")
Back to Journals » Biologics: Targets and Therapy » Volume 15
An Overview of the Safety and Efficacy of Monoclonal Antibodies for the Chronic Obstructive Pulmonary Disease
Authors Cazzola M , Ora J , Cavalli F, Rogliani P , Matera MG
Received 25 July 2021
Accepted for publication 19 August 2021
Published 27 August 2021 Volume 2021:15 Pages 363—374
DOI https://doi.org/10.2147/BTT.S295409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Doris Benbrook
Mario Cazzola,1 Josuel Ora,2 Francesco Cavalli,1 Paola Rogliani,1,2 Maria Gabriella Matera3
1Chair of Respiratory Medicine, Department of Experimental Medicine, University of Rome Tor Vergata, Rome, Italy; 2Division of Respiratory Medicine, University Hospital Tor Vergata, Rome, Italy; 3Chair of Pharmacology, Department of Experimental Medicine, University of Campania Luigi Vanvitelli, Naples, Italy
Correspondence: Mario Cazzola Email [email protected]
Abstract: Several mAbs have been tested or are currently under clinical evaluation for the treatment of COPD. They can be subdivided into those that aim to block specific pro-inflammatory and pro-neutrophilic cytokines and chemokines, such as TNF-α, IL-1β, CXCL8 and IL-1β, and those that act on T2-mediated inflammation, respectively, by blocking IL-5 and/or its receptor, preventing IL-4 and IL-13 signaling, affecting IL-33 pathway and blocking TSLP. None of these approaches has proved to be effective, probably because in COPD there is no dominant cytokine or chemokine and, therefore, a single mAb cannot be effective on all pathways. With a more in-depth understanding of the numerous pheno/endotypic pathways that play a role in COPD, it may eventually be possible to identify those specific patients in whom some of these cytokines or chemokines might predominate. In this case, it will be possible to implement a personalized treatment, but the use of each mAb will only be reserved for a very limited number of subjects.
Keywords: COPD, monoclonal antibodies, pheno/endotypic pathways, anti–T1-mediated inflammation mAbs, anti–T2-mediated inflammation mAbs
Introduction
Although COPD is a disorder characterized by the presence of chronic inflammation that is predominantly neutrophilic, there is a rather large proportion of patients with a predominantly eosinophil-mediated inflammation or in whom neutrophilic inflammation is combined to varying degrees with eosinophilic inflammation.1 Other inflammatory cells including macrophages and CD4+ and CD8+ T lymphocytes are also involved, but the extent of their participation varies depending on the patient’s endotype.2 In addition, inflammatory cells are variably distributed in the bronchial tissue between and within individuals with COPD and over time,3 which is certainly critical when treating a patient with COPD, but unfortunately still poorly studied.Table 1
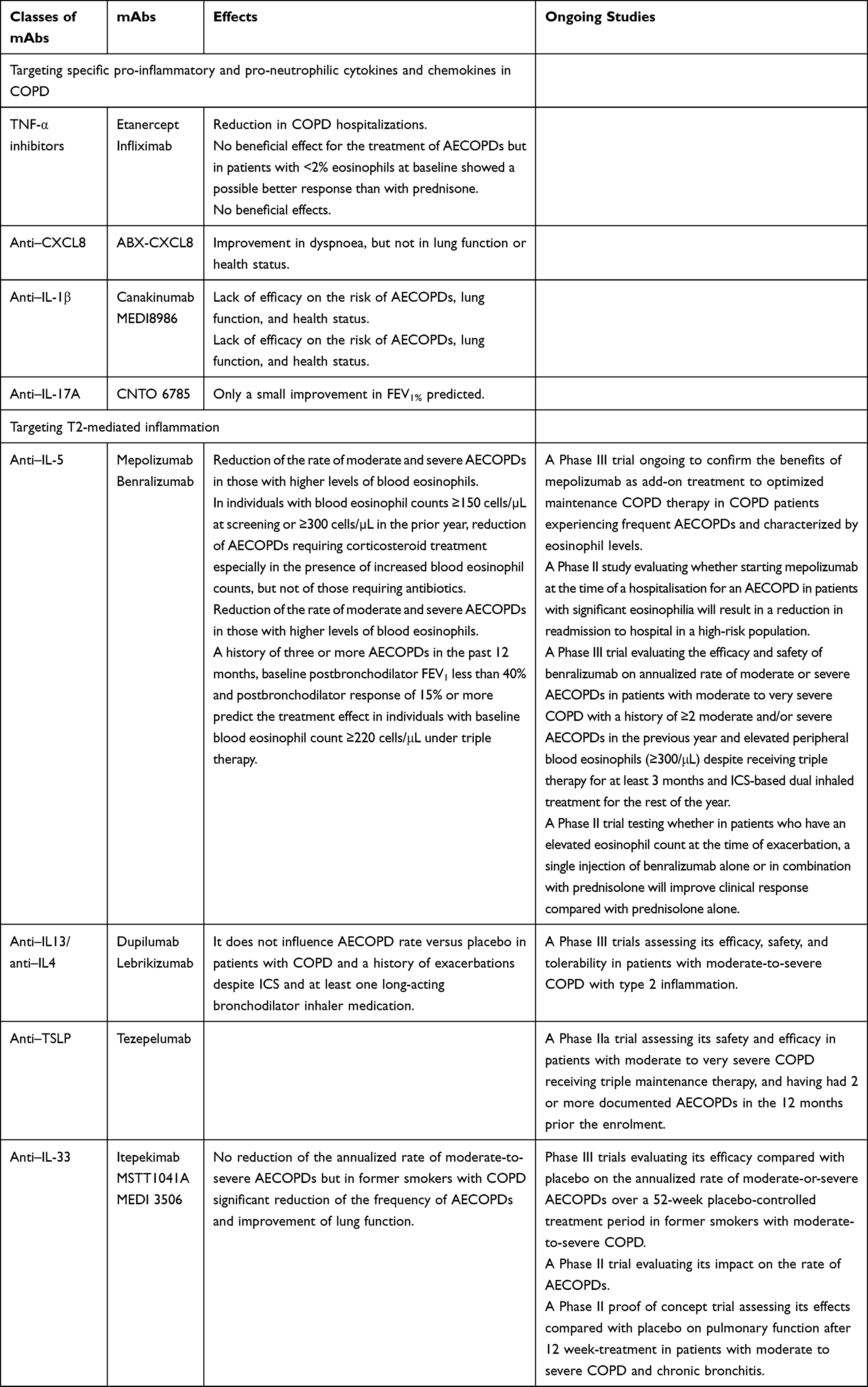 |
Table 1 Trials That Have Evaluated or are Evaluating mAbs in COPD |
Unfortunately, inflammation in COPD lungs responds poorly to corticosteroid treatment. For this reason, over the past two decades, pharmacological research has been moving toward finding new anti-inflammatory pharmacological approaches to treat COPD patients more adequately.4–6 The focus was on finding new targets that inhibit the recruitment and activation of inflammatory cells involved in COPD.7 This is leading to the synthesis of new drugs that should block the inflammatory mediators that recruit or activate these cells or are released by them.7 Unfortunately, many of these approaches have failed to reach the clinical development stage or have failed in the clinic.8
Nevertheless, the improved understanding of the inflammatory process underlying COPD is suggesting the use of biologic agents or biological response modifiers, which act by neutralizing or modulating the function of certain inflammatory mediators such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-4, IL-5, IL-6, IL-8, IL-13, IL-18, IL-23, IL-33, eotaxin-1 (CCL-11), thymic stromal lymphopoietin (TSLP), and transforming growth factor (TGF)-β, in order to achieve a more specific anti-inflammatory action.9 This should implement targeted treatment that improves disease control and decreases exacerbation rates.
Most biological therapies are based on the administration of antibodies against these mediators or their receptors (table 1), although inhibitors, mostly of kinases, are also used.9 Monoclonal antibodies (mAbs) offer several advantages such as high affinity and specificity binding against a wide variety of proteins depending on the target, relative metabolic stability that allows them to remain active for long periods of time with a duration of action of weeks or even months, and low toxicity because their degradation products are amino acids and therefore are not converted into toxic metabolites.10
Targeting Specific Pro-Inflammatory and Pro-Neutrophilic Cytokines and Chemokines in COPD
In neutrophil-associated COPD with activation of the inflammasome, T1 and T17 immunity is the most common phenotype.11,12 Several cytokines are thought to drive neutrophilic inflammation, such as TNF-α, IL-8, IL-17, and IL-23. Human bronchial epithelial cells also express IL-6, IL-17 and IL-22/IL-22R. IL-1β and TNF levels are increased in COPD and are associated with macrophage activation and neutrophilic inflammation.11,12 Macrophages are recruited with activation of the NOD-like receptor protein-3 (NLRP3) inflammasome and caspase-1-dependent release of pro-inflammatory IL-1-like cytokines IL-1α, IL-1β, IL-33, and IL-18.12 Although COPD is primarily characterized by neutrophilic inflammation, studies on targeted biologic therapy for neutrophilic inflammation have been disappointing to date.
TNF-α Inhibitors
A recent systematic review and meta-analysis performed to evaluate the correlation between TNF-α level and COPD reported that TNF-α level was increased in patients with COPD compared with healthy controls.13 Several pollutants, including cigarette smoke, induce TNF-α production by alveolar macrophages, neutrophils, T cells, mast cells, and epithelial cells.14 TNF-α augments neutrophil chemotaxis and migration by inducing IL-8 expression and increasing endothelial adhesion molecules.15 Furthermore, it is a potent activator of NF-κB, capable of amplifying the inflammatory response.16
However, TNF-α inhibitors have shown only limited clinical efficacy. In controlled trials, infliximab, an immunoglobulin (Ig)G mAb that can quickly form stable complexes with the human soluble or membrane form of TNF, and thus terminate the biological activity of TNF, had no beneficial effects in patients with mild, moderate, or severe COPD.17,18 Conversely, in patients with rheumatoid arthritis and COPD, etanercept, a receptor blocker that binds to free-floating and cell-bound TNF, and not infliximab, reduced COPD hospitalizations, but only 16 of 1205 patients (1.3%) in the study were on etanercept.19 In any case, in a double-blind randomized controlled trial, etanercept had no beneficial effect for the treatment of acute exacerbations of COPD (AECOPDs) although patients treated with etanercept and with <2% eosinophils at baseline showed a possibly better response than those treated with prednisone.20
The reason for the failure of anti–TNF therapies in patients with COPD is presumably because other proinflammatory cytokines drive the inflammatory process.15 However, preclinical studies suggest that TNF-α inhibitors could reverse corticosteroid insensitivity by restoring the broad attenuation effects of corticosteroids on inflammation and airway remodelling21 and, also, when combined with corticosteroids, they could induce synergistic effects in controlling airway remodelling.22
Anti–CXCL8 mAbs
IL-8 is a pro-inflammatory chemokine from the CXC family. It is also known as CXCL8 and is a chemokine with potent neutrophil chemotactic and activation properties.23 Patients with COPD have high levels of CXCL8 in sputum and BAL fluid.23 However, results from a Phase II pilot study in COPD patients with ABX-CXCL8, a fully human mAb that recognized only free CXCL8, suggested that neutralization of CXCL8 may improve dyspnoea, but fails to improve lung function or health status.24 It has been suggested that the clinical failure of this mAb is likely due to the active form of CXCL8 is bound to proteoglycans on the endothelial surface.4
PA401 or dnCXCL8, which is a modified recombinant human CXCL8 characterized by an increased binding affinity to glycosaminoglycans that allows it to displace endogenous CXCL8 from glycosaminoglycans expressed at the site of inflammation, can reduce BAL neutrophils and systemic inflammatory markers in a mouse lung inflammation model.25 HuMax-IL8 (previously known as BMS-986253), a fully human IgG1k mAb that binds to free IL-8, has previously been tested in a Phase I clinical trial on cancer patients26 and is currently the object of an ongoing Phase 2 clinical trial in patients affected by severe COVID-19 (NCT04347226). Both PA401 and HuMax-IL8 have not been evaluated in patients with COPD.
Anti–IL-1β mAbs
The IL-1 family consists of two related polypeptides, IL-1α and IL-1β that bind to the same primary receptor IL-1R1, which is expressed by almost all cell types, and can recruit and/or activate a variety of immune cells and immunocompetent cells, such as macrophages, endothelial cells, neutrophils, and epithelial cells.27 The activity of IL-1β in the lung induces a phenotype with typical COPD features, consisting of pulmonary inflammation, emphysema, and airway fibrosis.27 IL-1β secretion is increased in stable and exacerbating COPD28 and is likely involved in the initiation and persistence of inflammation. In COPD IL-1β serum level correlates with clinical aspects of disease severity.29
Because specific pharmacologic blockade of IL-1 activity in COPD may be relevant for limiting inflammation and exacerbations in COPD, canakinumab, which is an anti–IL-1β mAb, and MEDI8986, a fully human IgG2 mAb that binds selectively to IL-1R1, were tested in patients with COPD; however, the results demonstrated an acceptable safety profile but a lack of efficacy at least with regard to impact on the risk of AECOPDs, lung function, and health-related quality of life (HRQoL).30 Nevertheless, it has been highlighted the need for better stratification and targeting of specific disease cohorts and for studies with specific mAbs against IL-1α and IL-1β using lung-specific biomarkers to understand whether targeting these cytokines is a viable therapeutic approach for patients with COPD.31 In addition, it has been suggested that interfering with IL-1 may reduce mucosal inflammatory responses to microbes, which could predispose patients to respiratory tract infections and pneumonia.32
Anti–IL-17A mAbs
The IL-17 family of cytokines has six members, of which IL-17A and IL-17F are highly homologous and bind to a complex receptor of IL-17RA and IL-17RC, so that they share similar biological effects.33 IL-17A and IL-17F are involved in COPD pathophysiology, although a study found evidence of increased production of IL-17A but not IL-17F in the bronchial submucosa of COPD patients.34 IL-17 activates many signalling pathways, which in turn leads to the production of many other cytokines (such as IL-6, IL-1β, TNF-α, granulocyte colony-stimulating factor [G-CSF], granulocyte-macrophage colony-stimulating factor [GM-CSF], and TGF-β) and chemokines (including IL-8 and monocyte chemoattractant protein [MCP1]) from many alveolar cell types (endothelial cells, epithelial cells, and macrophages).35 The increase in the production of IL-17A causes neutrophil recruitment, leading to chronic inflammation.36 IL-17A can also promote airway remodelling by increasing TGF-β1 and inhibiting cell autophagy in inflammatory lung tissue.36 IL-17 airway activity positively correlates with disease severity in COPD patients.36
Even though different models of experimental COPD suggest that IL-17A drives lung inflammation and lung damage, targeting IL-17A has, so far, not been proven to be useful in the treatment of stable COPD patients.37 CNTO 6785, a mAb that binds to human IL-17A, caused a small improvement in forced expiratory volume in 1 s (FEV1)% predicted in patients with moderate-to-severe symptomatic COPD, but failed to show any statistically or clinically significant difference in primary or secondary end points between the intervention and placebo arms. In addition, there were more protocol defined AECOPDs in the CNTO 6785 arm compared with placebo at week 16 (18 vs 11).38
A number of anti–IL-17A and anti–IL-17RA mAbs are under development to decrease neutrophil recruitment and airway inflammation.8 However, most studies have been and are aimed at testing the effects of these mAbs on asthma rather than COPD, likely because IL-17 secreted by Th17 cells, plays a key role in the pathogenesis of neutrophilic asthma, which is unresponsive to high dose inhaled corticosteroids (ICSs), and probably to precision novel anti-IgE, and IL mAb therapies.39 A blockade of IL-17RA resulting in the inhibition of the effects of both IL-17A and IL-17F has been attempted in patients with severe asthma using brodalumab, which is a mAb that targets IL-17RA.40 Although it did not induce any improvement in lung function, symptoms, or QoL, the study population was not screened for neutrophils in sputum or mediators of the IL-17 pathway. A study (NCT01902290) that aimed to assess whether brodalumab was safe and effective compared to placebo as measured by changes in Asthma Control Questionnaire (ACQ) composite scores in inadequately controlled asthma subjects with high bronchodilator reversibility but not selected based on matching endotype was stopped due to lack of efficacy. Conversely, secukinumab, a fully human anti–IL-17A of the IgG1κ isotype, seemed to work in those patients suffering from not adequately controlled severe asthma despite high doses of ICSs and LABAs.41 These patients had low IgE (<150/uL) and increased nasal epithelial neutrophilic inflammation. Responders were defined as >5% change from baseline of percent predicted FEV1. However, treatment with secukinumab was terminated, as it was not effective in the target population.42
CCJM112, a novel fully human anti–IL-17A IgG1κ mAb that, in contrast to secukinumab, binds with similar affinity to both human IL-17A and IL-17A, has already been studied in a phase II trial in patients with uncontrolled severe asthma and low blood IgE and eosinophil levels (NCT03299686). The study has been completed, but results have not been published yet.
Brodalumab, secukinumab and CCJM112 have not yet been tested in patients with COPD. The lack of interest in evaluating these mAbs in COPD is surprising. as there is documentation that a signature of IL-17-associated airway inflammation is upregulated in approximately a third of COPD participants and is associated with distinct inflammatory, physiologic, and clinical features.43 It has been suggested that targeting IL-17 in COPD may therefore be therapeutically useful if it is aimed at well-characterised neutrophilic patients who may respond well to these drugs, possibly identified by nasal sampling performed to detect those with high IL-17 levels.44 However, it must be highlighted that secukinumab was unable to inhibit ozone-induced acute neutrophilic airway inflammation in healthy volunteers.45 Furthermore, targeting the IL-17 pathway may increase the risk of AECOPD by diminishing host defense.8 Neutralizing IL-17 activity can induce immunosuppression, which is problematic in patients with COPD given their susceptibility to pulmonary infections.43 Not insignificant, in this regard, is the concern that blocking the IL-17/IL-23 axis, which plays an important role in defending the lungs against bacterial infections through the release of antimicrobial peptides from airway epithelial cells, may cause reduced immunity to infections.46
Targeting T2-Mediated Inflammation Using mAbs
In COPD, patients may have increased eosinophil counts in peripheral blood and/or sputum that might predict a clinical response to ICSs, with an improvement in post-bronchodilator FEV1 following treatment.47,48 Although the T2 immune response, which encompasses eosinophilic inflammation and is mediated by IL-4, IL-5 and IL-13,2 typically decreases with aging, tobacco smoking may attenuate the age-related decrease in total serum IgE levels and eosinophilic inflammation in populations with and also without asthma.49 This immunologic picture in a smoking subject is termed asthma-COPD overlap (ACO), although controversy exists regarding the precise definition of ACO also because there are patients with neutrophil predominant ACO.50 The high percentage of patients with COPD who show a T2 inflammation explains the interest in evaluating the effects of specific mAbs able to interfere with eosinophils.6 However, it should always be considered that blood eosinophil counts can be variable in individual COPD patients.51 Consequently, classifying COPD as noneosinophilic, with intermediate eosinophilic picture, or clearly eosinophilic considering the three thresholds <100 cells/mm3, 100–299 cells/mm3, and ≥300 cells/mm3, respectively, suggested by the Global Initiative for Chronic Obstructive Lung Disease52 may be incorrect if repeated counts of eosinophils in the blood over time are not performed.
Anti–IL-5 mAbs
IL-5 modulates the proliferation, maturation, and activation of eosinophils and eosinophilic airway inflammation and appears to modulate the development and functions of human basophils and mast cells owing to the common expression of several crucial receptors in these cells.53 Its biological effects are mediated by its selective interaction with the IL-5 receptor (IL-5R), consisting of a specific α subunit (IL-5Rα) and a non-specific βc heterodimer.53
It has been reported that sputum concentrations of IL-5 are correlated with sputum eosinophil numbers in COPD patients with eosinophilia.54 Furthermore, soluble IL-5Rα is increased during virus-induced AECOPDs.55 Since eosinophils rapidly undergo apoptosis in the absence of IL-5, it is believed that blocking IL-5 may be a potential therapeutic approach to prevent or dampen eosinophil-mediated inflammation.56 The efficacy and safety of mepolizumab, a humanized mAb of the IgG1k class that blocks free IL-5, and benralizumab, a fully humanized afucosylated IgG1κ mAb that binds to a specific epitope within the extracellular domain on IL-5Rα, which is in close proximity to the IL-5 binding site and thus inhibits IL-5R signalling, independent of ligand, which are currently approved for treatment of severe eosinophilic asthma, have now been examined in large clinical trials in the eosinophilic COPD population. Both mepolizumab and benralizumab had a similar safety profile that was not significantly different compared to placebo.30
A recent Cochrane systematic review that included six studies involving a total of 5542 participants concluded that mepolizumab and benralizumab probably reduce the rate of moderate and severe AECOPDs in the highly selected group of people who have both COPD and higher levels of blood eosinophils (patients with blood eosinophils at the higher end of the normal range as well as those with true blood eosinophilia).57
However, it should be noted that it is still unknown which subpopulation of COPD patients is most likely to respond to anti-IL-5 mAbs,58 although they are most likely to be those with a higher disease burden and a higher degree of eosinophilic inflammation. A pre-specified meta-analysis of data of individuals with blood eosinophil counts ≥150 cells/µL at screening or ≥300 cells/µL in the prior year from two trials that looked at mepolizumab in COPD showed that mepolizumab had primarily reduced AECOPDs requiring corticosteroid treatment especially in the presence of increased blood eosinophil counts, while the effect on exacerbations requiring antibiotics was less pronounced.59 An analysis of two Phase 3 clinical trials testing the effects of benralizumab in patients with moderate to very severe COPD found that a history of three or more AECOPDs in the past 12 months, baseline postbronchodilator FEV1 less than 40% and postbronchodilator response of 15% or more were the strongest and most consistent baseline clinical characteristics that appeared to predict the treatment effect with this mAb in individuals with baseline blood eosinophil count ≥220 cells/μL and who were receiving triple therapy.60
However, the interest in correctly positioning anti–IL-5 Abs in the treatment of COPD has not yet died down. A Phase III trial is ongoing to confirm the benefits of mepolizumab as add-on treatment to optimized maintenance COPD therapy in COPD patients experiencing frequent AECOPDs and characterized by high eosinophil levels (NCT04133909 or MATINEE), while a Phase II study is evaluating whether starting mepolizumab at the time of a hospitalisation for an AECOPD in patients with significant eosinophilia will result in a reduction in readmission to hospital (NCT04075331 or COPD-HELP).
A Phase III trial that aims to evaluate the efficacy and safety of benralizumab on annualized rate of moderate or severe AECOPDs in patients with moderate to very severe COPD with a history of ≥2 moderate and/or severe AECOPDs in the previous year and elevated peripheral blood eosinophils (≥300/μL) despite receiving triple (ICS/LABA/LAMA) background therapy for at least 3 months and ICS-based dual inhaled treatment for the rest of the year is ongoing (NCT04053634 or RESOLUTE). In contrast, a Phase II trial is testing whether in patients who have an elevated eosinophil count at the time of exacerbation, a single injection of benralizumab alone or in combination with prednisolone will improve clinical response compared with prednisolone alone (NCT04098718 or ABRA).
In any case, the presence of resident human eosinophils in the lungs that are independent of IL-561 prevents massive removal of eosinophils from the airways from inducing any significant clinical benefit in many patients with COPD.62 Therefore, we cannot exclude that airway eosinophilia in COPD may be driven in an IL-5-independent manner and be different from that found in asthma.
Anti–IL13/Anti–IL4 mAbs
IL-4 and IL-13, which are made by mast cells, T2 cells, and innate lymphoid 2 cells (ILC2s), bind to the type 2 receptor complex, IL-4Rα/IL-13Rα1, on airway epithelial and smooth muscle cells, eosinophils, and mast cells.63 IL-4 also binds to the type 1 receptor complex found on T cells, which consists of IL-4Rα and a γc-chain. This leads to upregulation of T2 responses with accumulation of eosinophils and downregulation of T1 responses. IL-13 directly affects airway contractility. It also increases mucus production, and by stimulating periostin release from airway epithelial cells, may contribute to remodelling.
Experimentally in a transgenic mouse model, it has been observed that eosinophil-derived IL-13 stimulates alveolar macrophages to produce matrix metalloprotease (MMP)-12 and, thus, plays a role in alveolar destruction.64 At the same time, it was documented that in patients with eosinophilic COPD and coexisting emphysema, pulmonary eosinophilia was associated with elevated levels of MMP-12, predictive of emphysema.64
Two pivotal studies (NCT03930732 or BOREAS and NCT04456673 or NOTUS) are assessing the efficacy, safety, and tolerability of dupilumab, a fully human IgG4 mAb that targets IL4Rα thus inhibiting signalling of both IL-4 and IL-13, in patients with moderate-to-severe COPD with type 2 inflammation.
Lebrikizumab, a humanised mAb that binds to soluble IL-13 and blocks activation of IL-4Rα and IL-13Rα1 heterodimers, has been evaluated in patients with COPD and a history of exacerbations despite ICS and at least one long-acting bronchodilator inhaler medication (NCT02546700 or VALETA). The full results are yet to be released, but preliminary data suggest that they do not influence AECOPD rate versus placebo.55 There is no information on the use of tralokinumab, a human IgG4 mAb that neutralizes IL-13 and prevents interaction with its receptor, in COPD patients.
Anti–TSLP mAbs
Thymic stromal lymphopoietin (TSLP), an IL-7-like cytokine, is an upstream epithelial T2 cytokine known to exert multipotential pathogenic effects also beyond T2 inflammation.65 It initiates the intracellular T2 signalling by binding to its high-affinity heterodimer receptor complex, which consists of its specific receptor, TSLPR, and an IL-7Rα subunit in cells co-expressing TSLPR and IL-7Rα. TSLP initially binds to TSLPR, followed by recruiting of the IL-7Rα chain.65 TSLP is produced by airway epithelial cells during inflammation and induces T2 immune response by directly stimulating T cells, mast cells, and natural killer cells and indirectly by activation of CD11c+ dendritic cells located within the lung epithelium that then migrate to the lymph nodes where they prime CD4+ T cells to produce T2 inflammatory cytokines.66
Constitutive and in vivo expression of TSLP has been documented in the airway smooth muscle of COPD patients.67 It is likely that this TSLP expression could influence immune regulation by interacting with and influencing local immune cells in the COPD airways.68 Furthermore, viruses can induce overproduction of TSLP in COPD epithelial cells,69 suggesting a role for TSLP in AECOPDs. However, also T1 cytokines can induce TSLP production in COPD and it has been speculated that dendritic cell-derived TSLP acts as an important molecular checkpoint to limit IL-1β-mediated effector responses through a negative feedback loop that may limit the magnitude of the inflammatory response to injury.70
Tezepelumab, a human IgG2 mAb that binds to TSLP, preventing its interaction with the TSLP receptor complex,71 is under investigation in a phase IIa, multicentre, double-blind, randomised trial in patients with moderate to very severe COPD receiving triple inhaled maintenance therapy, and having had 2 or more documented AECOPDs in the 12 months prior the enrolment (NCT04039113 or COURSE).
Targeting IL-33 Pathway
IL-33, an alarmin cytokine from the IL-1 family, is released from the epithelium due to damage to epithelial cells and exerts its pro-inflammatory biological functions via its receptor, which is a heterodimeric complex that comprises suppression of tumorigenicity 2 (ST2) and IL-1 receptor accessory protein (IL-1RAcP).72 The resulting signal leads to the induction of transcription of downstream inflammatory and anti-inflammatory genes. IL-33 has been implicated in eosinophil recruitment to the airway and maturation in the bone marrow largely via ILC2, and thus it promotes the ILC2 response, which secretes a large amount of IL-5 and IL-13 in allergic inflammation.73
It is believed that IL-33 could play a role in the pathogenesis and progression of COPD because substantial increases in IL-33 levels have been documented in serum or plasma and sputum as well as in lung biopsy specimens, epithelial and endothelial cells of COPD patients.74 The expression of IL-33 and the ST2 receptor increases in this disorder probably due to the inducing action of cigarette smoke, with levels that are associated with the degree of airway and systemic inflammation.75 In effect, a reduction in COPD risk associated with IL33 (rs146597587) loss of function and, on the contrary, gains of function in IL33 and IL1RL1 variants with increased risk have been documented.73
A Phase II study has evaluated the impact of itepekimab, an anti–IL-33 human IgG4 mAb, as an add-on to the standard of care on the annualized rate of moderate-to-severe AECOPDs over up to 52 weeks of treatment.76 Compared with placebo, itepekimab did not significantly reduce the annualized rate of moderate-to-severe AECOPDs. However, in former smokers with COPD, it significantly reduced the frequency of exacerbations and improved lung function, again compared with placebo.
Another Phase II trial has evaluated the impact of MSTT1041A, an anti-ST2 mAb, administered subcutaneously by an infusion pump at 490 mg every 4 weeks over a 48-week treatment period, on the rate of AECOPDs (NCT03615040 or COPD-ST2OP), but no result has been posted.
Other RCTs are ongoing. Itepekimab is now under further investigation in two Phase III trials to evaluate its efficacy compared with placebo on the annualized rate of moderate-or-severe AECOPDs over a 52-week placebo-controlled treatment period in former smokers with moderate-to-severe COPD (NCT04701983 or AERIFY-1 and NCT04751487 or AERIFY-2). MEDI 3506, another anti–IL-33 mAb, is in a Phase II proof-of-concept trial that is assessing its effects compared with placebo on pulmonary function after 12 weeks of treatment in patients with moderate-to-severe COPD and chronic bronchitis (NCT04631016 or FRONTIER-4).
Administration of mAbs by Inhalation
It is possible that the low efficacy of mAbs is, at least in part, due to the fact that they are large molecules that are administered systemically and reach the lung only in a small percentage of the administered dose.10,77 It is therefore conceivable that their administration by inhalation may increase the proportion of active drug in the lung with limited passage of the drug into the bloodstream.78 However, the pulmonary delivery of mAbs is challenging in terms of aerosol technology and the formulation of biological agents for inhalation.78 It is important that mAbs remain stable during aerosolization. It is likely that mesh nebulizers allow for delivery of high amounts of drugs (often required for mAbs) and better preserve the molecular integrity of proteins by being less harsh regarding chemical and physical constraints.79 Obviously, the addition of surfactant to maintain the molecular integrity and, thus, the pharmacological activity of mAbs during vibrating net nebulizing is necessary.77 It is therefore desirable that trials comparing the effects of a systemically administered mAb with those induced by the same mAb administered by inhalation be performed as soon as possible. However, only mAbs with very high doses potency are suitable for pulmonary delivery because only small volumes of fluid can be administered.80
Conclusion
As we have repeatedly pointed out, there are different pheno/endotypes of COPD whose presence makes a personalized therapeutic approach to the COPD patient crucial and the generalization of results of clinical trials that have not considered this issue of no real value.6,81,82
It is likely, therefore, that the lack or near lack of therapeutic effect of the various mAbs tested in different RCTs reflects the complexity of COPD with its numerous pheno/endotypic pathways playing a role in COPD.6,82 In fact, in COPD, there is no dominant cytokine or chemokine and, therefore, a single mAb cannot be effective on all pathways. This makes it essential to evaluate these mAbs in specific well-identified pheno/endotypes in which some of these cytokines or chemokines might predominate, such as in eosinophilic COPD.6,82
The redundancy of signal-induced effects, particularly the possibility that other pathways can still induce or maintain the inflammatory state even when a specific pathway is turned off, represents a high critical point that must always be considered when evaluating the effects of mAbs in COPD.83 It is the most likely cause of failure when blocking a single specific pathway and is, furthermore, a major obstacle to the development of targeted therapies.6
In general, almost all available studies on the use of mAbs in COPD focus on the prevention of AECOPDs. However, there are several other outcomes that also need to be considered when treating a patient with COPD.84 Cochrane's analysis of anti-IL-5 therapies for COPD has concluded that there was probably little or no difference between the intervention and placebo for quality-of-life measures.57 Apparently, there is also no major impact of these mAbs on lung function.85 For other classes of mAbs, information is even more scarce. There is no doubt that in the future it will be necessary to study in detail the impact of mAbs on other key outcomes such as pulmonary function, as important differences may also be detected. For example, an experimental study showed that benralizumab was more potent than mepolizumab in increasing levels of cAMP in the airways,86 which may mean a different impact on lung function. Another experimental study documented that activation of the IL-4/IL-13 pathway can promote profound airway smooth muscle hyperreactivity, while neither IL-5 nor IL-17A enhanced airway contractile responses.87 Thus, the impact of blocking these cytokines with mAbs may result in completely different bronchial tone responses.
Although we completely agree that mAbs could be the future of personalized treatment in COPD,82 we are fully aware that there is a long way to go before they become part of everyday practice in COPD.88 In any case, our opinion is that mAbs will be reserved for a very limited number of patients suffering from COPD.82 Perhaps, however, if the therapeutic approach is centered on treatable traits as suggested for the treatment of COPD in general89 and eosinophilic forms in particular,90 new studies focusing on outcomes other than exacerbations may provide information that could broaden the range of those who may benefit from these drugs.
Funding
No funding was utilized in the creation of this review.
Disclosure
Professor Mario Cazzola reports grants and personal fees from Almirall, Boehringer Ingelheim, Novartis, and Zambon and personal fees from ABC Farmaceutici, AstraZeneca, Biofutura, Chiesi Farmaceutici, Cipla, Edmond Pharma, GlaxoSmithKline, Lallemand, Menarini, Mundipharma, Ockham Biotech, Pfizer, Sanofi, and Verona Pharma, outside the submitted work. Professor Paola Rogliani reports grants, personal fees, non-financial support from Almirall, AstraZeneca, Biofutura, Boehringer Ingelheim, Chiesi Farmaceutici, GlaxoSmithKline, Menarini Group, Mundipharma, and Novartis, and her department was funded by Almirall, Boehringer Ingelheim, Chiesi Farmaceutici Novartis, and Zambon. Professor Maria Gabriella Matera reports grants and personal fees from GlaxoSmithKline, and Novartis and personal fees from ABC Farmaceutici, AstraZeneca, and Chiesi Farmaceutici, outside the submitted work The authors declare no other competing interests in this work.
References
1. David B, Bafadhel M, Koenderman L, De Soyza A. Eosinophilic inflammation in COPD: from an inflammatory marker to a treatable trait. Thorax. 2021;76(2):188–195. doi:10.1136/thoraxjnl-2020-215167
2. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74(7):1249–1256.
3. Gamble E, Qiu Y, Wang D, et al. Variability of bronchial inflammation in chronic obstructive pulmonary disease: implications for study design. Eur Respir J. 2006;27(2):293–299. doi:10.1183/09031936.06.00027705
4. Cazzola M, Page CP, Calzetta L, Matera MG. Emerging anti-inflammatory strategies for COPD. Eur Respir J. 2012;40(3):724–741. doi:10.1183/09031936.00213711
5. Barnes PJ. Identifying molecular targets for new drug development for chronic obstructive pulmonary disease: what does the future hold? Semin Respir Crit Care Med. 2015;36(4):508–522. doi:10.1055/s-0035-1555611
6. Matera MG, Calzetta L, Annibale R, Russo F, Cazzola M. Classes of drugs that target the cellular components of inflammation under clinical development for COPD. Expert Rev Clin Pharmacol. 2021;14(8):1015–1027. doi:10.1080/17512433.2021.1925537
7. Cazzola M, Matera MG. Is it time to look beyond bronchodilators and corticosteroids in treating COPD? Future Drug Discov. 2021;3(2):FDD61. doi:10.4155/fdd-2021-0001
8. Matera MG, Cazzola M, Page C. Prospects for COPD treatment. Curr Opin Pharmacol. 2021;56:74–84. doi:10.1016/j.coph.2020.11.003
9. Gea J. The future of biological therapies in COPD. Arch Bronconeumol. 2018;54(4):185–186. doi:10.1016/j.arbr.2018.02.013
10. Matera MG, Page C, Rogliani P, Calzetta L, Cazzola M. Therapeutic monoclonal antibodies for the treatment of chronic obstructive pulmonary disease. Drugs. 2016;76(13):1257–1270. doi:10.1007/s40265-016-0625-9
11. Lange P, Ahmed E, Lahmar ZM, Martinez FJ, Bourdin A. Natural history and mechanisms of COPD. Respirology. 2021;26(4):298–321. doi:10.1111/resp.14007
12. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. 2019;54(2):1900651. doi:10.1183/13993003.00651-2019
13. Yao Y, Zhou J, Diao X, Wang S. Association between tumor necrosis factor-α and chronic obstructive pulmonary disease: a systematic review and meta-analysis. Ther Adv Respir Dis. 2019;13:1753466619866096. doi:10.1177/1753466619866096
14. Churg A, Wang RD, Tai H, Wang X, Xie C, Wright JL. Tumor necrosis factor-alpha drives 70% of cigarette smoke-induced emphysema in the mouse. Am J Respir Crit Care Med. 2004;170(5):492–498. doi:10.1164/rccm.200404-511OC
15. Matera MG, Calzetta L, Cazzola M. TNF-a inhibitors in asthma and COPD: we must not throw the baby out with the bath water. Pulm Pharmacol Ther. 2010;23(2):121–128. doi:10.1016/j.pupt.2009.10.007
16. Zhu NL, Li C, Huang HH, et al. TNF-alpha represses transcription of human Bone Morphogenetic Protein-4 in lung epithelial cells. Gene. 2007;393(1–2):70–80. doi:10.1016/j.gene.2007.01.016
17. van der Vaart H, Koëter GH, Postma DS, Kauffman HF, Ten Hacken NH. First study of infliximab treatment in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(4):465–469. doi:10.1164/rccm.200501-147OC
18. Rennard SI, Fogarty C, Kelsen S, et al. COPD Investigators. The safety and efficacy of infliximab in moderate to severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(9):926–934. doi:10.1164/rccm.200607-995OC
19. Suissa S, Ernst P, Hudson M. TNF-alpha antagonists and the prevention of hospitalisation for chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2008;21(1):234–238. doi:10.1016/j.pupt.2007.03.003
20. Aaron SD, Vandemheen KL, Maltais F, et al. TNFα antagonists for acute exacerbations of COPD: a randomised double-blind controlled trial. Thorax. 2013;68(2):142–148. doi:10.1136/thoraxjnl-2012-202432
21. Dejager L, Dendoncker K, Eggermont M, et al. Neutralizing TNFα restores glucocorticoid sensitivity in a mouse model of neutrophilic airway inflammation. Mucosal Immunol. 2015;8(6):1212–1225. doi:10.1038/mi.2015.12
22. Yilmaz O, Karaman M, Bagriyanik HA, et al. Comparison of TNF antagonism by etanercept and dexamethasone on airway epithelium and remodeling in an experimental model of asthma. Int Immunopharmacol. 2013;17(3):768–773. doi:10.1016/j.intimp.2013.08.021
23. Mukaida N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am J Physiol Lung Cell Mol Physiol. 2003;284(4):L566–77. doi:10.1152/ajplung.00233.2002
24. Mahler DA, Huang S, Tabrizi M, Bell GM. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest. 2004;126(3):926–934. doi:10.1378/chest.126.3.926
25. Adage T, Del Bene F, Fiorentini F, et al. PA401, a novel CXCL8-based biologic therapeutic with increased glycosaminoglycan binding, reduces bronchoalveolar lavage neutrophils and systemic inflammatory markers in a murine model of LPS-induced lung inflammation. Cytokine. 2015;76(2):433–441. doi:10.1016/j.cyto.2015.08.006
26. Bilusic M, Heery CR, Collins JM, et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019;7(1):240. doi:10.1186/s40425-019-0706-x
27. Rogliani P, Calzetta L, Ora J, Matera MG. Canakinumab for the treatment of chronic obstructive pulmonary disease. Pulm Pharmacol Ther. 2015;31:15–27. doi:10.1016/j.pupt.2015.01.005
28. Fu JJ, McDonald VM, Baines KJ, Gibson PG. Airway IL-1β and systemic inflammation as predictors of future exacerbation risk in asthma and COPD. Chest. 2015;148(3):618–629. doi:10.1378/chest.14-2337
29. Singh B, Arora S, Khanna V. Association of severity of COPD with IgE and interleukin-1 beta. Monaldi Arch Chest Dis. 2010;73(2):86–87.
30. Rogliani P, Matera MG, Puxeddu E, et al. Emerging biological therapies for treating chronic obstructive pulmonary disease: a pairwise and network meta-analysis. Pulm Pharmacol Ther. 2018;50:28–37. doi:10.1016/j.pupt.2018.03.004
31. Osei ET, Brandsma CA, Timens W, Heijink IH, Hackett TL. Current perspectives on the role of interleukin-1 signalling in the pathogenesis of asthma and COPD. Eur Respir J. 2020;55(2):1900563. doi:10.1183/13993003.00563-2019
32. Brusselle G, Bracke K. Targeting immune pathways for therapy in asthma and chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2014;11(Suppl 5):S322–8. doi:10.1513/AnnalsATS.201403-118AW
33. Le Rouzic O, Pichavant M, Frealle E, Guillon A, Si-Tahar M, Gosset P. Th17 cytokines: novel potential therapeutic targets for COPD pathogenesis and exacerbations. Eur Respir J. 2017;50(4):1602434. doi:10.1183/13993003.02434-2016
34. Di Stefano A, Caramori G, Gnemmi I, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316–324. doi:10.1111/j.1365-2249.2009.03965.x
35. Shibabaw T. Inflammatory Cytokine: IL-17A Signaling Pathway in Patients Present with COVID-19 and Current Treatment Strategy. J Inflamm Res. 2020;13:673–680. doi:10.2147/JIR.S278335
36. Liu M, Wu K, Lin J, et al. Emerging biological functions of IL-17A: a new target in chronic obstructive pulmonary disease? Front. Pharmacol. 2021;12:695957.
37. Ritzmann F, Beisswenger C. Preclinical studies and the function of IL-17 cytokines in COPD. Ann Anat. 2021;237:151729. doi:10.1016/j.aanat.2021.151729
38. Eich A, Urban V, Jutel M, et al. A randomized, placebo-controlled phase 2 trial of CNTO 6785 in chronic obstructive pulmonary disease. COPD. 2017;14(5):476–483. doi:10.1080/15412555.2017.1335697
39. Seys SF, Lokwani R, Simpson JL, Bullens DMA. New insights in neutrophilic asthma. Curr Opin Pulm Med. 2019;25(1):113–120. doi:10.1097/MCP.0000000000000543
40. Kolls JK, Kanaly ST, Ramsay AJ. Interleukin-17: an emerging role in lung inflammation. Am J Respir Cell Mol Biol. 2003;28(1):9–11. doi:10.1165/rcmb.2002-0255PS
41. Khokhlovich E, Grant S, Kazani S, et al. The biological pathways underlying response to anti-IL-17A (AIN457; secukinumab) therapy differ across severe asthmatic patients [abstract]. Eur Respir J. 2017;50(suppl61):OA2897.
42. Shastri MD, Chong WC, Dua K, et al. Emerging concepts and directed therapeutics for the management of asthma: regulating the regulators. Inflammopharmacology. 2021;29(1):15–33.
43. Christenson SA, van den Berge M, Faiz A, et al. An airway epithelial IL-17A response signature identifies a steroid-unresponsive COPD patient subgroup. J Clin Invest. 2019;129(1):169–181. doi:10.1172/JCI121087
44. Baker JR, Donnelly LE. Leukocyte Function in COPD: clinical Relevance and Potential for Drug Therapy. Int J Chron Obstruct Pulmon Dis. 2021;16:2227–2242. doi:10.2147/COPD.S266394
45. Kirsten A, Watz H, Pedersen F, et al. The anti-IL-17A antibody secukinumab does not attenuate ozone-induced airway neutrophilia in healthy volunteers. Eur Respir J. 2013;41(1):239–241. doi:10.1183/09031936.00123612
46. Barnes PJ. Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap syndromes. J Allergy Clin Immunol. 2015;136(3):531–545. doi:10.1016/j.jaci.2015.05.052
47. Rogliani P, Ora J, Puxeddu E, Cazzola M. Airflow obstruction: is it asthma or is it COPD? Int J Chron Obstruct Pulmon Dis. 2016;11:3007–3013. doi:10.2147/COPD.S54927
48. Brightling CE, McKenna S, Hargadon B, et al. Sputum eosinophilia and the short term response to inhaled mometasone in chronic obstructive pulmonary disease. Thorax. 2005;60(3):193–198. doi:10.1136/thx.2004.032516
49. Nagasaki T, Matsumoto H, Nakaji H, et al. Smoking attenuates the age-related decrease in IgE levels and maintains eosinophilic inflammation. Clin Exp Allergy. 2013;43(6):608–615.
50. Cazzola M, Rogliani P. Do we really need asthma-chronic obstructive pulmonary disease overlap syndrome? J Allergy Clin Immunol. 2016;138(4):977–983. doi:10.1016/j.jaci.2016.04.028
51. Van Rossem I, Vandevoorde J, Hanon S, Deridder S, Vanderhelst E. The stability of blood eosinophils in stable chronic obstructive pulmonary disease: a retrospective study in Belgian primary care. BMC Pulm Med. 2020;20(1):200. doi:10.1186/s12890-020-01234-3
52. Singh D, Agusti A, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease: the GOLD science committee report 2019. Eur Respir J. 2019;53(5):1900164. doi:10.1183/13993003.00164-2019
53. Varricchi G, Bagnasco D, Borriello F, Heffler E, Canonica GW. Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr Opin Allergy Clin Immunol. 2016;16(2):186–200. doi:10.1097/ACI.0000000000000251
54. Eltboli O, Mistry V, Barker B, Brightling CE. Relationship between blood and bronchial submucosal eosinophilia and reticular basement membrane thickening in chronic obstructive pulmonary disease. Respirology. 2015;20(4):667–670. doi:10.1111/resp.12475
55. Caramori G, Adcock IM, Di Stefano A, Chung KF. Cytokine inhibition in the treatment of COPD. Int J Chron Obstruct Pulmon Dis. 2014;9:397–412.
56. Narendra DK, Hanania NA. Targeting IL-5 in COPD. Int J Chron Obstruct Pulmon Dis. 2019;14:1045–1051. doi:10.2147/COPD.S155306
57. Donovan T, Milan SJ, Wang R, Banchoff E, Bradley P, Crossingham I. Anti-IL-5 therapies for chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2020;12(12):CD013432.
58. Yousuf A, Ibrahim W, Greening NJ, Brightling CE. T2 biologics for chronic obstructive pulmonary disease. J Allergy Clin Immunol Pract. 2019;7(5):1405–1416. doi:10.1016/j.jaip.2019.01.036
59. Pavord ID, Chapman KR, Bafadhel M, et al. Mepolizumab for eosinophil-associated COPD: analysis of METREX and METREO. Int J Chron Obstruct Pulmon Dis. 2021;16:1755–1770. doi:10.2147/COPD.S294333
60. Criner GJ, Celli BR, Singh D, et al. Predicting response to benralizumab in chronic obstructive pulmonary disease: analyses of GALATHEA and TERRANOVA studies. Lancet Respir Med. 2020;8(2):158–170. doi:10.1016/S2213-2600(19)30338-8
61. Mesnil C, Raulier S, Paulissen G, et al. Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest. 2016;126(9):3279–3295. doi:10.1172/JCI85664
62. Brightling CE, Bleecker ER, Panettieri RA, et al. Benralizumab for chronic obstructive pulmonary disease and sputum eosinophilia: a randomised, double-blind, placebo-controlled, phase 2a study. Lancet Respir Med. 2014;2(11):891–901. doi:10.1016/S2213-2600(14)70187-0
63. Saco TV, Pepper A, Casale TB. Uses of biologics in allergic diseases: what to choose and when. Ann Allergy Asthma Immunol. 2018;120(4):357–366. doi:10.1016/j.anai.2018.02.029
64. Doyle AD, Mukherjee M, LeSuer WE, et al. Eosinophil-derived IL-13 promotes emphysema. Eur Respir J. 2019;53(5):1801291. doi:10.1183/13993003.01291-2018
65. Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H. The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol. 2013;66:129–155.
66. Borowski A, Vetter T, Kuepper M, et al. Expression analysis and specific blockade of the receptor for human thymic stromal lymphopoietin (TSLP) by novel antibodies to the human TSLPRα receptor chain. Cytokine. 2013;61(2):546–555. doi:10.1016/j.cyto.2012.10.025
67. Zhang K, Shan L, Rahman MS, Unruh H, Halayko AJ, Gounni AS. Constitutive and inducible thymic stromal lymphopoietin expression in human airway smooth muscle cells: role in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L375–82. doi:10.1152/ajplung.00045.2007
68. Redhu NS, Gounni AS. Function and mechanisms of TSLP/TSLPR complex in asthma and COPD. Clin Exp Allergy. 2012;42(7):994–1005. doi:10.1111/j.1365-2222.2011.03919.x
69. Calvén J, Yudina Y, Hallgren O, et al. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG-I-like helicases. J Innate Immun. 2012;4(1):86–99. doi:10.1159/000329131
70. Elder MJ, Webster SJ, Williams DL, Gaston JS, Goodall JC. TSLP production by dendritic cells is modulated by IL-1β and components of the endoplasmic reticulum stress response. Eur J Immunol. 2016;46(2):455–463. doi:10.1002/eji.201545537
71. Matera MG, Rogliani P, Calzetta L, Cazzola M. TSLP Inhibitors for asthma: current status and future prospects. Drugs. 2020;80(5):449–458. doi:10.1007/s40265-020-01273-4
72. Gabryelska A, Kuna P, Antczak A, Białasiewicz P, Panek M. IL-33 mediated inflammation in chronic respiratory diseases-understanding the role of the member of IL-1 superfamily. Front Immunol. 2019;10:692. doi:10.3389/fimmu.2019.00692
73. Cayrol C, Girard JP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31–37. doi:10.1016/j.coi.2014.09.004
74. Donovan C, Hansbro PM. IL-33 in chronic respiratory disease: from preclinical to clinical studies. ACS Pharmacol Transl Sci. 2019;3(1):56–62. doi:10.1021/acsptsci.9b00099
75. Xia J, Zhao J, Shang J, et al. IL-33 expression in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2015;308(7):L619–27. doi:10.1152/ajplung.00305.2014
76. Rabe KF, Celli BR, Wechsler ME, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial. Lancet Respir Med. 2021. doi:10.1016/S2213-2600(21)00167-3
77. Cazzola M, Cavalli F, Usmani OS, Rogliani P. Advances in pulmonary drug delivery devices for the treatment of chronic obstructive pulmonary disease. Expert Opin Drug Deliv. 2020;17(5):635–646. doi:10.1080/17425247.2020.1739021
78. Maillet A, Guilleminault L, Lemarié E, et al. The airways, a novel route for delivering monoclonal antibodies to treat lung tumors. Pharm Res. 2011;28(9):2147–2156. doi:10.1007/s11095-011-0442-5
79. Respaud R, Marchand D, Parent C, et al. Effect of formulation on the stability and aerosol performance of a nebulized antibody. MAbs. 2014;6(5):1347–1355. doi:10.4161/mabs.29938
80. Matera MG, Calzetta L, Rogliani P, Cazzola M. Monoclonal antibodies for severe asthma: pharmacokinetic profiles. Respir Med. 2019;153:3–13. doi:10.1016/j.rmed.2019.05.005
81. Cazzola M, Calzetta L, Rogliani P, Matera MG. The challenges of precision medicine in COPD. Mol Diagn Ther. 2017;21(4):345–355. doi:10.1007/s40291-017-0266-z
82. Cazzola M, Rogliani P, Stolz D, Matera MG. Pharmacological treatment and current controversies in COPD. F1000Res. 2019;8:1533. doi:10.12688/f1000research.19811.1
83. Nucera F, Lo Bello F, Shen SS, et al. Role of Atypical chemokines and chemokine receptors pathways in the pathogenesis of COPD. Curr Med Chem. 2021;28(13):2577–2653. doi:10.2174/0929867327999200819145327
84. Cazzola M, MacNee W, Martinez FJ, et al. Outcomes for COPD pharmacological trials: from lung function to biomarkers. Eur Respir J. 2008;31(2):416–469. doi:10.1183/09031936.00099306
85. Maselli DJ, Hanania NA. Management of asthma COPD overlap. Ann Allergy Asthma Immunol. 2019;123(4):335–344. doi:10.1016/j.anai.2019.07.021
86. Calzetta L, Ritondo BL, Matera MG, Facciolo F, Rogliani P. Targeting IL-5 pathway against airway hyperresponsiveness: a comparison between benralizumab and mepolizumab. Br J Pharmacol. 2020;177(20):4750–4765. doi:10.1111/bph.15240
87. Manson ML, Säfholm J, James A, et al. IL-13 and IL-4, but not IL-5 nor IL-17A, induce hyperresponsiveness in isolated human small airways. J Allergy Clin Immunol. 2020;145(3):808–817. doi:10.1016/j.jaci.2019.10.037
88. Siafakas N. Monoclonal antibodies for chronic obstructive pulmonary disease. Pulmonology. 2020;26(2):61–62. doi:10.1016/j.pulmoe.2019.11.002
89. McDonald VM, Fingleton J, Agusti A. participants of the Treatable Traits Down Under International Workshop; Treatable Traits Down Under International Workshop participants: Treatable traits: a new paradigm for 21st century management of chronic airway diseases: treatable Traits Down Under International Workshop report. Eur Respir J. 2019;53(5):1802058.
90. Bel EH, Ten Brinke A. New anti-eosinophil drugs for asthma and COPD: targeting the Trait! Chest. 2017;152(6):1276–1282. doi:10.1016/j.chest.2017.05.019
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.