Back to Journals » Drug Design, Development and Therapy » Volume 13
An Optimized Surfactant-Based PEG-PLCL In Situ Gel Formulation For Enhanced Activity Of Rosuvastatin In Poloxamer-Induced Hyperlipidemic Rats
Authors Ahmed TA, Mussari MA, Abdel-Hady SES, El-Say KM ![]()
Received 24 July 2019
Accepted for publication 28 October 2019
Published 3 December 2019 Volume 2019:13 Pages 4035—4051
DOI https://doi.org/10.2147/DDDT.S224442
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Tarek A Ahmed,1,2 Mohammed A Mussari,1 Seham El-Sayed Abdel-Hady,1 Khalid M El-Say1,2
1Department Of Pharmaceutics, Faculty Of Pharmacy, King Abdulaziz University, Jeddah, Kingdom of Saudi Arabia; 2Department Of Pharmaceutics And Industrial Pharmacy, Faculty Of Pharmacy, Al-Azhar University, Cairo 11651, Egypt
Correspondence: Tarek A Ahmed
Department Of Pharmaceutics, Faculty Of Pharmacy, King Abdulaziz University, PO. Box 80200, Jeddah 21589, Kingdom of Saudi Arabia
Email [email protected]
Background: Injectable in situ gel (ISG) systems suffer from high initial drug release that may result in toxic effects.
Objective: This work aimed to develop an injectable sustained release rosuvastatin (RSV) ISG formulation with minimum initial drug burst and improved hyperlipidemic efficacy.
Methods: Six formulation factors that affect RSV release after 0.5, 2 and 24 hrs have been screened and the significant ones were optimized utilizing an experimental design tool. The optimum ISG formulation components were physicochemically characterized. Kinetic treatment, dissolution efficiency and mean dissolution time were investigated for the developed ISG formulations. Pharmacodynamic effects of the optimized ISG formulation were studied and compared to ISG formulation loaded with free RSV and to a marketed oral drug product.
Results: The concentration polylactide-co-ϵ-caprolactone (25: 75), the surfactant hydrophilic lipophilic balance (HLB) and the ratio of surfactant to polyethylene glycol 400 were significantly affecting the release of RSV during the first 24 h. Physicochemical characterization demonstrated complete dispersion of RSV in the polymeric matrix with slight changes in the drug crystalline structure. The optimized formulation demonstrated acceptable syringeability, good flow rate and was able to extend the in vitro drug release for 34 days. The ISG formulations complied with Weibull model. Pharmacodynamic study revealed a sustained reduction in the lipid profile that lasted for 21 days with a marked decrease in the lipid level during the first 24 hrs from the ISG system loaded with free RSV.
Conclusion: The optimized RSV ISG formulation could be considered a promising strategy due to a reduction in dosing frequency and enhancement in hypolipidemic efficacy.
Keywords: hypolipidemic activity, in situ gel, initial burst, optimization, rosuvastatin
Introduction
In situ gel (ISG) systems are formulations existed in the liquid state but transformed into a gel once in contact with the biological environment of the body, allowing the drug to be released in a sustained/controlled manner. The process of in situ gelation can be triggered by multiple factors such as pH change, temperature change, availability of ions, UV radiation, and others.1 The main drawback of these systems is the accompanied initial drug release (burst effect) which may cause severe toxicity and tissue irritation.2,3 Initial drug burst effect is attributed to escape of the drug to the surrounding before complete hardening of the polymer and also due to uneven distribution of the drug within the polymeric matrix.4–6 ISG systems depend on the presence of smart polymers which are macromolecules that demonstrate a dramatic change in response to a certain stimulus such as temperature, pH, magnetic field, light, or solvent exchange. Smart polymers have been utilized in a pharmaceutical formulation of many drug delivery systems due to their biodegradability, biocompatibility, ease of application, site-specific drug delivery actions and extended release mechanisms. These polymers may be either natural, such as alginate, dextran, cellulose, chitosan, and lipids, or synthetic such as poly(amides), poly(amino acids), poly (anhydrides), poly(cyanoacrylates), polyesters, poly(ε-caprolactone) (PCL), poly(glycolic acids), and poly(lactic acids).7,8 Some smart polymers have been investigated in recent years, but most of the marketed ISG products are based on poly(lactic acid) and poly(lactic-co-glycolic acid) (PLGA).9 Polylactide-co-ε-caprolactone (PLCL) is a copolymer made of L-lactide and ε-caprolactone which has been approved by the United States Food and Drug Administration (FDA).10 It is a flexible, elastic, durable and biodegradable polymer. The elasticity of this polymer is provided by the caprolactone moiety and the toughness by the lactide part. This polymer has been used in many medical applications, especially in tissue engineering, due to its good biocompatibility, biodegradability, high elasticity and slow polymer degradation.11,12 PLCL is degraded in the body into lactic acid and caproic acid which are then metabolized by the tricarboxylic acid cycle and execrated through the kidney.13
Initial drug burst is the major drawback of the ISG systems. In this phenomenon, a high amount of the drug loaded is released during the early release phase, in the first 24 hrs, that may result in a plasma toxic drug level. This is mainly caused by the presence of lag time between the ISG administration and complete solidification.14,15 Other possible causes for this behavior are attributed to the amount of drug adsorbed on the ISG surface and the unequal drug distribution in the polymeric matrix which may cause rapid and high initial drug diffusion into the release medium.5,6 Different strategies have been reported to overcome this drawback such as the use of hydrophobic solvents, change in the polymer hydrophilic–lipophilic characters, polymer concentration, polymer molecular weight, incorporation of surfactant and/or plasticizer, development of in situ microparticles and in situ microglobules.16
Formulation scientists face many challenges and obstacles during the development of an effective dosage form due to the high number of process variables, multiple responses, cost considerations, insufficient drug quantity and drug cost used.17 These challenges always demonstrate the need for new tools that keep the formulators' efforts efficient and more focused by reducing the number of experiments.18 Design of experiments (DOE) is an effective tool that focuses on utilizing statistical analysis to facilitate proper decision-making during all the formulation stages.18 This tool can analyze the effect of formulation-related variables on the specific response(s) to provide more efficient research work. DOE starts with a screening of the possible formulation variables to identify the significant ones, followed by optimization of the significant ones to establish a polynomial model equation that interprets the relationship between the variables and the studied responses utilizing the response surface methodology (RSM). RSM uses a sequence of designed experiments to predict the optimum conditions that achieve the study goals. Statistical analysis of the obtained data is employed to maximize the results and optimize the operational factors through the study of the interaction between process variables.19,20 Using BB and RSM provides a smaller number of experiments with efficient decision-making.
Statins are a class of lipid-lowering agents that reduce cardiovascular disease in those people at high risk. They reduce the cholesterol level in the body by inhibiting the enzyme 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase which plays a key role in the production of cholesterol. Suppression of this enzyme leads to a reduction of atherosclerosis probability and the accompanied cardiovascular events. Statins have been investigated for their effectiveness in the management of many diseases such as Parkinson’s, Alzheimer’s, irritable bowel syndrome, sclerosis, stroke, and chronic obstructive pulmonary diseases.21 HMG-CoA reductase inhibitors are generally classified into hydrophilic and lipophilic ones. The former tends to be distributed to the hepatic and extrahepatic tissues, whereas the latter group is more hepato-selective.21 Poor bioavailability is a general criterion of the statin members due to their low water solubility, low permeability, high molecular weight and extensive first-pass metabolism. Statins are mainly formulated as oral dosage form despite being poorly available after oral intake. This poor bioavailability encouraged many researchers and formulation scientists to develop many statin formulations with improved bioavailability. Rosuvastatin (RSV) is a member of this class that is mainly distributed to liver cells with low peripheral tissue distribution. The bioavailability of RSV is 20% and it is 88% bound to plasma protein with 19-hr half-life. The drug is poorly soluble in water (0.0886 mg/mL), marginally soluble in ethanol and soluble in organic solvents such as DMSO and dimethyl formamide (DMF).22 RSV reaches the maximum plasma level after 3 to 5 hrs, and the drug plasma level is proportional to the amount of RSV administered. Food does not affect the amount of RSV absorbed.23
Currently, no injectable RSV is available in the market especially in the form of ISG formulation. So, the aim of this study was to use the experimental design to develop a long-acting parenteral RSV ISG formulation with minimum initial drug burst. The in vitro release data were treated using different mathematical models to investigate the drug release kinetics and release mechanism and correlate results to the possible causes of initial drug burst. The developed RSV ISG formulation is expected to improve the drug efficacy and achieve a sustained release drug action.
Materials And Methods
Materials
Polylactide-co-ε-caprolactone (PLCL) of 25:75 and 80:20 ratios were purchased from DURETEC Cooperation (Cupertino, CA, USA). N-methyl-2-pyrrolidone (NMP) was supplied from Acros organics (Fair Lawn, New Jersey, USA). Rosuvastatin calcium was a kind gift from the Saudi Arabian Japanese Pharmaceuticals Co. Ltd (SAJA) (Jeddah, KSA). Polyethylene glycol (PEG), Mw 400 and 4000, were purchased from Fischer Scientific (Bartlesville, OK, USA). Potassium dihydrogen orthophosphate was purchased from BDH Chemicals Ltd (Poole, England). Sodium hydroxide was supplied from VWR International Ltd. (Poole, England). Tween 80 and Span 40 were obtained from Sigma-Aldrich (St. Louis, MO). Blood lipid diagnostic kit was purchased from Crescent diagnostics (Jeddah, KSA).
All the above materials were of analytical grade and were used without further purification.
Methodology
Preliminary Study
This section aimed to investigate the initial RSV release from a polymeric drug in situ gel (ISG) system that contains PLCL (25:75) and (80:20) without surfactant and/or plasticizer. Six ISG formulations were prepared by adding 20%, 30% and 40% w/v of PLCL to NMP. The calculated amounts of the polymer were placed in scintillation glass vials containing the specified volume of NMP. Vials were subjected to probe sonication, to dissolve the polymer in NMP, using Sonics Vibra cell, VCX 750; Sonics & Materials, Inc. (Newtown, CT, USA) for 10 mins at an amplitude of 60% under ice cooling to prevent polymer degradation. The prepared formulations were kept in a shaking water bath (Model 1031; GLF Corp; Burgwedel, Germany) for 24 hrs at 25 °C until clear solutions were obtained. Known weight of RSV powder was subsequently added to each vial to yield a concentration of 40 mg/mL. Finally, the prepared vials were kept under shaking until the complete dissolving of RSV.24
A quantity of each formulation equivalent to 20 mg RSV was injected into 900 mL phosphate buffer of pH 7.4 in a USP dissolution tester apparatus II at a rotation speed of 100 rpm and a temperature of 37 ± 0.5 °C. The in vitro drug release was studied for 24 hrs. Samples of 2 mL were withdrawn at 0.5, 2, and 24 hrs and replaced with an equivalent volume of phosphate buffer after each withdrawal. The concentration of RSV in the collected samples was measured spectrophotometrically at 242 nm using 6705 UV/Vis spectrophotometer (JENWAY). The accuracy of the spectrophotometric method and its freedom from any possible interference by the formulation excipients were verified. Recovery testing of the drug concentration in solutions containing different concentrations of RSV and the studied excipients was verified. The experiment was done in triplicate. During the in vitro test, the dissolution tester was covered with aluminum foil to protect RSV from being degraded by light.
Plackett–Burman Screening Design
Six formulation factors that may affect the initial RSV release after 0.5 hr (Y1), 2 hrs (Y2) and 24 hrs (Y3) were screened using StatGraphics Centurion XV software (StatPoint Technologies Inc, Warrenton, VA, USA). PLCL concentration (X1), polylactide-to-polycaprolactone ratio (X2), molecular weight of PEG (X3), PEG concentration (X4), surfactant hydrophilic–lipophilic balance (HLB) (X5) and surfactant concentration (X6) were studied as independent variables. Based on our previously published work and the literature review, 0.5, 2 and 24 hrs of drug release were selected as dependent variables to optimize the release of rosuvastatin from the prepared ISG formulations. The composition of the ISG formulations obtained from the Plackett–Burman (PB) screening design is depicted in Table 1.
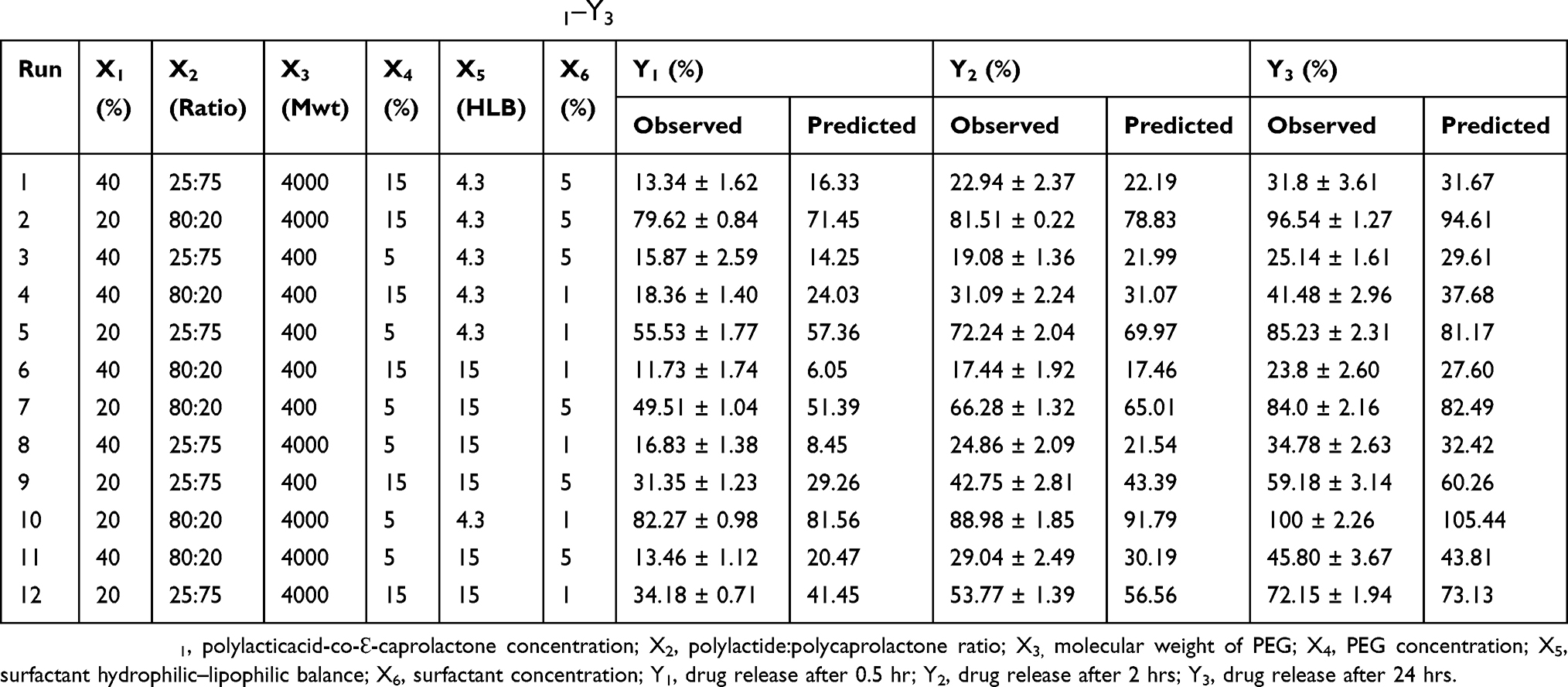 |
Table 1 Composition Of The Prepared Rosuvastatin In Situ Gel Formulations Generated By The Plackett–Burman Screening Design, And The Observed And Predicted Values Of Y1–Y3 |
Preparation And In Vitro Release Characterization
The PB ISG formulations were prepared by adding the accurately weighed amount of the studied polymer to a scintillation glass vials containing the specified volume of NMP. The calculated weight of surfactant (Tween 80 or Span 40) and PEG (400 or 4000) were subsequently added. Vials were subjected to probe sonication under ice cooling and vials were kept shaking in a shaking water bath (Model 1031; GLF Corp; Burgwedel, Germany) until clear solutions were obtained. RSV (40 mg/mL) was finally added to each vial and mixed until the complete dissolving of the drug.
Each ISG formulation was subjected to an in vitro release study utilizing the same dissolution conditions previously mentioned. Samples that withdrawn were quantified for RSV content after 0.5, 2, and 24 hrs.
Plackett–Burman Design Statistical Analysis
The observed values for Y1, Y2, and Y3, were statistically analyzed and a p-value ˂ 0.05 was considered significant. The significant factors that affected the initial RSV burst from the prepared ISG formulations were identified.
Box–Behnken Optimization Design
Three different independent variables that affect the release of RSV after 0.5 hr (Y1), 2 hrs (Y2) and 24 hrs (Y3) were optimized using StatGraphics Centurion XV software (StatPoint Technologies Inc, Warrenton, VA, USA). The Box–Behnken design was implemented to study the effect of PLCL (25:75) concentration (X1`), the surfactant HLB (X2`), and the surfactant to PEG 400 ratios (X3`). The studied levels for X1`, X2` and X3` were 35–45%, 13–15% and 0–100%, respectively. The aim was to produce RSV ISG formulation with a minimum initial drug burst.
Preparation And In Vitro Release Of The Box–Behnken ISG Formulations
Fifteen different ISG formulations were prepared and the in vitro drug release profile was conducted as described above. A concentration of 10% PEG–surfactant mixture was added to each formulation.
Box–Behnken Statistical Analysis And Optimum Desirability
The observed values for Y1, Y2 and Y3 were statistically analyzed at p-value < 0.05 to indicate the correlation between the independent and dependent variables. The optimum desirability was estimated, and the optimized formulation that contains the optimum levels of X1`, X2` and X3` was prepared, characterized for Y1, Y2 and Y3 as described above. The residual values for the predicted and observed responses were calculated.
Characterization Of The Optimized Formulation
A quantity of each ISG formulation equivalent to 20 mg RSV was added to phosphate buffer of pH 7.4 utilizing the same conditions described above. Aliquots were withdrawn for 34 days and the release profile was constructed by plotting the cumulative percent of drug released versus time.
The ability of the optimized RSV ISG formulation to successfully flow through a syringe needle, syringeability, was also estimated.24,25 This character was evaluated by measuring the magnitude of the force required to push the formulation through the needle using a universal syringe rig (Stable Micro Systems, Surrey, UK). The formulation flow rate (mL/second) was estimated using an apparatus designed in our laboratory. A known volume of the formulation was withdrawn in a plastic syringe of 20-G needle, and a specified weight, equivalent to the predetermined force, was applied to the syringe plunger. The color, clarity, and pH of this formulation were also examined.
Kinetic Treatment Of The In Vitro Release Data
The data obtained for the in vitro release of RSV from the fifteen Box–Behnken ISG formulations and the optimized ISG formulation were fitted to different mathematical models to investigate the drug release kinetics and release mechanism. The models used were Zero,26 First,26 Weibull,27 Hixson–Crowell,28 Higuchi,29 Korsmeyer–Peppas,30,31 and Baker–Lonsdale.32 The highest value of the coefficient (R2) was used to identify the goodness of fit and the appropriate release model. Equations used for the studied kinetic orders/models are as follows:
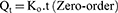

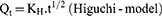




where Qt is the amount of RSV released at time (t), Qo is the initial amount of RSV released (mostly equal zero), K0 and K1 are the zero-order and first-order release constants and KH is the Higuchi diffusion rate constant. Qt/Qα is the fraction of RSV released at time t. Mt/Mα represents the fraction of RSV released at time t, K is a constant comprising the structural and geometric characteristics of the prepared ISG formulation, and n is the release exponent that depends on the drug release mechanism. The parameter a is the scale parameter that defines the timescale of the process, while β is the shape parameter, respectively. Wo and Wt are the initial and remaining amount of drug in the ISG formulation, respectively. Ks is a constant incorporating the surface–volume relation. KL is the release constant that corresponds to the slope.
Kinetic treatment for the fifteen Box–Behnken ISG formulations was considered up to 24 hrs, while that of the optimized ISG formulation was calculated separately for the studied two drug release phases, namely during the initial 24 hrs (Phase I) and after 24 hrs until 34 days (Phase II).
Dissolution Efficiency And Mean Dissolution Time
The dissolution efficiency (DE) and mean dissolution time (MDT) of the fifteen Box–Behnken ISG formulations and the optimized ISG formulation were calculated using Kinet DS 3.0 software (Free license software Rev. 2010, sourceforge.net). The following equations were employed:


where j is the sample number, n the number of time increments considered, tˆj is the time at midpoint between tj and tj − 1, and ∆Qj is the additional amount of drug dissolved in the period of time tj and tj – 1.
Physicochemical Characterization
Differential Scanning Calorimetry (DSC)
The thermal analysis of pure RSV, PLCL and the optimized formulation was investigated using Shimadzu DSC-TA-50 ESI (Tokyo, Japan). A sample of 2 mg from each component was transferred to aluminum crucibles and heated at a heating rate of 10 °C/min under a dynamic nitrogen atmosphere.
Fourier Transform Infrared (FT-IR) Spectrum
Samples used in the DSC analysis were also studied using a Nicolet Is10 (Thermo Scientific, Inc., Waltham, MA). The FT-IR spectra of the studied samples were recorded in the range of 4000–400 cm–1.
X-Ray Powder Diffraction (XRPD)
The diffraction pattern of free RSV was recorded using (D/max 2500; Rigaku, Tokyo, Japan) and compared to that of the freeze-dried drug-loaded optimized ISG formulation. The scan speed was set at 0.5 degree/min.
Pharmacodynamics And Biochemical Hypolipidemic Efficacy
The protocol for this work received a prior approval from the animal research ethical committee, Faculty of Pharmacy, King Abdulaziz University, Saudi Arabia. It was performed in accordance with the guidelines that ensure the care and use of laboratory animals conformed to the declaration of Helsinki and to the “Principles of Laboratory Animals Care” (NIH publication #85-23, revised in 1985).
Male Wistar rats of an average weight of 300 g were used. Hyperlipidemia was induced in the studied animals by intraperitoneal injection of Poloxamer 407 dissolved in 0.9% saline in a dose of 0.25 g/kg.25,33 Animals were divided into three groups (n= 6). The first group was subjected to intramuscular injection, into the right Gluteus Maximus muscle, of the optimized ISG formulation (Test group). The second group was intramuscularly administered an ISG formulation containing 45% PLCL (25:75) loaded with free RSV without surfactant and PEG (positive control group). Group three received marketed oral RSV tablets, Rosavi® 20 mg tablets, SAJA Pharmaceuticals Co. Ltd., (Jeddah, Saudi Arabia), that were crushed, dissolved in 1% carboxymethylcellulose and administered using a gastric tube (reference group). Formulation of the positive control group was prepared utilizing the same procedure mentioned above during the development of the ISG system except that no surfactant or plasticizer was added. The test and positive control groups were compared to study the effect of incorporation of the surfactant and plasticizer on the drug initial burst and the profile of drug release in vivo. A dose of 20 mg/kg was administered to each rat.34,35 Blood samples were withdrawn at 0.5, 1, 2, 24, 48 hrs and then after 3, 5, 7, 10, 14, 17 and 21 days. The serum was separated from each blood sample by centrifugation at 10,000 rpm for 5 mins. The collected serum samples were evaluated for total cholesterol and triglycerides using a specified lipid kit (Crescent Diagnostics Industry, Jeddah, Saudi Arabia).
Statistical Analysis Of The Pharmacodynamics Data
The data obtained from the pharmacodynamics study were statistically analyzed at p-value < 0.05 using Graph Prism 6 software (San Diego, CA). Two-way analysis of variance followed by Tukey’s multiple comparison test was used to indicate the difference between the studied groups.
Results And Discussion
Preliminary Study
In this section, the initial RSV release from ISG formulations containing different concentrations of PLCL without surfactant and plasticizer was investigated. The selection of the proper polymer is fundamental to obtain the desired drug release profile from the ISG system. The characteristics of PLCL are dependent on the molecular weight and the ratio of both lactic and caprolactone in the polymer chain. PLCL with higher polycaprolactone ratio results in a more hydrophobic polymer.36 Two PLCL grades (80:20 and 25:75) with different lactide and caprolactone ratios were investigated in this study. PLCL (25:75) and (80:20) were used in a concentration range of 20–40% w/v, in NMP, to prepare six different ISG formulations. A polymeric concentration of less than 20% often gives a fast drug release and high initial drug burst as previously stated for ISG systems loaded with poly lactide-co-glycolide.4,15,16,24,37 Viscus polymeric solutions of poor syringeability and injectability are often obtained from ISG systems that contain greater polymer concentration (above 50–60%).24,25
Table 2 demonstrates the obtained results for the in vitro release of RSV from the prepared six ISG formulations after 0.5, 2 and 24 hrs. Lower initial drug release was observed from the ISG formulation prepared utilizing PLCL (25:75) and higher polymer concentration. This finding may be attributed to the increase in viscosity of the polymeric drug system when higher polymer concentration and more hydrophobic ISG system were used.38 Xin et al studied the release of hydrochloric thiothixene from polylactic acid ISG containing different polymeric concentrations and reported higher initial drug release from ISG containing low polymer concentration and attributed this behavior to the polymeric matrix viscosity.39 Moreover, low initial drug release from PLCL (25:75) ISG formulations may be attributed to the higher molecular weight and lower water uptake of this polymer when compared to PLCL (80:20). Dalmoro et al reported the same finding for the release of α-tocopherol from ISG systems containing different copolymers of ε-caprolactone and L-lactide and mentioned lower drug release from the more hydrophobic PLCL (10:90) when compared to PLCL containing higher ratios of lactide.36 The studied ISG formulations exhibited RSV release of 40.24 ± 2.16–60.01 ± 3.01, 46.32 ± 1.87–76.14 ± 2.52 and 59.60 ± 1.20–88.57 ± 0.92% after 0.5, 2 and 24 hrs, respectively. Accordingly, we have studied the incorporation of different surfactants of different HLB and PEG of different molecular weights to decrease the amount of drug released during the first 24 hrs and so avoid the expected toxic drug effect.
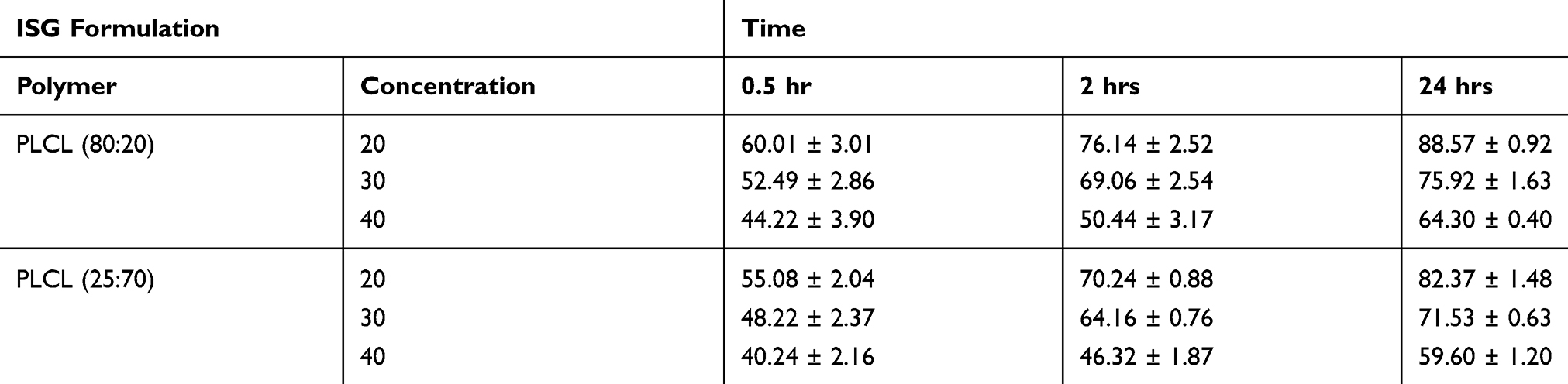 |
Table 2 Percent Of Rosuvastatin Released From In Situ Gel Formulations Containing Different Concentrations Of PLCL (80:20) And (25:75) In Phosphate Buffer Of pH 7.4 |
Plackett–Burman Screening Design
After preparation and investigation of the in vitro release profile of the Placket–Burman ISG formulations, the observed values for Y1, Y2 and Y3 were calculated and the obtained data were tabulated in Table 1.
Analysis of variance (ANOVA) for the effect of the studied variables on Y1 indicated a significant effect of X1, X2 and X5 as graphically illustrated in the Pareto chart (Figure 1). A reference line at a P-value of 0.05 is observed in this chart. Any factor effect that exceeds this line is significantly affecting the studied response. P-values of 0.0257, 0.0003 and 0.0119 were obtained for X1, X2 and X5, respectively. The obtained R-squared value indicated that the model as fitted explains 95.54% of the variability on the release after 0.5 hr, while the adjusted R-squared value, which is more suitable for comparing models with different numbers of independent variables, was 90.18 %. The model indicated that to obtain a minimum value for Y1 of 8.54%, the independent variables X1, X2 and X5 should be 25:75, 40% and 14.75, respectively. After analyzing the independent variables by multiple regressions, the obtained polynomial equation of the fitted model for the release after 0.5 hr was found to be as follows:
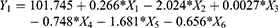
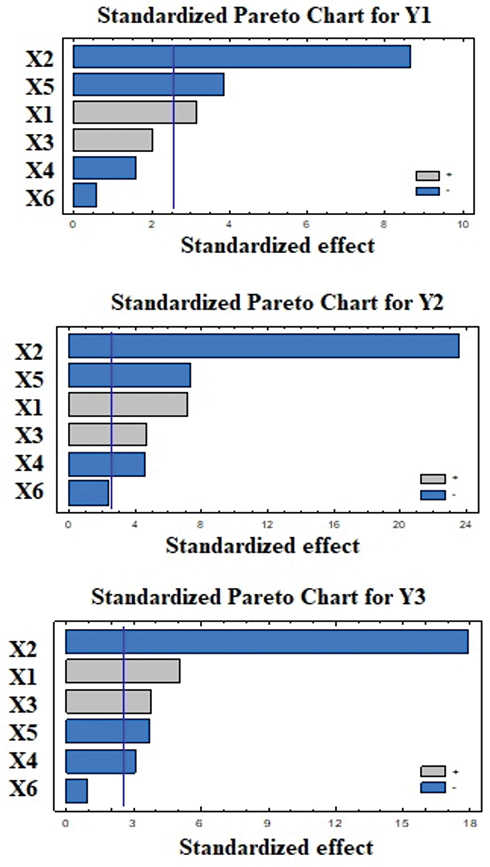 |
Figure 1 Standardized Pareto charts for the effect of the independent variables on Y1–Y3 for the Plackett–Burman design ISG formulations. Abbreviations: X1, Polylactide-co-ε-caprolactone concentration; X2, polylactide-to-polycaprolactone ratio; X3, molecular weight of polyethylene glycol; X4, polyethylene glycol concentration; X5, surfactant hydrophilic–lipophilic balance; X6, surfactant concentration; Y1, rosuvastatin release after 0.5 h; Y2, rosuvastatin release after 2 hrs; Y3, rosuvastatin release after 24 hrs. |
The effect of polymer concentration (X1) and polylactic:polycaprolactone ratio (X2) has been discussed in the preliminary study section. The concept of incorporation of surfactant during the development of the ISG system has not gained much attention in the literature. Poly(ethylene oxide)/poly(propylene oxide)/poly(ethylene oxide) triblock copolymers (pluronic) have been added to polylactic-co-glycolic acid (PLGA) and poly D, L-lactide-based ISG systems for delivery of Hen egg protein and fluorescein (low-molecular-weight model drug molecule), respectively.40,41 Also, Span 20, Span 80, Tween 20, Tween 60, Cremophor EL, and Cremophor RH 40 have been added to different PLGA in situ implant systems loaded with tamsulosin. The authors mentioned that the incorporated surfactants successfully lowered the in vitro release rate of tamsulosin from the prepared formulations.42 They attributed this behavior to the formation of a phase boundary between the polymeric matrix and the surrounding aqueous phase. The surfactant hydrophobic part is directed toward the hydrophobic polymer matrix while the hydrophilic part of the surfactant is extended in the aqueous phase. In our work, we have explored the effect of surfactant HLB and surfactant concentration and found that the former (X5) had a marked significant effect on Y1. Formation of a phase boundary at higher surfactant HLB value was found to have a significant effect on the drug release, the effect which could be attributed to the establishment of a well-developed barrier between the polymeric matrix and the external aqueous phase when using a more hydrophilic surfactant.
Statistical analysis for the effect of the studied factors on Y2 showed the significant effect of X1, X2, X3, X4, and X5 at P-values of 0.0009, 0.00008, 0.0053, 0.0059 and 0.0007, respectively. The Pareto chart (Figure 1) confirmed this finding. The obtained values for R-squared and adjusted R-squared were 99.2963% and 98.4518%, respectively. The polynomial equation of the fitted model for the release after 2 hrs, after analyzing the independent variables by multiple regressions, was found to be:
It is noteworthy to mention that the significant factors affecting Y2 were comparative to that obtained for Y1. Both were affected by X1, X2 and X5, but Y2 was also affected by X3 and X4. These results indicate that the molecular weight and the concentration of PEG showed a marked significant effect on Y2. In this study, PEG was added due to its plasticizing action and solubilizing power. Plasticizer is a substance that is added to decrease the attraction force between polymer chains and so make them more flexible. It has been previously mentioned that PEG affects the initial drug release from the ISG system possibly by its solubilizing power that permits uniform distribution of the drug particles inside the polymeric matrix and decreases the adsorption of any drug particles at the surface. This effect was recently mentioned by Ibrahim et al who studied the effect of incorporation of PEG on the initial release of meloxicam from the PLGA-based ISG system.37 Ahmed et al recently stated that the release of atorvastatin from the PLGA ISG system was significantly affected by the addition of PEG.24 They attributed this behavior to the plasticizing action of PEG which decreased the polymer glass transition temperature and so facilitated congealing of the polymer.
Moreover, the obtained results for RSV release from the prepared ISG formulations after 24 hrs was markedly affected by the same factors affecting the drug release after 2 hrs. ANOVA demonstrated that X1, X2, X3, X4 and X5 were significantly affecting Y3 at P-values of 0.039, 0.0002, 0.0128, 0.0288 and 0.0143, respectively, as graphically illustrated in the pareto chart of Figure 1. The R-squared statistic indicates that the model as fitted explains 98.7134% of the variability. The adjusted R-squared value was found to be 97.1695%. The following polynomial equation that describes the effect of variables on Y3 was obtained:
Data obtained from the screening study denoted that PLCL concentration, polylactic: polycaprolactone ratio and surfactant HLB had a pronounced effect on the drug release after 0.5 hr while the same factors in addition to PEG molecular weight and PEG concentration had a significant effect on the drug release after 2 and 24 hrs. So, it could be concluded that PLCL concentration that affects the polymeric matrix density and viscosity, hydrophobicity of the polymer and surfactant concentration promotes the process of in situ gelling formation by solvent exchange mechanism during the first 24 hrs possibly by decreasing the number and size of pores in the matrix that occurs during solvent exchange process. In addition, the formation of a phase boundary between the polymeric matrix and the surrounding aqueous phase brought about by the incorporation of surfactant also decreased the drug release. This boundary phase is more efficient at higher surfactant HLB and is not affected by the surfactant concentration. Finally, addition of PEG decreased the drug release from the ISG system after 2 hrs and 24 hrs. This effect was noticeable with low-molecular-weight PEG that is liquid or low-melting solid, which makes the polymeric matrix more flexible (plasticizing action) and promotes uniform drug distribution inside the polymeric matrix, yet this effect was not effective during the first 0.5 hrs of the drug release.
Among the design points, maximum desirability was achieved at run 6. To prepare an ISG formulation characterized by minimum drug release after 0.5 hr, 2 hrs and 24 hrs, factors X1–X5 need to be optimized to achieve the optimum level of hydrophobic PLCL (25:75), the optimum level of low-molecular-weight PEG at high surfactant HLB. These factors were subsequently optimized, using the Box–Behnken design, at three levels of PLCL 25:75 concentrations, three levels of high surfactant HLB values and three levels of surfactant to PEG 400 ratios. A fixed concentration of PEG-surfactant mixture (10% w/v based on the total formulation) at varied ratios was added to study the effect of presence, absence and a combination of both components.
Box–Behnken Optimization Design
Based on the screening study, three formulation factors were optimized for their effect on RSV release from the proposed ISG Box–Behnken formulations. These formulations were characterized for the in vitro drug release after 0.5 hr (Y1), 2 hrs (Y2) and 24 hrs (Y3) as described earlier. The in vitro release profiles of these formulations are demonstrated graphically in Figure 2. The observed and predicted values for RSV release at the specified time points are listed in Table 3.
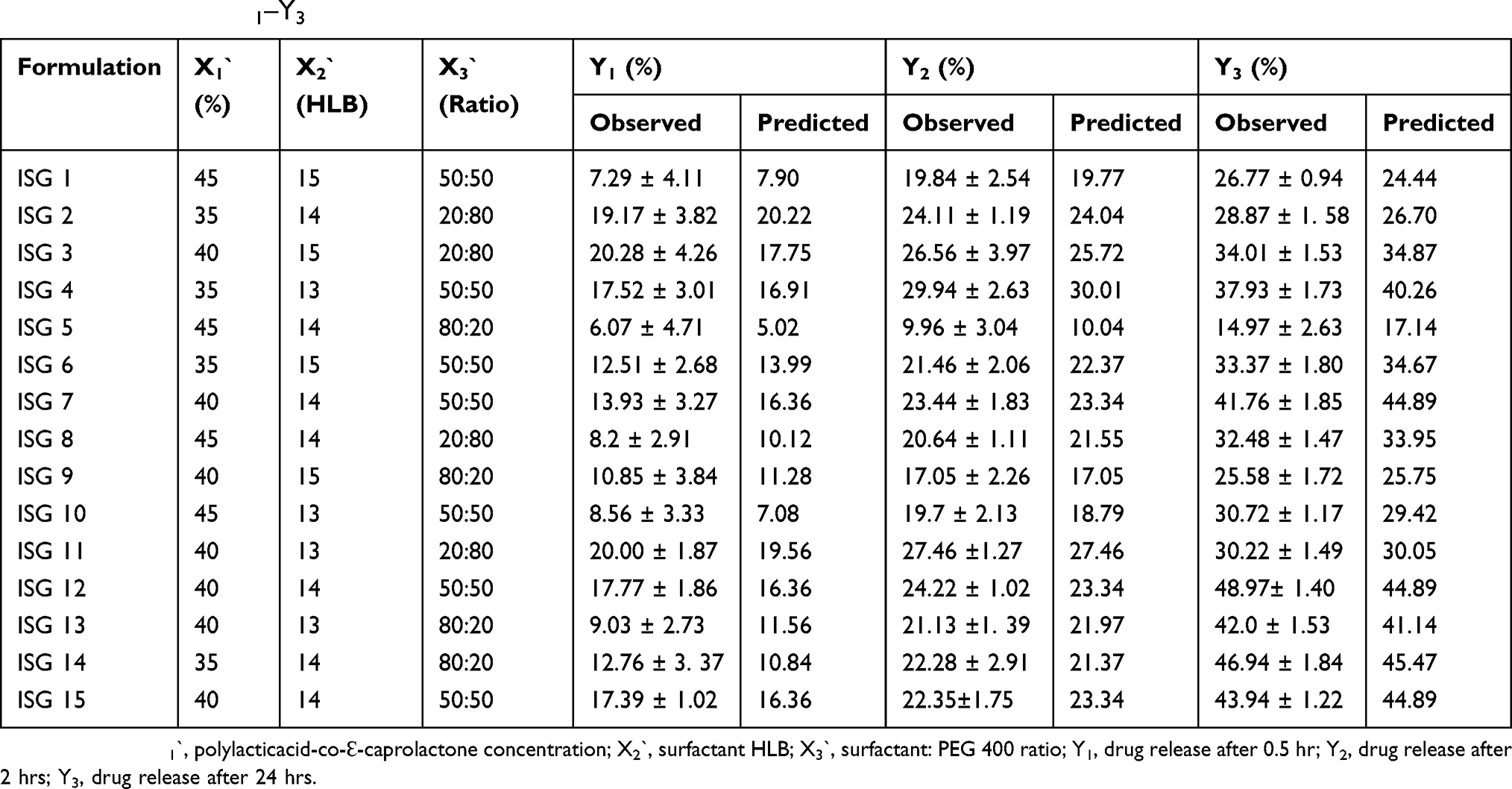 |
Table 3 Compositions Of Rosuvastatin In Situ Gel Formulations Generated By The Box–Behnken Design And The Observed And Predicted Values Of Y1–Y3 |
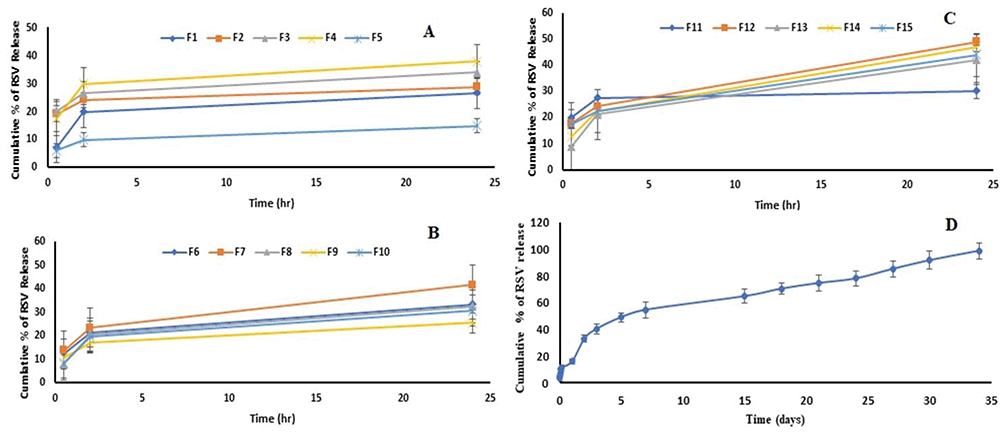 |
Figure 2 Percent of rosuvastatin released into phosphate buffer solution of pH 7.4 as a function of time from the Box–Behnken (A–C) and the optimized ISG formulation (D). |
The in vitro release of RSV from the prepared Box–Behnken ISG formulations after 0.5, 2 and 24 hrs was in the range of 6.07 ± 4.71–20.28 ± 4.26, 9.96 ± 3.04–29.94 ± 2.63 and 14.97 ± 2.63–48.97 ± 1.40, respectively. These results are considerably lower than their corresponding ISG formulations used in the preliminary study and in the Plackett–Burman screening design.
Effect Of The Independent Variables On Y1–Y3
Results of RSV in vitro release were statistically analyzed, and the obtained results for the estimated effects, F-ratios and P-values are demonstrated in Table 4. Accordingly, X1`, X3` and X1`X1` were significantly affecting Y1 while, X1`, X2`, X3`, X1`X1`, X1`X2`, X1`X3`, X2`X2` and X3`X3` had a marked significant effect on Y2. Also, X1`, X1`X1`, X1`X3`, X2`X2`, X2`X3` and X3`X3`were significantly affecting Y3. When the estimated effect has a positive sign value, this is an indication of a synergistic effect of this factor on the selected response, while an antagonistic effect is expected when a negative sign is obtained. The F-ratio is used to compare between the actual and the expected variation of variable averages, an F-ratio that is greater than 1 is an indication of a location effect, and hence, the P-value is used to notify the significant level. A factor is considered significant if the P-value differs from 0 and is less than 0.05. Three-dimensional response surface plots were constructed to explain the effect of changing two independent variables, when the third variable was kept at its intermediate level, on a studied response. These plots are graphically illustrated in Figure 3. The polynomial equations that relate the studied factors and drug release after 0.5, 2 and 24 hrs were found to be:
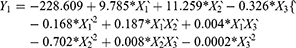
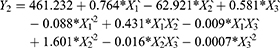

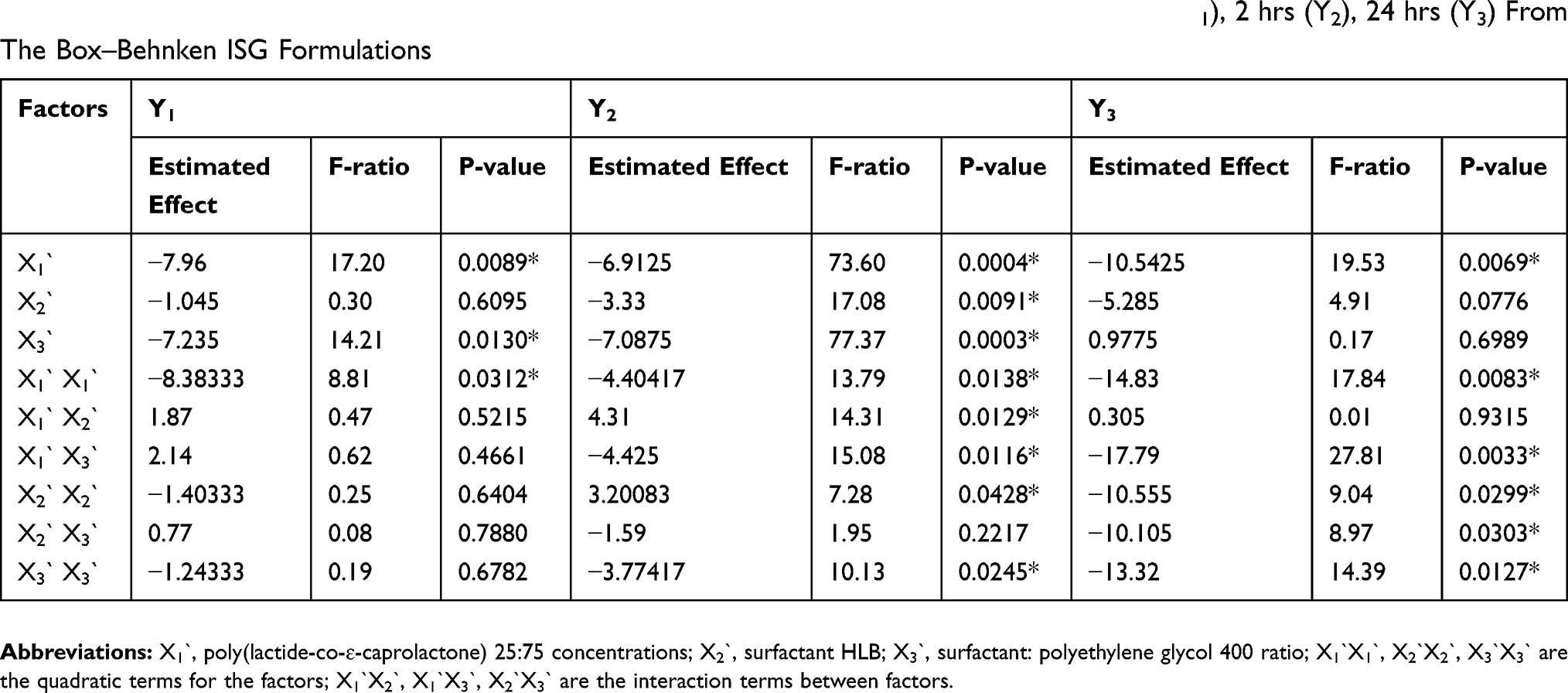 |
Table 4 Estimated Effects Of Factors, F-Ratio, And P-Values For Rosuvastatin Release After 0.5 hr (Y1), 2 hrs (Y2), 24 hrs (Y3) From The Box–Behnken ISG Formulations |
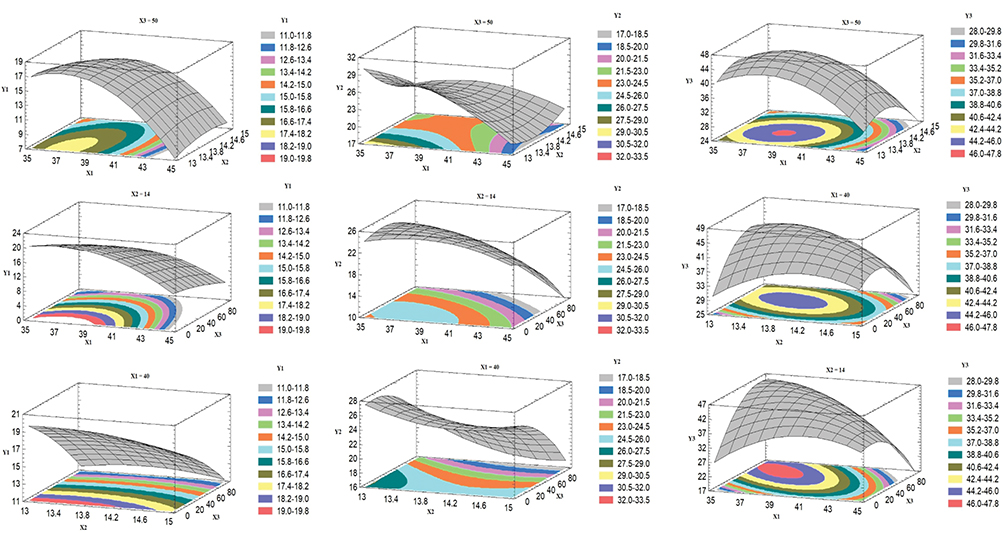 |
Figure 3 Estimated response surface plots for the effect of the independent variables on the drug release after 0.5 hr (Y1), 2 hrs (Y2) and 24 hrs (Y3) from the Box–Behnken ISG formulations. |
Based on the above finding, PLCL (25: 75) concentration (X1) had a significant antagonistic effect on the drug release at all the studied time points (Y1–Y3). This effect may be attributed to the increase in the polymeric drug ISG system viscosity, as described earlier. The surfactant HLB (X2`) only affected the drug release after 2 hrs (Y2), which could be attributed to the establishment of an efficient boundary phase by the surfactant molecules at the outer layer of the hydrophobic polymeric matrix after 2 hrs of ISG injection in the buffer medium. The drug release from this system at 0.5 hr and 24 hrs was not affected by X2 possibly due to partial localization of the surfactant molecules at the boundary and so incomplete formation of the boundary phase (effect on Y1), while the interaction effect of this factor (X2X2) markedly affected Y3. Similarly, surfactant: PEG 400 ratio (X3`) affected RSV release in the same way but with a different mechanism, as explained in the above sections.
To prepare an optimized ISG formulation characterized by RSV release of 5.17%, 10.05% and 14.97% after 0.5 hr, 2 hrs and 24 hrs, respectively, the optimum levels of X1`, X2` and X3` should be 45%, 14.25 and 80:20, respectively.
Characterization Of The Optimized RSV ISG Formulation
The in vitro release of RSV from the optimized ISG formulation is illustrated in Figure 2D. During the first 24 hrs, an initial fast release phase was observed that was followed by a second controlled (slow) release phase that lasts for days. In the first phase, a high initial drug release is expected from this system since the ISG formulation is administered as a liquid. Accordingly, a lag time is present between the injection of the liquid and the development of a semi-solid gel by a solvent exchange mechanism. We tried to decrease the drug release during this period and successfully achieved observed values of 5.51± 2.34, 10.48± 2.01 and 16.26± 1.94% for Y1, Y2 and Y3, respectively. The predicted values for the same responses were 5.17%, 10.05% and 14.96%, respectively. In the second phase, a diffusion-controlled slower drug release from the semi-solid polymeric drug matrix was observed. It has been previously mentioned that the drug release from ISG systems is mostly triphasic but biphasic release was also reported.15 In the present study, the second phase lasts for about 34 days. Therefore, the screening and the optimization techniques have been successfully implemented to develop an RSV ISG system with minimum initial drug burst that controls the drug release for 34 days.
The prepared optimized formulation was clear of slightly white color and neutral pH (7.43). Acceptable syringeability (39.98 ± 3.12 N) and flow rate (0.134 mL/second) were obtained. Kurakula and Ahmed reported syringeability values in the range of 37.8 ± 2.3–44.8 ± 2.4 for atorvastatin PLGA-based ISG formulations.25 They reported good syringeability of the system within this range.
Kinetic Treatment Of The In Vitro Release Data
According to the R2 values presented in Table 5, kinetic analysis of the in vitro release data of the Box–Behnken formulations and the release of RSV from the optimized formulation during the first 24 hrs (first Phase) were found to follow the Weibull model. Pharmaceutical systems complied with this model demonstrate a linear plot when the logarithm of the amount of drug released is plotted versus the logarithm of time.43 The computed β values for these formulations were greater than 1, indicating a sigmoid curve of complex release mechanism.44 This finding is in a good agreement with our interpretation for the reason of the drug initial burst from this system. The drug release from the optimized formulation after the first 24 hrs and up to 34 days (second phase) was found to obey zero-order model with a β value of 0.9779 indicating Fickian diffusion with a contribution of another release mechanism.
 |
Table 5 In Vitro Release Kinetics, Dissolution Efficiency And Mean Dissolution Time Of Rosuvastatin From The Box–Behnken And The Optimized Formulations |
The β value was used to identify the release mechanism since there is a linear relationship between this value and the n exponent used in the Peppas model.44 Unlike the n exponent which is valid only for the first 60% of the release, this value can be successfully applied to almost all kinds of dissolution/release curves.43 As illustrated in Table 5, the calculated values indicated a linear relationship between the β and the n exponent used in the Peppas model to determine the drug release mechanism. This finding was also mentioned by Papadopoulou et al, during their discussion of the use of Weibull function for the discernment of the possible mechanism of drug release.44
Dissolution Efficiency And Mean Dissolution Time
Results of the DE for the Box–Behnken formulations and the optimized formulation during the first 24 hrs (first phase) were in the range 12.60–36.92%. The MDT for the same formulations were 1.486–5.13 hrs. The wide range of values obtained for DE and MDT could be attributed to the difference in the composition of the prepared ISG formulations. Ibrahim et al previously illustrated that the polymer concentration and its intrinsic viscosity had a negative effect on the DE of meloxicam from PLGA/NMP ISG system. They also mentioned that the NMP level has a positive effect on the DE from the same system.37 The DE and MDT for the second release phase of optimized formulation (after 24 hrs and up to 34 days) were 67.131% and 1.084 hrs, respectively.
Physicochemical Characterization
The DSC thermogram of RSV (Figure 4) showed a sharp characteristic endothermic peak at about 155 ºC.45 The thermogram of pure PLCL illustrated a distinguished peak at 70°C identical to the melting point of polycaprolactone and another peak at about 160 ºC corresponding to polylactic acid melting temperatures.46 A small melting endotherm for polylactic acid was observed due to its relatively low ratio compared to polycaprolactone in the studied polymer (PLCL 25: 75). The drug peak was not fully recognized in the DSC thermogram of the optimized drug-loaded ISG formulation except for a small peak around 130–140 °C which could be attributed to some change in the drug properties during development of the ISG system. This finding supports complete entrapment and molecular dispersion of RSV in the polymeric matrix. Moreover, the melting points of polycaprolactone and polylactic acid in the optimized ISG formulation seem to be affected by the incorporation of PEG 400 and the studied surfactants.
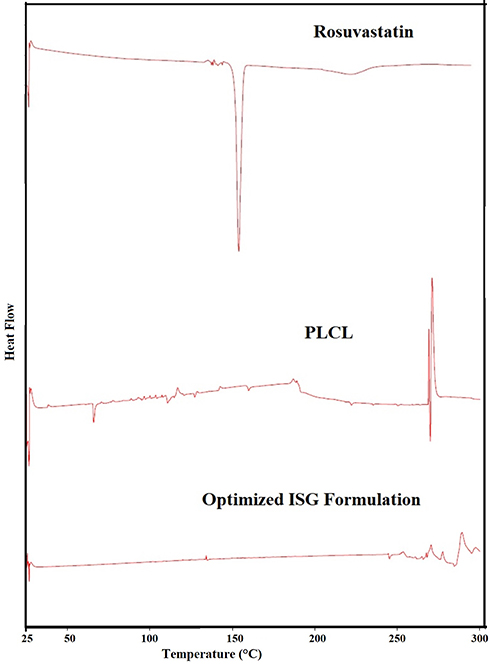 |
Figure 4 DSC thermal analysis of pure RSV, PLCL, and the optimized ISG formulation. |
The FT-IR spectrum of RSV (Figure 5) revealed a broad band for O–H stretching at 3380 cm,–1 a band at 2920 cm–1 for =C–H stretching, a peak at 1550 cm–1 for C=C stretching, and a peak at 1515 cm–1 for N–H bending. Asymmetric and symmetric bending vibrations of CH3 group were detected at 1485 cm–1 and 1380 cm–1, respectively. The asymmetric vibration of S=O was noticed at 1330 cm–1. The bending vibrations for C–H and C–F stretching vibrations were detected at 1230 cm–1 and 1155 cm–1, respectively. The FT-IR spectrum of PEG illustrated polymer peaks at 1464 and 1343 cm–1 for C–H bending vibrations. Another PEG peaks at 1279 and 1094 cm–1 corresponding to O–H and C–O–H stretching. The FT-IR spectrum of PLCL revealed a broad polymer peak at 2919 cm–1 corresponding to CH stretching vibration (CH2). Another PLCL peak was found at 1724 cm–1 corresponding to the C=O stretching vibration (non-conjugated). Also, a characteristic PLCL peak was found at 1164 cm–1 corresponding to C–O–C (esters) vibration. The FT-IR spectrum of the optimized drug-loaded ISG formulation showed the characteristic drug peaks of S=O and C–F stretching vibration while other RSV characteristic peaks were overlapped with the PEG and PLCL peaks.
 |
Figure 5 FT-IR spectroscopy of pure RSV, PEG 400, PLCL and the optimized ISG formulation. |
The X-ray diffraction patterns of pure RSV and the optimized drug-loaded ISG formulation are illustrated in Figure 6. RSV demonstrated a broad and numerous distinct diffraction peaks which indicate the crystalline nature of the pure rosuvastatin calcium. The diffraction spectrum of the optimized drug-loaded ISG formulation demonstrated halo diffractograms with some changes in the drug characteristic peaks that was confirmed by the absence of the drug characteristic peak at 2Ө diffraction angle of 15.5 ° and appearance of two sharp peaks at 22 °C and 24 °C. This behavior might be attributed to the complete dissolving of the pure drug in the PLCL-NMP ISG system with subsequent gelation of the polymeric matrix after injection in the buffer. The above finding indicates that rosuvastatin calcium exhibited slight changes in its crystalline form in the ISG formulation but did not confirm complete transformation into the amorphous form.
 |
Figure 6 X-ray diffraction patterns of pure RSV and the optimized ISG formulation. |
Pharmacodynamic And Biochemical Hypolipidemic Efficacy
To investigate the hypolipidemic efficacy of RSV after intramuscular injection of the optimized ISG formulation, the pharmacodynamic and biochemical effects were studied. Results obtained were compared to other groups that were given ISG formulation containing 45% PLCL (25: 75) loaded with free RSV (positive control) and to oral marketed drug product (reference group). Serum samples were collected from all rats 24 hrs before the induction of hyperlipidemia using Poloxamer 407. Induction of hyperlipidemia was initially confirmed by the change in the appearance of the rats’ serum from normal clear to milky white. The studied formulations were given to the assigned groups after induction of hyperlipidemia and blood samples were withdrawn from all animals at the predetermined time intervals. The total cholesterol and triglycerides were quantified in the collected samples and data obtained are graphically illustrated in Figure 7.
 |
Figure 7 Total cholesterol level (A) and triglyceride level (B) versus time following administration of RSV in the studied animals. Notes: *Statistical significance between test group and positive control group, $statistical significance between test group and reference group, #statistical significance between positive control group and reference group. |
Based on the obtained data, high levels of cholesterol and triglycerides were noticed before the administration of RSV (time zero). Administration of the ISG formulations and the marketed drug product affected the serum lipid level that was altered by Poloxamer 407. High initial drug release from the ISG formulation loaded with free RSV induced a very low total cholesterol and triglyceride levels in the first 24 hrs. Similar behavior was noticed from the group treated with the optimized ISG formulation but to a marked lesser extent which is an indication of low initial drug burst from this group. After 24 hrs of treatment, the group treated with the marketed drug product reduced the total cholesterol and triglycerides by 12.99% and 34.16%, respectively. Groups treated with the ISG formulations showed a sustained reduction in lipid profile that lasted for 21 days. This effect was not observed in the reference group that demonstrated a return to the original high lipid profile after 72 hrs of RSV treatment. The effect of the optimized ISG formulation on serum lipids was statistically significant (P <0.05) when compared with ISG loaded with free RSV and the marketed drug product groups.
Conclusion
The Plackett–Burman screening and Box–Behnken optimization designs have been successfully implemented to investigate and optimize the significant factors affecting the initial drug burst from PLCL-based ISG formulations. An optimized ISG formulation characterized by an initial RSV release of 5.17%, 10.05% and 14.97% after 0.5 hr, 2 hrs and 24 hrs, respectively, was developed. The optimized formulation illustrated acceptable syringeability, good flow rate and was able to extend the in vitro drug release for 34 days. Pharmacodynamic study revealed a sustained reduction in the lipid profile that lasts for 21 days with a marked decrease in the serum lipid level from the ISG system loaded with free RSV in the first 24 hrs.
Acknowledgment
This project was funded by the Deanship of Scientific Research (DSR) at King Abdulaziz University, Jeddah, under grant no. RG-2-166-39. The authors, therefore, acknowledge with thanks DSR technical and financial support.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Al-Tahami K, Singh J. Smart polymer based delivery systems for peptides and proteins. Recent Pat Drug Deliv Formul. 2007;1(1):65–71. doi:10.2174/187221107779814113
2. Huang X, Brazel CS. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J Control Release. 2001;73(2–3):121–136. doi:10.1016/S0168-3659(01)00248-6
3. Huang X, Brazel CS. Analysis of burst release of proxyphylline from poly(vinyl alcohol) hydrogels. Chem Eng Commun. 2003;190(4):519–532. doi:10.1080/00986440302081
4. Ahmed TA, Ibrahim HM, Samy AM, Kaseem A, Nutan MTH, Hussain MD. Biodegradable injectable in situ implants and microparticles for sustained release of montelukast: in vitro release, pharmacokinetics, and stability. AAPS PharmSciTech. 2014;15(3):772–780. doi:10.1208/s12249-014-0101-3
5. Mallapragada SK, Peppas NA, Colombo P. Crystal dissolution-controlled release systems. II. Metronidazole release from semicrystalline poly(vinyl alcohol) systems. J Biomed Mater Res. 1997;36(1):125–130. doi:10.1002/(ISSN)1097-4636.
6. Pekarek KJ, Jacob JS, Mathiowitz E. Double-walled polymer microspheres for controlled drug release. Nature. 1994;367(6460):258–260. doi:10.1038/367258a0
7. Pandey SK, Haldar C, Patel DK, Maiti P. Biodegradable polymers for potential delivery systems for therapeutics. Adv Polym Sci. 2013;254:169–202. doi:10.1007/12_2012_198
8. Bhise K, Kotwal V, Saifee M, Inamdar N. Biodegradable polymers: which, when and why? Indian J Pharm Sci. 2007;69(5):616. doi:10.4103/0250-474X.38465
9. Qi F, Wu J, Li H, Ma G. Recent research and development of PLGA/PLA microspheres/nanoparticles: a review in scientific and industrial aspects. Front Chem Sci Eng. 2018;1–14. doi:10.1007/s11705-018-1729-4
10. Daranarong D, Chan RTH, Wanandy NS, Molloy R, Punyodom W, Foster LJR. Electrospun polyhydroxybutyrate and poly(L-lactide-co-ε-caprolactone) composites as nanofibrous scaffolds. Biomed Res Int. 2014;2014:741408. doi:10.1155/2014/741408
11. Jeong SI, Kim SH, Kim YH, et al. Manufacture of elastic biodegradable PLCL scaffolds for mechano-active vascular tissue engineering. J Biomater Sci Polym Ed. 2004;15(5):645–660. doi:10.1163/156856204323046906
12. Jung Y, Kim SH, You HJ, Kim S-H, Ha Kim Y, Min BG. Application of an elastic biodegradable poly(L-lactide-co-ε-caprolactone) scaffold for cartilage tissue regeneration. J Biomater Sci Polym Ed. 2008;19(8):1073–1085. doi:10.1163/156856208784909336
13. Díaz E, Sandonis I, Valle MB. In vitro degradation of poly(caprolactone)/nHA composites. J Nanomater. 2014;2014:1–8. doi:10.1155/2014/802435
14. Ibrahim HM, Ahmed TA, Hussain MD, et al. Development of meloxicam in situ implant formulation by quality by design principle. Drug Dev Ind Pharm. 2014;40:1. doi:10.3109/03639045.2012.746360
15. Ahmed TA, Ibrahim HM, Ibrahim F, et al. Development of biodegradable in situ implant and microparticle injectable formulations for sustained delivery of haloperidol. J Pharm Sci. 2012;101(10):3753–3762. doi:10.1002/jps.23250
16. Ahmed TA. Approaches to develop PLGA based in situ gelling system with low initial burst. Pak J Pharm Sci. 2015;28(2):657–665.
17. Gurpreet S, Tanurajvir K, Ravinder Kaur AK. Recent biomedical applications and patents on biodegradable polymer-PLGA. Int J Pharmacol Pharm Sci. 2014;1(2):30–42.
18. K KM, Mohanty D, Bakshil DV. An overview on design of experiment in product formulation. Res Rev J Pharm Nanotechnol. 2016:61–67.
19. Asadi N, Zilouei H. Optimization of organosolv pretreatment of rice straw for enhanced biohydrogen production using Enterobacter aerogenes. Bioresour Technol. 2017;227:335–344. doi:10.1016/J.BIORTECH.2016.12.073
20. Maged A, Haridy S, Shamsuzzaman M, Alsyouf I, Zaied R. Statistical monitoring and optimization of electrochemical machining using Shewhart Charts and response surface methodology. Int J Eng Mater Manuf. 2018;3(2):68–77. doi:10.26776/ijemm.03.02.2018.01
21. Davies JT, Delfino SF, Feinberg CE, et al. Current and emerging uses of statins in clinical therapeutics: a review. Lipid Insights. 2016;9:13–29. doi:10.4137/LPI.S37450
22. Karasulu HY, Gundogdu E, Turk UO, et al. Enhancing solubility and bioavailability of rosuvastatin into self nanoemulsifying drug delivery system. Curr Drug Deliv. 2018;15(7):1072–1082. doi:10.2174/1567201815666180226114545
23. Li J, Yang M, Xu W. Development of novel rosuvastatin nanostructured lipid carriers for oral delivery in an animal model. Drug Des Devel Ther. 2018;12:2241–2248. doi:doi:10.2147/DDDT.S169522
24. Tarek A Ahmed, Yasser A Alharby, Abdel-Rahim M El-Helw, Khaled M Hosny, Khalid M El-Say. Depot injectable atorvastatin biodegradable in situ gel: development, optimization, in vitro, and in vivo evaluation. Drug Des Dev Ther. 2016:10 405–415.
25. Kurakula M, Ahmed TA. Co-delivery of atorvastatin nanocrystals in PLGA based in situ gel for anti-hyperlipidemic efficacy. Curr Drug Deliv. 2016;13:1–11. doi:10.2174/1567201813666151109102718
26. Wagner JG. Interpretation of percent dissolved-time plots derived from in vitro testing of conventional tablets and capsules. J Pharm Sci. 1969;58(10):1253–1257. doi:10.1002/jps.2600581021
27. Langenbucher F. Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. doi:10.1111/j.2042-7158.1972.tb08930.x
28. Hixson AW, Crowell JH. Dependence of reaction velocity upon surface and agitation. Ind Eng Chem. 1931;23(8):923–931. doi:10.1021/ie50260a018
29. Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52:1145–1149. doi:10.1002/jps.2600521210
30. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
31. Peppas NA. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60(4):110–111.
32. Baker RW, Lonsdale HS. Controlled release: mechanisms and release. In: Taquary AC, Lacey RE, editors. Controlled Release of Biological Active Agents. New York: Plenum Press; 1974: 15–71. https://www.popline.org/node/495093:
33. Chaudhary HR, Brocks DR. The single dose poloxamer 407 model of hyperlipidemia; systemic effects on lipids assessed using pharmacokinetic methods, and its effects on adipokines. J Pharm Pharm Sci. 2013;16(1):65–73. doi:10.18433/J37G7M
34. Reisin E, Liao J, Lee BS, et al. Effect of the HMG-CoA reductase inhibitor rosuvastatin on early chronic kidney injury in obese zucker rats fed with an atherogenic diet. Am J Med Sci. 2009;338(4):301–309. doi:10.1097/MAJ.0b013e3181b27195
35. Georgieva-Kotetarova MT, Kostadinova II. Effect of atorvastatin and rosuvastatin on learning and memory in rats with diazepam-induced amnesia. Folia Med (Plovdiv). 2013;55(2):58–65. doi:10.2478/folmed-2013-0018
36. Dalmoro A, Barba AA, Lamberti M, Mazzeo M, Venditto V, Lamberti G. Random l-lactide/ε-caprolactone copolymers as drug delivery materials. J Mater Sci. 2014;49(17):5986–5996. doi:10.1007/s10853-014-8317-x
37. Ibrahim HM, Ahmed TA, Hussain MD, et al. Development of meloxicam in situ implant formulation by quality by design principle. Drug Dev Ind Pharm. 2013;9045:1–8. doi:10.3109/03639045.2012.746360
38. Tefas LR, Tomuţă I, Achim M, Vlase L. Development and optimization of quercetin-loaded PLGA nanoparticles by experimental design. Clujul Med. 2015;88(2):214–223. doi:10.15386/cjmed-418
39. Xin C, Lihong W, Qiuyuan L, Hongzhuo L. Injectable long-term control-released in situ gels of hydrochloric thiothixene for the treatment of schizophrenia: preparation, in vitro and in vivo evaluation. Int J Pharm. 2014;469(1):23–30. doi:10.1016/j.ijpharm.2014.04.044
40. DesNoyer JR, McHugh AJ. The effect of Pluronic on the protein release kinetics of an injectable drug delivery system. J Control Release. 2003;86(1):15–24. doi:10.1016/S0168-3659(02)00293-6
41. Patel RB, Carlson AN, Solorio L, Exner AA. Characterization of formulation parameters affecting low molecular weight drug release from in situ forming drug delivery systems. J Biomed Mater Res A. 2010;94(2):476–484. doi:doi:10.1002/jbm.a.32724
42. Elias-Al-Mamun M, Khan HA, Dewan I, Jalil R-U. In vitro study on tamsulosin release kinetics from biodegradable PLGA in situ implants. Pak J Pharm Sci. 2009;22(4):360–367.
43. Sousa Lobo JM, Costa P. Modeling and comparison of dissolution profiles. Eur J Pharm Sci. 2001;13:123–133. doi:10.1016/S0928-0987(01)00095-1
44. Papadopoulou V, Kosmidis K, Vlachou M, Macheras P. On the use of the Weibull function for the discernment of drug release mechanisms. Int J Pharm. 2006;309(1–2):44–50. doi:10.1016/j.ijpharm.2005.10.044
45. Kapure VJ, Pande VV, Deshmukh PK. Dissolution enhancement of rosuvastatin calcium by liquisolid compact technique. J Pharm. 2013;2013:315902. doi:10.1155/2013/315902
46. Chavalitpanya K, Phattanarudee S. Poly(lactic acid)/polycaprolactone blends compatibilized with block copolymer. Energy Procedia. 2013;34:542–548. doi:10.1016/j.egypro.2013.06.783
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.