria for the clinical diagnosis of the CF in Venezuela, and to create a national reference center for the molecular diagnosis of the CF.
In either case, both new and complex alleles found in this study widen our knowledge of the CF causing mutations in general, and their inclusion in future molecular diagnostic screens in Venezuelan patients with CF will allow a more complete analysis, which should improve clinic diagnostic confirmation, carrier analysis, and genetic counseling, and can also guide the development of a cost-effective newborn screening program.
Acknowledgments
We are grateful to our many clinician colleagues, especially Dr Luz Mindiola and Dr Isabel Tovar of the Children’s Medical Center Hospital, as well as the physicians at the other medical centers who referred the patients and patients’ families to our team. We also thank César Paz for help with the final format editions.
Disclosure
The authors report no conflicts of interest in this work.
References
Cystic Fibrosis Centre. Cystic Fibrosis Mutation Database [database on the Internet]. Available from: http://www.genet.sickkids.on.ca/app. Accessed October 7, 2014. | |
Zielenski J, Rozmahel R, Bozon D, et al. Genomic DNA sequence of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Genomics. 1991;10(1):214–228. | |
McCarthy VA, Harris A. The CFTR gene and regulation of its expression. Pediatr Pulmonol. 2005;40(1):1–8. | |
Shrimpton AE. Molecular diagnosis of cystic fibrosis. Expert Rev Mol Diagn. 2002;2(3):240–256. | |
Riordan JR, Rommens JM, Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. | |
Quinton PM. Physiological basis of cystic fibrosis: a historical perspective. Physiol Rev. 1999;79(1 Suppl):S3–S22. | |
WHO. Genes and Human Disease. Geneva: Genomic Resource Center, World Health Organization; 2014. Available from: http://www.who.int/genomics/public/geneticdiseases/en/index2.html#CF. Accessed December 21, 2014. | |
Sánchez K, Arcia O, Matute X, Mindiola L, Chaustre I, Takiff H. Frequency of common CFTR gene mutations in Venezuelan patients with cystic fibrosis. Invest Clín. 2014;55(1):44–54. | |
Farrell PM, Rosenstein BJ, White TB, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: cystic fibrosis foundation consensus report. J Pediatr. 2008;153(2):S4–S14. | |
Cuppens H, Marynen P, De Boeck C, Cassiman JJ. Detection of 98.5% of the mutations in 200 Belgian cystic fibrosis alleles by reverse dot-blot and sequencing of the complete coding region and exon/intron junctions of the CFTR gene. Genomics. 1993;18(3):693–697. | |
Pérez MM, Luna MC, Pivetta OH, Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent. J Cyst Fibros. 2007;6(3):194–208. | |
Shekdar K, Langer J, Venkatachalan SP, Sawchuk DJ, inventors; Chromocell Corporation, assignee. Cell lines expressing cftr and methods of using them. United States patent US WO2010088630 A2. 2010 Aug 1. Available from: http://www.google.com/patents/WO2010088630A2. Accessed October 7, 2014. | |
The Clinical and Functional Translation of CFTR (CFTR2). Baltimore, MD:Johns Hopkins University. Available from:http://www.cftr2.org/browse.php. Accessed October 7, 2014. | |
Lucarelli M, Grandoni F, Rossi T, Mazzilli F, Antonelli M, Strom R. Simultaneous cycle sequencing assessment of (TG)m and Tn tract length in CFTR gene. Biotechniques. 2002;32(3):540–542, 544–547. | |
Larralde áR, Guerra de C, Coira MG, Morales J. Frecuencia génica y porcentaje de mezcla en diferentes áreas geográficas de Venezuela, de acuerdo a los grupos RH y ABO. Interciencia. 2001;26(1):8–12. | |
Horaitis R. HGVS recommendations for the description of DNA sequence variants – v2.0. 2013. Available from: http://www.hgvs.org/mutnomen/recs-DNA.html. Accessed October 7, 2014. | |
CFGAC. Population variation of common cystic fibrosis mutations. The Cystic Fibrosis Genetic Analysis Consortium. Hum Mutat. 1994;4(3):167–177. doi:10.1002/humu.1380040302. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.