Back to Journals » Journal of Asthma and Allergy » Volume 16
Analysis of ceRNA Regulatory Mechanism of Rape Pollen Allergy Based on Whole-Transcriptome Sequencing of Peripheral Blood Mononuclear Cells
Authors Wang Y, Li J ![]() , Wang F, Cui Y, Song L, Ruan B, Yu Y
, Wang F, Cui Y, Song L, Ruan B, Yu Y
Received 11 April 2023
Accepted for publication 12 July 2023
Published 27 July 2023 Volume 2023:16 Pages 775—788
DOI https://doi.org/10.2147/JAA.S416772
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Luis Garcia-Marcos
Ya Wang,1 Jianhua Li,2 Fang Wang,1 Yunhua Cui,1 Li Song,1 Biao Ruan,1 Yongmei Yu1
1Department of Otolaryngology, the First Affiliated Hospital of Kunming Medical University, Kunming, People’s Republic of China; 2Department of Otolaryngology, the Sixth Affiliated Hospital of Kunming Medical University, Yuxi, People’s Republic of China
Correspondence: Yongmei Yu, Email [email protected]
Background: Rape pollen allergy is a common allergic reaction disorder that affects the health and life of patients seriously. The research on ceRNA regulatory network in rape pollen allergy is poor.
Methods: High throughput whole-transcriptome sequencing was conducted on rape pollen allergic samples and non-allergic samples. Differentially expressed microRNAs (DEmiRNAs), circRNAs (DEcircRNAs), long non-coding RNA (DElncRNAs), mRNA (DEmRNAs) were identified and a ceRNA regulatory network was constructed by Cytoscape. Functional enrichment analyses were performed on DEmRNAs in the ceRNA network. Then, the least absolute shrinkage and selection operator (LASSO) regression model was used to identify characteristic genes for rape pollen allergy. The receiver operating characteristic (ROC) curve was used to evaluate the diagnostic ability of characteristic genes.
Results: A total of 25 DEmiRNAs, 258 DEcircRNAs, 304 DElncRNAs, and 383 DEmRNAs in the allergic group compared with the non-allergic group were uncovered, respectively. A ceRNA network containing 21 miRNAs, 57 circRNAs, 28 lncRNAs, and 33 mRNAs was generated with 139 nodes and 160 edges. The signal transduction-related processes, immune-related processes, the ion, inorganic substance, and hormone regulation processes were associated with mRNAs in the ceRNA network. The results of pathway enrichment illustrated that mRNAs in the ceRNA were significantly linked to IL-17 signaling pathway, inflammatory mediator regulation of trp channels, GMP-PKG signaling pathway, signaling by GPCR, and GPCR downstream signaling pathway. Then, five characteristic genes (KCNQ3, CCR5, FOSB, CFAP43, and PRKG1) were defined by the LASSO algorithm. The AUC values of these genes indicated that these genes had a powerful discrimination ability in discriminating allergic samples from non-allergic samples.
Conclusion: Taken together, we revealed the ceRNA regulatory network in rape pollen allergy and excavated five characteristic genes (KCNQ3, CCR5, FOSB, CFAP43, and PRKG1) with the diagnostic value that may be a potential target in diagnosis and treatment.
Keywords: rape pollen allergy, ceRNA, characteristic genes, GO, KEGG, IPA
Introduction
Allergic rhinitis (AR) is a type I allergic reaction caused by allergens entering the nasal mucosa and stimulating the local production of corresponding IgE in the nasal cavity.1,2 In the pathological process, a variety of immune cells and immune factors are involved, such as mast cells, eosinophils/basophils, T cells, histamine, prostaglandins, and interleukin.1,2 It is mainly characterized by nasal itching, sneezing, clear nasal discharge and nasal congestion. AR is divided into perennial AR and seasonal AR. Seasonal AR is mainly allergic to inhalation of airborne plant pollen, also known as pollinosis, which has obvious seasonality. Rape flower pollen is one of the most common seasonal AR allergenic plant pollen in Yunnan Province. People with rape flower pollen allergy have repeated symptoms every spring. In the initial stage, the patients mainly have symptoms, such as nasal itching, sneezing, clear nose, nasal congestion, eye itching, tears and so on. In severe cases, they can be complicated with bronchial asthma. The diagnosis can be made through the clinical manifestations, signs and allergen test results. Currently, immunotherapy is the only cause-related treatment method, but immunotherapy takes 3–5 years, with long treatment times and poor medical adherence by patients. The purpose of this study is to mine the characteristic genes of rape flower pollen allergy which can distinguish rape flower allergy from non-rape flower allergy through whole-transcriptome sequencing and bioinformatics analysis, so as to provide new ideas for the treatment of rape flower pollen allergy patients, and provide a new reference for clinical diagnostic biomarker and gene targeted therapy.
In addition to mRNA, there are many RNAs that do not encode proteins in organisms, called noncoding RNAs (ncRNAs), including microRNA, lncRNA and circRNA. These ncRNAs have very important functions in biological genetics, of which the core role is regulation.3 CeRNA regulation mechanism means that lncRNA and circRNA can regulate gene (mRNA) expression by competitively binding miRNAs. Building a ceRNA network diagram was significant to mine key genes and further study the molecular regulation mechanism and disease pathogenesis. More and more studies have shown that miRNA plays a key role in skin inflammation and allergic diseases,4 transcriptome sequencing is also used in asthma, dermatitis and rhinitis.5 However, the research on the ncRNAs in rape pollen is quite limited, and the ceRNA regulatory network in rape pollen allergy remains to be explored.
Hence, in order to investigate the regulation mechanism of ceRNA in rape pollen allergy, we constructed and analyzed the ceRNA regulation network based on the whole-transcriptome sequencing data, which is of great significance to discover the candidate diagnostic markers, therapeutic targets and molecular mechanism of rape pollen allergy.
Materials and Methods
People and Sample Information
We collected the peripheral blood mononuclear cell (PBMC) samples from 8 people allergic to rape pollen and 8 people without rape pollen allergy. The patients and healthy volunteers were collected at The Sixth Affiliated Hospital of Kunming Medical University, Yuxi, China. At present, the clinical diagnosis basis of rape pollen allergy is as follows: first, patients have symptoms, such as itching, sneezing and runny nose in spring, which can be accompanied by itchy eyes, tears and other symptoms. Second, the physical examination shows that the nasal mucosa is pale and swollen. Third, rape pollen skin prick test was positive, and wind clusters and erythema appeared in the inner skin prickles of 20min. Compared with the negative control, the average diameter of wind masses was >3mm, and the other spring pollen prick tests were negative. Inclusion criteria: ① adult patients; ② Clearly diagnosed as seasonal AR (diagnosis and treatment guidelines for AR (2022, revised version) - AR patients with spring pollen allergy (and rape pollen allergy in skin prick test was positive); ③ None of the enrolled samples in the control group has ever been diagnosed with AR, and all allergen skin prick tests are negative; Exclusion criteria: ① Subjects whose skin is seriously damaged and cannot undergo skin prick test; ② Subjects who have recently taken drugs that affect the results of skin prick test; ③ Subjects with immune deficiency or autoimmune diseases; ④ Subjects with a history of severe systemic anaphylaxis (None of the above conditions shall be included in the group experiment). After the completion of case enrollment, 5–10mL of whole blood was collected from each patient, and PBMC was obtained after centrifugation and treatment of Phosphate Buffered Saline (PBS) solution, PBMC was stored in -80 °C refrigerator. The study was approved by the Ethics Committee of the Sixth Affiliated Hospital of Kunming Medical University (Approval No: kmykdx6f). All patients and healthy volunteers had signed an informed consent form.
Whole-Transcriptome Sequencing
Total RNA from each sample was extracted using a Total RNA Purification Kit (TRK-1001, LC Sciences) according to the manufacturer’s instructions. The total RNA quantity, purity, and integrity were analyzed with NanoDrop ND-2000 (Thermo Scientific) and Bioanalyzer 2100 (Agilent Technologies). μParaflo microRNA microarray assay was performed by the service provider (LC Sciences) and conducted at LC Biotech in Hangzhou, China. The construction of the next-generation sequencing libraries and sequencing on the Illumina HiSeq 2500 platform were also performed at LC Biotech in Hangzhou, China.
Identification of Differentially Expressed microRNA (DEmiRNAs), circRNA (DEcircRNAs), Long Non-Coding RNA (DElncRNAs), mRNA (DEmRNAs)
The microRNA microarray data was analyzed by first subtracting the background, and then normalizing the signals using a LOWESS filter (locally weighted regression).6 The DEmiRNAs between the allergic group and non-allergic group based on the normalized signal were analyzed via the “limma” R package.7 The transcriptome clean data was obtained and aligned against the human genome (hg19) using the HISAT2 software.8 Samtools was used to sort reads in binary alignment map (bam) file by chromosome.9 Then, we used the featureCount program to count the reads and quantify mRNA and lncRNA.10 To quantify circRNA, we extracted the anchor_sequence from the sequences not matched to the human genome by using find_circ software.11 Then we matched the anchor sequence to the human genome (hg19) using bowtie2 software.12 Finally, the circRNAs were annotated based on the circbase database. The DEcircRNAs, DElncRNAs, and DEmRNAs were determined by using the “DEseq2” R package.13 The |log2 Fold Change (FC)| ≥ 1 and p-value < 0.05 were regarded as the cut-off criteria. The volcano maps were drawn to visualize the differences in gene expression levels between two groups. The heatmaps were plotted to display the expression of the top50 DEmiRNAs, DEcircRNAs, DElncRNAs, and DEmRNAs.
The Construction of ceRNA Regulatory Network
The lncBaseV2 database14 and circbank15 database were used to predict the miRNAs regulated by DElncRNAs and DEcircRNAs. The DElncRNA-DEmiRNA and DEcircRNA-DEmiRNA pairs were identified by taking the intersection of predicted miRNAs and DEmiRNAs. Subsequently, the miRDB database in the miRWalk website was used to predict the target mRNAs of the above DEmiRNAs.16 The final DEcircRNA-DEmiRNA-DEmRNA and DElncRNA-DEmiRNA-DEmRNA pairs were obtained by overlapping the predicted mRNAs and DEmRNAs. In addition, we selected the dysregulated target miRNA with a negative correlation of expression with the respective DElncRNA, DEcircRNA, and DEmRNA. The ceRNA networks were built with the Cytoscape software.17
Function and Pathway Enrichment
GO, KEGG pathway, and Reactome pathway enrichment analysis were conducted in Metascape software.18 Ingenuity canonical pathways and disease and function enrichment analysis were performed in IPA software.19
Screening the Characteristic Genes
The least absolute shrinkage and selection operator (LASSO) is an algorithm that can obtain a more refined model by constructing a penalty function.20 We used the LASSO algorithm to screen the characteristic genes in rape pollen allergy by the “glmnet” package. The area under the curve (AUC) value of receiver operating characteristic (ROC) curve was utilized to determine the diagnostic effectiveness of potential biomarkers in discriminating rape pollen allergic samples from normal samples and performed by using the pROC package.21
Results
Expression Profiles of miRNAs, circRNAs, lncRNAs, and mRNAs
DEmiRNAs, DEcircRNAs, DElncRNAs, and DEmRNAs between the allergic group and non-allergic group were shown using volcano plot (Figure 1A-D), and the expression of the top50 DEmiRNAs, DEcircRNAs, DElncRNAs, and DEmRNAs was displayed by heatmap (Figure 1E-H). The results showed that there were 25 DEmiRNAs (9 up-regulation and 16 down-regulation), 258 DEcircRNAs (123 up-regulation and 135 down-regulation), 304 DElncRNAs (138 up-regulation and 166 down-regulation), and 383 mRNAs (232 up-regulation and 151 down-regulation) in the allergic group compared with the non-allergic group, respectively. The detailed information of DEmiRNAs, DEcircRNAs, DElncRNAs, and DEmRNAs was listed in Tables S1-S4.
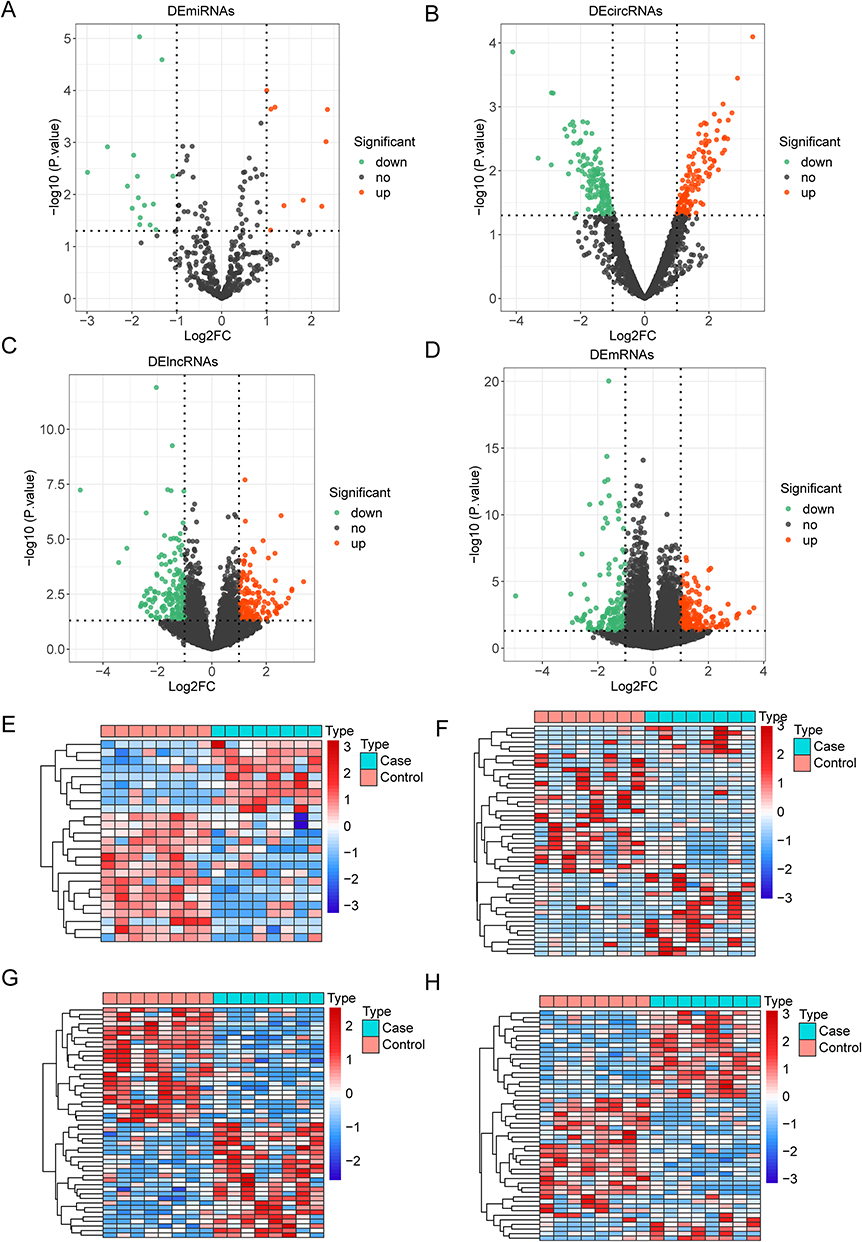 |
Figure 1 Differential expression analysis. The volcano plot (A-D) and heatmap (E-H) of DEmiRNAs (A and E), DEcircRNAs (B and F), DElncRNAs (C and G) and DEmRNAs (D and H). |
LncRNA/circRNA-miRNA-mRNA ceRNA Network
Using the method described above, a ceRNA network containing 21 miRNAs, 57 circRNAs, 28 lncRNAs, and 33 mRNAs was generated with 139 nodes and 160 edges (Figure 2, Table S5). To further explore the function of mRNAs in the ceRNA network, the function and pathway enrichment were performed. Forty-five GO terms, including 30 BP terms, 12 MF terms, 3 CC terms, and 10 pathways, including 6 KEGG pathways and 4 Reactome pathways, were enriched (Table S6, Figure 3A and B). The top 10 BP and MF terms are shown in Figure 3A. The signal transduction-related processes, such as cell–cell adhesion via plasma-membrane adhesion molecules, second-messenger-mediated signaling, and dephosphorylation, immune-related processes, such as leukocyte migration, chemotaxis, and transforming growth factor beta receptor signaling pathway were correlated to mRNAs in the ceRNA network. The ion, inorganic substance, and hormone regulation processes, such as regulation of body fluid levels, response to corticosteroid, response to steroid hormone, response to inorganic substance, and cellular ion homeostasis were also associated with mRNAs in the ceRNA network. Concerning MF, the mRNAs in the ceRNA were mainly involved in inorganic cation transmembrane transporter activity, metal ion transmembrane transporter activity, inorganic molecular entity transmembrane transporter activity, cation channel activity, GTP binding, protein kinase activity, and DNA-binding transcription activator activity. The results of pathway enrichment illustrated that mRNAs in the ceRNA were significantly linked to IL-17 signaling pathway, inflammatory mediator regulation of trp channels, GMP-PKG signaling pathway, signaling by GPCR, and GPCR downstream signaling pathway. In addition, the results of IPA show that one significant canonical pathway, adrenomedullin signaling pathway, was enriched with Z-score ≥ 2 (Table S7). Seventy-one disease and function terms were also enriched by IPA based on mRNAs in the ceRNA network (Table S7) and the top 20 terms are listed in Figure 3C. As to disease and function enrichment result, the cell-to-cell signaling and interaction, nervous system development and function, cardiovascular disease, organismal injury and abnormalities, cardiovascular system development and function, cell death and survival, gastrointestinal disease, cell signaling, respiratory disease, inflammatory response, immunological disease, immune cell trafficking, cell-mediated immune response, metabolic disease, cellular compromise, protein synthesis, DNA replication, recombination, and repair, post-translational modification, respiratory system development, and function may play an important role in the process of rape pollen allergy.
 |
Figure 2 The ceRNA network. The size of the point indicates the connectivity. The greater the point is, the greater the connectivity is. Circular nodes represent miRNA, triangular nodes represent circRNA, square nodes represent mRNA, and diamond nodes represent lncRNA. |
 |
Figure 3 The enrichment analysis results of mRNA in the ceRNA network. (A) The CC terms and, top 10 BP and MF terms. (B) The enriched KEGG pathways and Reactome pathways. (C) The top 20 disease and function terms enriched by mRNA in the ceRNA network. |
Identification of Characteristic Genes
To screen characteristic genes from the mRNA in the ceRNA network, we adopted the LASSO algorithm. Five characteristic genes (KCNQ3, CCR5, FOSB, CFAP43, and PRKG1) were discerned using the LASSO algorithm (Figure 4A). The ROC curve of the LASSO regression model was plotted and the AUC value = 1, indicating the model based on the five characteristic genes was able to predict sample grouping effectively (Figure 4B). Finally, we evaluated the diagnostic value of each characteristic gene by ROC analysis. The AUC values of these genes were all greater than 0.9, indicated that these genes had a powerful discrimination ability in discriminating allergic samples from non-allergic samples (Figure 4C). The sub-network related to the characteristic genes was extracted from the ceRNA network and shown in (Figure 5). In the ceRNA sub-network, the expression of FOSB were regulated by hsa-miR-30c-2-3p which may be sponged by hsa_circ_0062426, hsa_circ_0031569, hsa_circ_0000126, hsa_circ_0007161, hsa_circ_0007539 hsa_circ_0043428, HIPK1-AS1, LINC00051, or LINC00271. The hsa-miR-27a-5p targeted CCR5 and was regulated by IL21-AS1. The hsa-miR-618 regulated the expression of PRKG1 and was competed by hsa_circ_0057684, hsa_circ_0056560, IL21-AS1, or LINC00664. The expression of CFAP43 were regulated by hsa-miR-3120-3p which may be sponged by hsa_circ_0007161, hsa_circ_0000996, hsa_circ_0069606, hsa_circ_0027707, hsa_circ_0003677, hsa_circ_0028190, hsa_circ_0052575, hsa_circ_0001513, LINC00504, USP2-AS1, LINC00535, LINC00271, AP000708.1, or DNAJC27-AS1. Two miRNAs, hsa-miR-3157-5p and hsa-miR-1284, targeted KCNQ3. The hsa-miR-3157-5p was competed by hsa_circ_0057252, hsa_circ_0056560, hsa_circ_0007631, AC064874.1, or LINC00664. The hsa-miR-1284 was sponged by hsa_circ_0004511, hsa_circ_0057684, LINC00664, or LINC00943. Then, we used the TRRUST database to predict the potential transcription factor regulating the characteristic genes.22 As shown in Table 1, five transcription factors, namely RELA, YY1, NFKB1, KLF2, and NR3C2, regulated CCR5. Two transcription factors, REST and SP1, regulated KCNQ3. STAT6 regulated FOSB.
 |
Table 1 The List of Transcription Factors Regulating the Characteristic Genes |
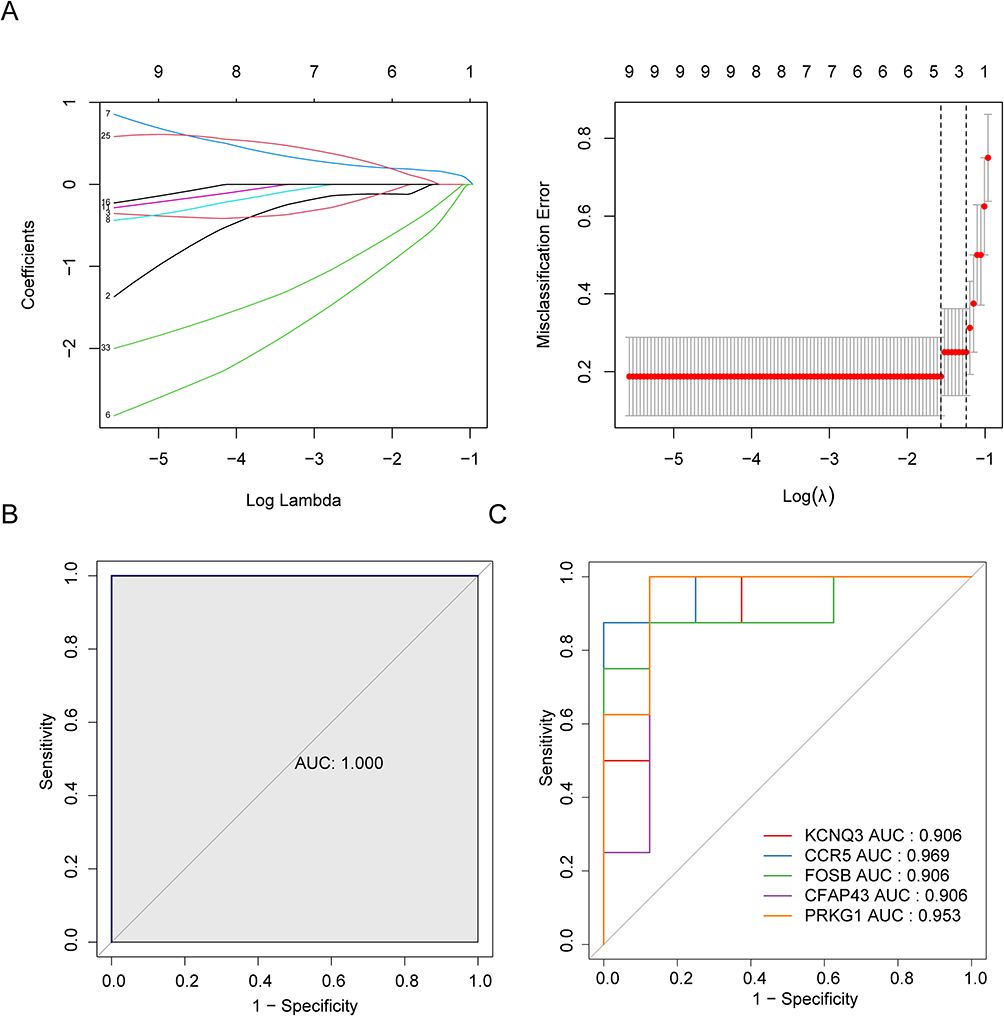 |
Figure 4 Identification of characteristic genes. (A) The characteristic genes were screened by LASSO regression analysis. Left image: The horizontal axis represents log (Lambda), and the vertical axis represents the coefficient of genes (left); Right image: The horizontal axis represents log (Lambda), while the vertical axis represents the error of cross validation (right). In practical analysis, we hope to find the location with the smallest cross validation error. In the right graph, the left dashed line position is the location with the smallest cross validation error. Based on this position (lambda. min), the corresponding horizontal coordinate log (Lambda) is determined. The number of feature genes is displayed on the top, and the optimal log (Lambda) value is found. The corresponding gene and its coefficient are found in the left graph. (B) The ROC curve of LASSO regression model. (C) The single gene ROC curve of characteristic gene. |
 |
Figure 5 The ceRNA network for characteristic genes. The size of the point indicates the connectivity. The greater the point is, the greater the connectivity is. Circular nodes represent miRNA, triangular nodes represent circRNA, square nodes represent mRNA, and diamond nodes represent lncRNA. |
The Gene Interaction Network of the Characteristic Genes
We further predicted the gene interaction network of the characteristic genes by IPA and presented in Figure 6A and B. Red indicated the up-regulated DEmRNA, green indicated the down-regulated DEmRNA, and colorless indicated the predicted component. The five characteristic genes were marked with red boxes. As shown in Figure 6A, CCR5 interacted with immunoglobulin and FOSB may interact with immunoglobulin. PRKG1 interacted with ERK1/2, GUCY1B1, and Tgf beta possibly. The result of Figure 6B indicated that KCNQ3 interacted with KCNQ5 and REST. TGFB1 may interact with KCNQ3. CFAP43 interacted with KPNA2.
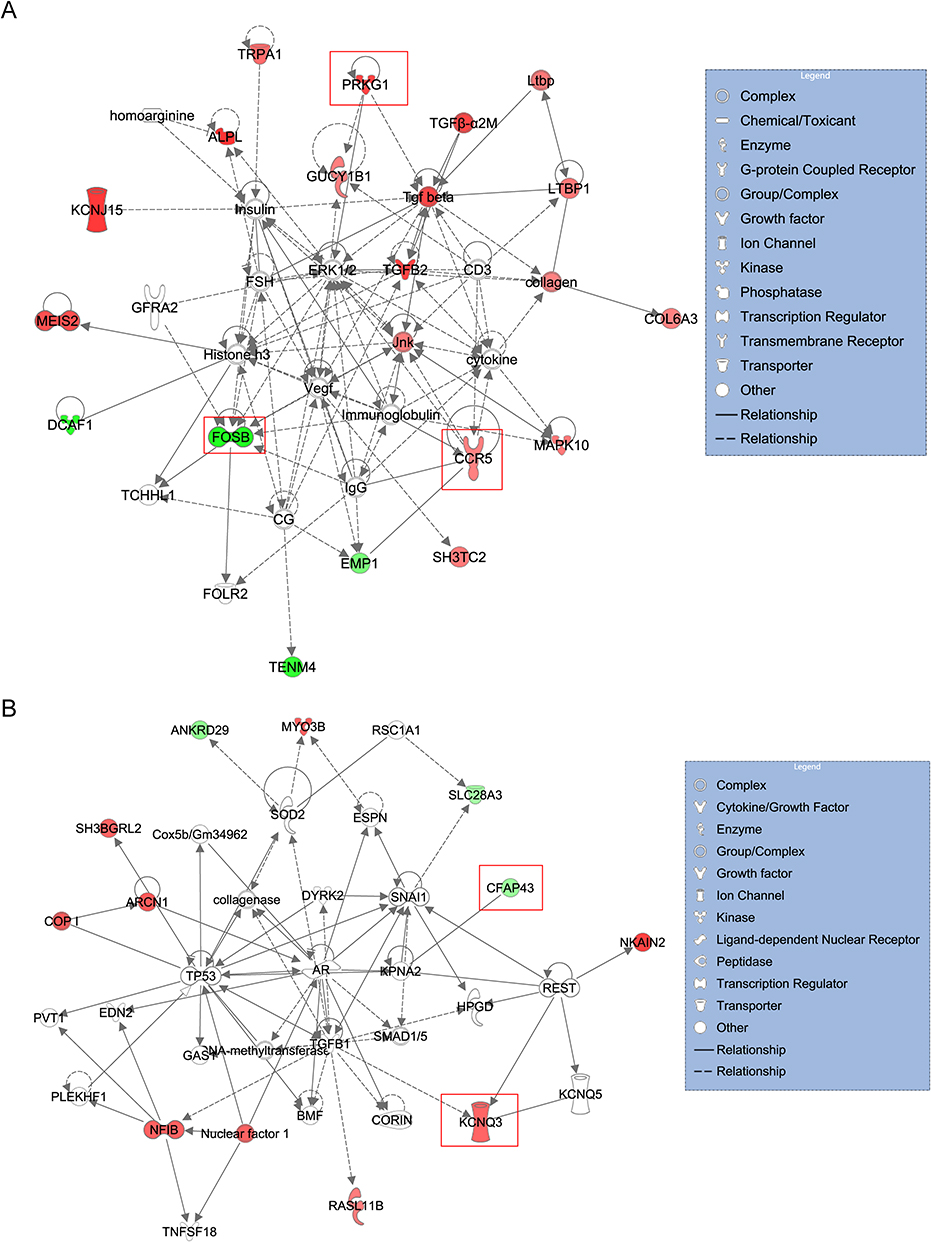 |
Figure 6 The gene interaction network of the characteristic genes predicted by IPA. (A) The gene interaction network of CCR5, FOSB and PRKG1. (B) The gene interaction network of CFAP43 and KCNQ3. Red indicated the up-regulated DEmRNA, green indicated the down-regulated DEmRNA, and colorless indicated the predicted component. The five characteristic genes were marked with red boxes. |
Discussion
Rape flower is one of the most representative ornamental flowers in Yunnan Province. It is planted in most areas of Yunnan Province. Therefore, there are not a few patients with rape flower pollen allergy in Yunnan Province. In order to provide more accurate diagnosis and treatment for patients with rape pollen allergy, we constructed a ceRNA regulatory network based on high-throughput whole-transcriptome sequencing data. The ceRNA mechanism provides effective information for the molecular regulatory mechanism of rape pollen allergy, and may provide candidate genes for revealing the pathogenesis, diagnostic biomarkers or mining potential therapeutic targets.
Through functional and pathway enrichment analysis, we found that the genes encoding proteins in the ceRNA network were mainly involved in some signal transduction processes, immune related processes, ion transport and steady-state processes. IL-17 signal pathway, the regulation of tryptophan channel by inflammatory mediators, and GMP-PKG signal pathway were also enriched. IL-17 cytokine, produced by Th17 cells, plays a pivotal role in host defense reactions, inflammatory diseases and allergic responses.23 Studies have found that IL-17 plays a role in allergic diseases, such as allergic asthma, asthma airway hyperresponsiveness, atopic dermatitis and allergic conjunctivitis. Ciprandi et al24 found that in persistent pollen AR, the expression level of serum IL-17 was positively correlated with the severity of rhinitis. Nasal congestion is thought to be caused by the expansion of cavernous plexus, while the process of vasodilation is mediated by NO, which is considered to be related to endothelium-dependent vascular smooth muscle response. By activating soluble guanylate cyclase (SGC) to release NO and increase the level of cyclic guanosine monophosphate-dependent protein (cGMP), the activity of cGMP-dependent protein kinase (PKG) is enhanced, resulting in the regulation (phosphorylation) of various intracellular proteins and vasodilation.25 Studies have shown that antigen-elicited upregulation of SGC and PKG plays a key role in non-induced nasal mucosal venous dilatation, which may be related to the occurrence of AR nasal obstruction.26 IPA enrichment analysis showed that the genes encoding proteins in ceRNA network were related to the functions of body injury and abnormality, inflammatory response, immune cell transport and respiratory diseases. AR was a slow inflammatory process of upper airway in which inflammatory mediators, immune cells and cytokines interacted.
Based on the genes encoding proteins in ceRNA network, five characteristic genes KCNQ3, CCR5, FOSB, CFAP43 and PRKG were screened by machine learning method. The five characteristic genes had good diagnostic ability and could distinguish between rape flower pollen allergy and non-rape flower pollen allergy samples. KCNQ potassium channel is a kind of voltage-gated potassium channel. There are five subtypes of KCNQ1 ~ KCNQ5, which jointly encode KV7 (KV channels) family (KV7.1-KV7.5).27 The effect of KV7 encoded by KCNQ gene on airway smooth muscle has been confirmed. In human and guinea pig airways, application of KV7 activators and blockers increased and decreased airway diameter, respectively.28 Therefore, KV7 can be regarded as a potential therapeutic target for asthma, but KCNQ3 is involved in coding Kv7, whether it can be speculated that KCNQ3 is also involved in the pathogenesis of asthma.
According to the hypothesis that AR and asthma are “the same airway, the same disease”, it can also be speculated that KCNQ also plays a role in AR, but further research is needed. As a receptor of chemokine, CCR5 is expressed on the cell membrane of T cells, monocytes and dendritic cells. It plays a key role in the infiltration and activation of immune cells in the immune response, regulates the proliferation and migration of T cells, promotes the migration of different subtypes of Th cells in the immune response, and acts as a pro-inflammatory agent.29 In previous studies, the role of CCR5 in Henoch Schonlein purpura and allergic contact dermatitis has been confirmed.30 Of course, its role in AR cannot be ignored. Research findings31 manifested that a large number of Th2 chemokines and their receptors played an important role in the pathogenesis of AR and participated in the induction of inflammation, inflammatory cell aggregation, sensitizing cell degranulation and other processes in allergic reaction. As an important member of AP-1 family, FOSB participates in the proliferation, apoptosis and migration of tumor cells and plays an important role in the occurrence and development of tumor,32 but its role in allergic diseases is not clear. CFAP43 is a highly conserved motor ciliary axon protein, which is closely related to motor cilia and participates in the regulation of the beating frequency of tracheal cilia. The lack of CFAP43 will lead to severe mucus accumulation in the nasal cavity.33 Whether this performance is related to the clinical manifestation of a large amount of clear water like nose in AR needs further research. The role of PRKG1 in AR is expected to be further studied. AR is a process involving goblet cells, T lymphocytes, eosinophils, plasma cells, mast cells and other cells. The miRNA and lncRNA almost participate in the whole process of AR pathogenesis and persistence, including the injury reaction of nasal epithelial cells, antigen presentation of dendritic cells, differentiation and activation of T helper cells, development and recruitment of eosinophils, and degranulation of mast cells in the early stage.34 Studies have shown that35 miR-126 may be involved in the pathogenesis of AR by positively regulating the expression of Treg cytokines and negatively regulating the expression of the Th1 and Th2 cytokines. It can be inferred that these miRNAs also play a key role in AR. LncRNA can regulate immune cell differentiation by interacting with transcription factors, participate in the expression of inflammatory factors in immune response and control inflammatory response.34 At present, the role of circRNA in the pathogenesis of AR has not been clearly reported.
In order to further understand the action mechanism of characteristic genes, we analyzed the proteins interacting with characteristic gene through IPA. DCAF1 is a highly up-regulated factor in activated T cells and plays a key role in controlling T cell function and T cell-mediated antiviral and autoimmune response.36 However, the specific potential mechanism and how DCAF1 regulate T cell-mediated immune response need to be further studied. ERK1/2, as a downstream effector molecule of IL-33, is involved in inducing the inflammatory response of Th2 cells, mast cells, eosinophils, macrophages and dendritic cells.37 Some research results show that38 nasal mucosal epithelial cells in AR model promote the release of cytokines through ERK1/2 pathway. Lnc-PVT1 is a biomarker and its abnormal expression is related to disease severity and Th1/Th2 imbalance. Studies have shown that its overexpression is related to Th1/Th2 imbalance and increased disease severity in AR children.39 TGF-b1 can be either pro-inflammatory or anti-inflammatory in the pathophysiology of allergic diseases. It is important in chemotaxis for inflammatory cells.40 As an inflammatory mucosal disorder of the nose, AR is characterized by the accumulation of mast cells.2 In a preliminary study, we found that TGFB1 gene promoter polymorphism–509C/T is associated with the susceptibility and the severity of persistent AR of Han Chinese.41 Ouyang et al demonstrated that TGF-β1 was activated and mast cells were accumulated during AR progression in mice.42 Salib et al found that epithelial immune response mediated by TGF-β1 was enhanced, intraepithelial mast cells increased, and TGF-β1 receptor co-located in AR patients, suggesting that there was an interaction between TGF-β1 expression and mast cells in AR.43 The relationship between other interacting proteins and AR needs to be further studied.
To sum up, the five characteristic genes screened and the interaction network of related proteins predicted by five genes, combined with the reported related proteins, all play an important role in the pathogenesis of allergic diseases and allergic rhinitis. So far, there is no research on these five genes in rape pollen allergy. Seasonal allergic rhinitis caused by rape pollen allergy is a kind of allergic rhinitis, and it can be boldly speculated that these five genes play a key role in the mechanism of rape pollen allergy. However, this is the first time that we have found the characteristic gene of rape pollen allergy, and its specific pathogenesis needs to be further studied.
Through high-throughput whole-transcriptome sequencing data, we revealed the ceRNA regulatory network of rape pollen allergy, and mined five characteristic genes with diagnostic value, which may become potential targets for diagnosis and treatment. But at the same time, our research also has shortcomings. First, our sample size is small; Secondly, although key genes have been screened, further experiments and clinical verification are needed; Additionally, we will continue to pay attention to the role of these genes.
Abbreviation
AR, allergic rhinitis; ncRNAs, noncoding RNAs; DEmiRNAs, differentially expressed microRNAs; DEcircRNAs, differentially expressed circRNAs; DElncRNAs, differentially expressed long non-coding; DEmRNAs, differentially expressed mRNAs; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; LASSO, least absolute shrinkage and selection operator; ROC, receiver operating characteristic; PBMC, peripheral blood mononuclear cell; PBS, Phosphate Buffered Saline; ceRNA, competing endogenous RNA; AUC, area under the curve; sGC, soluble guanylate cyclase; cGMP, cyclic guanosine monophosphate-dependent protein.
Data Sharing Statement
The data that support the findings of this study are available on request from the corresponding author.
Ethics Approval and Informed Consent
The study was conducted in accordance with the principles outlined in the Helsinki Declaration. The study was approved by the Ethics Committee of the Sixth Affiliated Hospital of Kunming Medical University (Approval No: kmykdx6f). All patients and healthy volunteers had signed an informed consent form.
Consent for Publication
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient/parent/guardian/relative of the patient.
Acknowledgments
This work was supported by the Joint Special Fund for Applied Basic Research of Kunming Medical University (40220136).
Funding
This work was supported by the Joint Special Fund for Applied Basic Research of Kunming Medical University (40220136).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Okubo K, Kurono Y, Ichimura K, et al. Japanese Society of Allergology. Japanese guidelines for allergic rhinitis 2020. Allergol Int. 2020;69(3):331. doi:10.1016/j.alit.2020.04.001
2. Cheng L, Chen J, Fu Q, et al. Chinese Society of Allergy Guidelines for diagnosis and treatment of allergic rhinitis. Allergy Asthma Immunol Res. 2018;10:312–313.
3. Ghafouri-Fard S, Shoorei H, Taheri M, Sanak M. Emerging role of non-coding RNAs in allergic disorders. Biomed Pharmacother. 2020;130:110615. doi:10.1016/j.biopha.2020.110615
4. Specjalski K, Jassem E. MicroRNAs: potential biomarkers and targets of therapy in allergic diseases? Arch Immunol Ther Exp (Warsz). 2019;67(4):213–223. doi:10.1007/s00005-019-00547-4
5. Lemonnier N, Melén E, Jiang Y, et al. A novel whole blood gene expression signature for asthma, dermatitis, and rhinitis multimorbidity in children and adolescents. Allergy. 2020;75(12):3248–3260. doi:10.1111/all.14314
6. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi:10.1093/bioinformatics/19.2.185
7. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
8. Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi:10.1038/s41587-019-0201-4
9. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
10. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi:10.1093/bioinformatics/btt656
11. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. doi:10.1038/nature11928
12. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
13. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
14. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts. Nucleic Acids Res. 2016;44(D1):D231–8. doi:10.1093/nar/gkv1270
15. Liu M, Wang Q, Shen J, Yang BB, Ding X. Circbank: a comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019;16(7):899–905. doi:10.1080/15476286.2019.1600395
16. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020;48(D1):D127–d131. doi:10.1093/nar/gkz757
17. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
18. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
19. Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30(4):523–530. doi:10.1093/bioinformatics/btt703
20. Friedman J, Hastie T, Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22.
21. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011;12:77. doi:10.1186/1471-2105-12-77
22. Han H, Cho JW, Lee S, et al. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46(D1):D380–d386. doi:10.1093/nar/gkx1013
23. Sheha D, El-Korashi L, AbdAllah AM, El Begermy MM, Elzoghby DM, Elmahdi A. Lipid Profile and IL-17A in Allergic Rhinitis: correlation With Disease Severity and Quality of Life. J Asthma Allergy. 2021;14:109–117. doi:10.2147/JAA.S290813
24. Ciprandi G, De Amici M, Murdaca G, et al. Serum interleukin-17 levels are related to clinical severity in allergic rhinitis. Allergy. 2009;64(9):1375–1378. doi:10.1111/j.1398-9995.2009.02010.x
25. Waldman SA, Murad F.Cyclic GMP synthesis and function. Pharmacol Rev. 1987;39(3):163–196.
26. Sakai H, Hara T, Todoroki K, et al. Elevated guanylate cyclase and cyclic-guanosine monophosphate-dependent protein kinase levels in nasal mucosae of antigen-challenged rats. Microvasc Res. 2013;90:150–153. doi:10.1016/j.mvr.2013.08.009
27. Stott JB, Jepps TA, Greenwood IA. K(V)7 potassium channels: a new therapeutic target in smooth muscle disorders. Drug Discov Today. 2014;19(4):413–424. doi:10.1016/j.drudis.2013.12.003
28. Brueggemann LI, Kakad PP, Love RB, et al. Kv7 potassium channels in airway smooth muscle cells: signal transduction intermediates and pharmacological targets for bronchodilator therapy. Am J Physiol Lung Cell Mol Physiol. 2012;302(1):L120–32. doi:10.1152/ajplung.00194.2011
29. Rautenbach A, Williams AA. Metabolomics as an Approach to Characterise the Contrasting Roles of CCR5 in the Presence and Absence of Disease. Int J Mol Sci. 2020;21(4). doi:10.3390/ijms21041472
30. Marques RE, Guabiraba R, Russo RC, Teixeira MM. Targeting CCL5 in inflammation. Expert Opin Ther Targets. 2013;17(12):1439–1460. doi:10.1517/14728222.2013.837886
31. Nur Husna SM, Tan HT, Md Shukri N, Mohd Ashari NS, Wong KK. Allergic Rhinitis: a Clinical and Pathophysiological Overview. Front Med. 2022;9:874114. doi:10.3389/fmed.2022.874114
32. Gazon H, Barbeau B, Mesnard JM, Peloponese JM Jr. Hijacking of the AP-1 Signaling Pathway during Development of ATL. Front Microbiol. 2017;8:2686. doi:10.3389/fmicb.2017.02686
33. Rachev E, Schuster-Gossler K, Fuhl F, et al. CFAP43 modulates ciliary beating in mouse and Xenopus. Dev Biol. 2020;459(2):109–125. doi:10.1016/j.ydbio.2019.12.010
34. Cheng S, Tang Q, Xie S, et al. The Role of Noncoding RNA in Airway Allergic Diseases through Regulation of T Cell Subsets. Mediators Inflamm. 2022;2022:6125698. doi:10.1155/2022/6125698
35. Jia H, Zhang R, Liang X, Jiang X, Bu Q. Regulatory effects of miRNA-126 on Th cell differentiation and cytokine expression in allergic rhinitis. Cell Signal. 2022;99:110435. doi:10.1016/j.cellsig.2022.110435
36. Guo Z, Kong Q, Liu C, et al. DCAF1 controls T-cell function via p53-dependent and -independent mechanisms. Nat Commun. 2016;7:10307. doi:10.1038/ncomms10307
37. Ota K, Kawaguchi M, Matsukura S, et al. Potential involvement of IL-17F in asthma. J Immunol Res. 2014;2014:602846. doi:10.1155/2014/602846
38. Li YQ, Zhong Y, Xiao XP, et al. IL-33/ST2 axis promotes the inflammatory response of nasal mucosal epithelial cells through inducing the ERK1/2 pathway. Innate Immun. 2020;26(6):505–513. doi:10.1177/1753425920918911
39. Sun Y, Han J, Ma H, Ma J, Ren Z. Aberrant expression of long non-coding RNA PVT1 in allergic rhinitis children: correlation with disease risk, symptoms, and Th1/Th2 imbalance. J Clin Lab Anal. 2022;36(4):e24281. doi:10.1002/jcla.24281
40. Tirado-Rodriguez B, Ortega E, Segura-Medina P, et al. TGF-b: an important mediator of allergic disease and a molecule with dual activity in cancer development. J Immunol Res. 2014;2014:318481.
41. Zhu XJ, Lu MP, Chen RX, et al. Polymorphism -509C/T in TGFB1 Promoter Is Associated With Increased Risk and Severity of Persistent Allergic Rhinitis in a Chinese Population. Am J Rhinol Allergy. 2020;34(5):597–603. doi:10.1177/1945892420913441
42. Ouyang Y, Nakao A, Han D, Zhang L. Transforming growth factor-β1 promotes nasal mucosal mast cell chemotaxis in murine experimental allergic rhinitis. ORL J Otorhinolaryngol Relat Spec. 2012;74(3):117–123. doi:10.1159/000328587
43. Salib RJ, Kumar S, Wilson SJ, Howarth PH. Nasal mucosal immunoexpression of the mast cell chemoattractants TGF-beta, eotaxin, and stem cell factor and their receptors in allergic rhinitis. J Allergy Clin Immunol. 2004;114(4):799–806. doi:10.1016/j.jaci.2004.07.010
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.