")
Back to Journals » Drug Design, Development and Therapy » Volume 10
An open-label clinical trial of agalsidase alfa enzyme replacement therapy in children with Fabry disease who are naïve to enzyme replacement therapy
Authors Goker-Alpan O, Longo N, McDonald M, Shankar S , Schiffmann R, Chang P, Shen Y, Pano A
Received 18 December 2015
Accepted for publication 1 March 2016
Published 25 May 2016 Volume 2016:10 Pages 1771—1781
DOI https://doi.org/10.2147/DDDT.S102761
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Ozlem Goker-Alpan,1 Nicola Longo,2 Marie McDonald,3 Suma P Shankar,4,5 Raphael Schiffmann,6 Peter Chang,7 Yinghua Shen,7 Arian Pano7
1Lysosomal Disorders Unit, Fairfax, VA, 2University of Utah, Salt Lake City, UT, 3Department of Pediatrics, Duke University, Durham, NC, 4Department of Ophthalmology, 5Department of Human Genetics, Emory University School of Medicine, Atlanta, GA, 6Institute of Metabolic Disease, Baylor Research Institute, Dallas, TX, 7Shire, Lexington, MA, USA
Background: Following a drug manufacturing process change, safety/efficacy of agalsidase alfa were evaluated in enzyme replacement therapy (ERT)-naïve children with Fabry disease.
Methods: In an open-label, multicenter, Phase II study (HGT-REP-084; Shire), 14 children aged ≥7 years received 0.2 mg/kg agalsidase alfa every other week for 55 weeks. Primary endpoints: safety, changes in autonomic function (2-hour Holter monitoring). Secondary endpoints: estimated glomerular filtration rate, left ventricular mass index (LVMI), midwall fractional shortening, pharmacodynamic parameters, and patient-reported quality-of-life.
Results: Among five boys (median 10.2 [range 6.7, 14.4] years) and nine girls (14.8 [10.1, 15.9] years), eight patients experienced infusion-related adverse events (vomiting, n=4; nausea, n=3; dyspnea, n=3; chest discomfort, n=2; chills, n=2; dizziness, n=2; headache, n=2). One of these had several hypersensitivity episodes. However, no patient discontinued for safety reasons and no serious adverse events occurred. One boy developed immunoglobulin G (IgG) and neutralizing antidrug antibodies. Overall, no deterioration in cardiac function was observed in seven patients with low/abnormal SDNN (standard deviation of all filtered RR intervals; <100 ms) and no left ventricular hypertrophy: mean (SD) baseline SDNN, 81.6 (20.9) ms; mean (95% confidence interval [CI]) change from baseline to week 55, 17.4 (2.9, 31.9) ms. Changes in SDNN correlated with changes in LVMI (r=-0.975). No change occurred in secondary efficacy endpoints: mean (95% CI) change from baseline at week 55 in LVMI, 0.16 (-3.3, 3.7) g/m2.7; midwall fractional shortening, -0.62% (-2.7%, 1.5%); estimated glomerular filtration rate, 0.15 (-11.4, 11.7) mL/min/1.73 m2; urine protein, -1.8 (-6.0, 2.4) mg/dL; urine microalbumin, 0.6 (-0.5, 1.7) mg/dL; plasma globotriaosylceramide (Gb3), -5.71 (-10.8, -0.6) nmol/mL; urinary Gb3, -1,403.3 (-3,714.0, 907.4) nmol/g creatinine, or clinical quality-of-life outcomes.
Conclusion: Fifty-five weeks’ agalsidase alfa ERT at 0.2 mg/kg every other week was well tolerated. Disease progression may be slowed when ERT is started prior to major organ dysfunction.
Trial registration: https://ClinicalTrials.gov identifier NCT01363492.
Keywords: agalsidase alfa, efficacy, enzyme replacement therapy, Fabry disease, pediatric study, safety
Introduction
Fabry disease (FD) is an X-linked, glycosphingolipid storage disorder caused by deficient activity of the lysosomal enzyme alpha-galactosidase A, resulting from alterations in the alpha-galactosidase A gene.1 Alpha-galactosidase A deficiency is thought to cause progressive accumulation of globotriaosylceramide (Gb3) in the lysosomes of vascular endothelial cells of multiple organ systems: kidneys, heart, skin, and brain, leading to significant morbidity2 and premature death.3 More recently, the deacylated Gb3 globotriaosylsphingosine (lysoGb3) was also shown to occur at high concentrations in the plasma of patients with FD and was hypothesized to play a role in the pathogenesis of this disorder.4
The clinical onset of FD manifestations usually occurs during late childhood and adolescence, and signs and symptoms become more severe with increasing age. It was originally thought that females experienced a milder form of FD,5 but studies have shown that the disease can be just as severe as in males.6,7 Furthermore, age at both symptom onset and diagnosis has been found to be more variable in females, although may occur later than in males.8,9 The clinical phenotype of FD in pediatric patients has been described in several studies. Similar to studies in adults, it shows a higher incidence and an earlier onset of symptoms in male than in female patients. These include neurological manifestations (acroparesthesias, chronic neuropathic pain, hypo-anhidrosis, tinnitus, and hearing loss), gastrointestinal symptoms (abdominal pain and diarrhea), angiokeratomas, and ocular abnormalities (cornea verticillata, tortuous retinal vessels, and subcapsular cataracts). In addition, signs of major organ damage (microalbuminuria or proteinuria, urinary hyperfiltration, left ventricular hypertrophy [LVH], and stroke) are occasionally encountered in children with FD,10–12 although these generally manifest in adulthood. Some of the cardiac manifestations of FD observed in childhood include mitral valve prolapse, alterations in left ventricular geometry, and increased left ventricular mass (LVM) independently in both boys and girls.13
Enzyme replacement therapy (ERT) in children has the potential to halt disease progression and reverse some of the signs and symptoms. Previous studies determined an initial safety profile and efficacy follow-up of agalsidase alfa treatment in children with FD.14–17 An initial beneficial response of cardiac autonomic innervation and an improvement in heart rate variability (HRV) were previously reported with agalsidase alfa therapy.16
The method of manufacturing agalsidase alfa has changed from a roller bottle to a bioreactor process. Per International Conference on Harmonisation (ICH) Q5E guidance,18 the results of in vitro and in vivo analyses showed that agalsidase alfa manufactured using both processes is comparable in terms of pharmacokinetics, biodistribution, and pharmacodynamics. This study using agalsidase alfa manufactured via the bioreactor process was performed as a comparison to similar studies that used agalsidase alfa produced via the roller bottle process16,17 to satisfy a specific obligation to the European Medicines Agency to provide additional clinical data. The primary objective was to assess the safety of agalsidase alfa (0.2 mg/kg body weight every other week) in ERT-naïve children with FD. Efficacy and pharmacodynamic parameters are also reported.
Methods
Study design
In an open-label, multicenter clinical trial, treatment-naïve children (aged 7 to <18 years) with FD received agalsidase alfa from May 2011 to April 2013 (ClinicalTrials.gov identifier NCT01363492). FD diagnosis was confirmed by measuring alpha-galactosidase A activity in serum, leukocytes, or fibroblasts in male subjects, or through molecular analysis of alpha-galactosidase A gene mutations in both females and males. Written informed consent/assent was required from each patient, their parent(s), or legal guardian(s) before participation. The study complies with Good Clinical Practice guidelines and the Declaration of Helsinki, and was reviewed by local institutional review boards. The study procedures were approved and conducted according to Western Institutional Review Board, Olympia, WA; Emory University Institutional Review Board, Atlanta, GA; The University of Utah Institutional Review Board, Salt Lake City, UT; Copernicus Group IRB, Durham, NC; and Baylor Research Institute Office of Research Subject Protection, Dallas, TX clinical protocol.
Agalsidase alfa, at 0.2 mg/kg body weight intravenously (IV), was infused over 40 minutes (±10 minutes) every other week for up to 55 weeks.19 Infusions and clinical assessments, including height and weight measurements, were performed at the clinical sites for at least 13 weeks, with the option for home infusions after this period, dependent on investigator consent. A post hoc analysis calculated Z-scores for height and body mass index (BMI) using World Health Organization standards.20
Safety evaluation
Safety was a primary endpoint and assessments included monitoring adverse events (AEs), measuring anti-agalsidase alfa antibody formation, clinical laboratory testing, physical examinations, and 12-lead electrocardiograms (ECGs). All AEs that occurred from the time of enrollment to 30 (±7) days after the last treatment dose were recorded by the investigator in terms of potential relationship to agalsidase alfa and severity. Mild and moderate AEs were defined as causing none or some limitations to normal activities, respectively, and severe AEs as causing inability to carry out usual activities. Any AEs that occurred on or after the first agalsidase alfa infusion and within 37 days of the final infusion were regarded as treatment-emergent AEs. Any AEs that occurred within 12 hours after infusion started and were deemed possibly or probably related to agalsidase alfa were regarded as infusion-related AEs. Any AE that was life-threatening, required or prolonged hospitalization, or resulted in persistent disability was recorded as a serious AE.
Anti-agalsidase alfa antibody testing was performed at the Shire Bioanalytical and Biomarker Development laboratory using a tiered approach. Briefly, all samples were first screened using enzyme-linked immunosorbent assays (ELISAs) for the presence of anti-agalsidase alfa immunoglobulin G, A, M, E (IgG, IgA, IgM, and IgE) isotype antibodies. When a signal of any sample met the IgG, IgA, IgM, or IgE positive cut-point criteria, the same isotype-specific ELISA was performed again to confirm the presence of antibodies. Only confirmed positive samples were considered anti-agalsidase alfa antibody positive and subjected to titer determination using the same ELISA format by serial twofold dilution until the signal fell below the screening cut point. Samples that were confirmed positive for the presence of anti-agalsidase alfa IgG, IgA, IgM, or IgE antibodies were further evaluated for the presence of neutralizing antibodies using an enzyme activity inhibition assay.
Efficacy evaluation
Similar to previous pediatric studies of agalsidase alfa ERT,13,16,17 the primary efficacy objective herein was to assess changes in the autonomic nervous system using HRV in children with FD. Secondary efficacy objectives were to assess cardiac structure using left ventricular mass indexed to height (LVMI g/m2.7) and cardiac function via midwall fractional shortening (MFS), renal function (via estimated glomerular filtration rate [eGFR], assessing creatinine clearance from 8-hour urine samples and calculating serum creatinine using Counahan–Barratt and Cockcroft–Gault equations), agalsidase alfa pharmacodynamics (via plasma and urine levels of Gb3 at baseline and after ERT initiation), plus outcomes of clinical quality-of-life indices.
Autonomic function was evaluated via changes in the HRV parameters SDNN (standard deviation of all filtered RR intervals for the length of the analysis), r-MSSD (square root of the sum of squares of differences between adjacent filtered RR intervals over the length of the analysis), and pNN50 (percentage of differences between adjacent filtered RR intervals that is >50 ms for the whole analysis). Measurement of SDNN describes both long-term (sympathetic) and short-term (parasympathetic) variability, reflecting overall autonomic nervous system activity in the heart, whereas r-MSSD and pNN50 both describe short-term variability, primarily reflecting parasympathetic influences on heart rate. Reduced parasympathetic stimulation of the heart has been reported in male pediatric patients with FD.13 Two-hour continuous ECG (Holter) monitoring13 was performed at rest and heart rate was assessed at a central laboratory under normal ranges estimated using a published correlation formula for HRV and age.21
Cardiac structure and function were evaluated using echocardiography to determine LVMI and MFS (both calculated using published formulae);22 echocardiograms were analyzed in a central laboratory by a single, blinded reader. Normal reference values for LVMI were prospectively defined as <51 g/m2.7 in males and <48 g/m2.7 in females. These correspond to a conservative cutoff point that determines the presence of LVH in children and adolescents. However, this cutoff point also has established prognostic value in adults with hypertension,23 and is consistent with Fabry literature in both children and adults.24 Renal function was assessed using eGFR, calculated using the Counahan–Barratt equation, and urine protein and microalbumin values.
Finally, clinical quality-of-life outcomes were assessed using recommended methodologies for each of the following instruments: the Brief Pain Inventory (BPI),25 Health Utilities Index (HUI2 and HUI3),26 and Childhood Health Questionnaire (CHQ-PF50; physical [PhS] and psychosocial scores [PsS]).27 The 10-point BPI is used to measure pain intensity and its interference in various functional domains. The HUI and CHQ assess quality-of-life using both self- and proxy-assessed forms. For the HUI, a score of 1.0 indicates best health and 0.0 worst health; for the CHQ, scoring ranges from 0 to 100 and is standardized so that increments above or below 50 (average) indicate change. For all three patient-reported outcomes, the forms were to be filled out before infusions and patients had to be older than 12 years to complete the forms by themselves.
Pharmacodynamic evaluation
Plasma and urinary Gb3 levels were measured at prespecified time points in blood samples from fasted patients and 8-hour urine samples, respectively. Liquid chromatography-tandem mass spectrometry, performed at a central laboratory using an AB Sciex API 5000 triple quadrupole mass spectrometer (Concord, Canada) coupled with a Waters Acquity UPLC system (Milford, MA, USA), was used to detect and monitor eleven different isoforms of Gb3, which were then combined to calculate total values (method validated both in-house, Shire, and at the central laboratory).
Statistical analysis
The safety population comprised all patients who enrolled in the study and received at least one dose of agalsidase alfa. Descriptive statistics were used to summarize demographic and baseline characteristics (also stratified by sex), treatment duration and exposure, efficacy endpoints, clinical laboratory measurements, and immunogenicity results. The effects of anti-agalsidase alfa antibodies on infusion-related AEs and on the clinical assessments above were analyzed by anti-agalsidase alfa antibody status, where cases with at least one positive anti-agalsidase alfa antibody test during the study were considered positive, and cases with no positive test were considered negative.
Subgroup analyses by sex were performed for plasma and urinary Gb3, and HRV parameters and LVMI. Furthermore, changes in SDNN and LVMI over time were correlated in patients with baseline SDNN scores <100 ms as a post hoc analysis to determine the possibility of a concurrent treatment effect, and Z-scores were calculated for the HRV parameters as an ad hoc analysis to compare normal ranges. SAS Software version 9.3 was used for all statistical analyses of parameters reported herein (SAS Institute Inc., Cary, NC, USA).
The observed values and change from baseline for the BPI parameters, HUI2 and HUI3 scores, and CHQ-PF50 PhS and PsS were descriptively summarized.
Results
Clinical and demographic characteristics
Fourteen patients comprised the safety population (Table 1) and all 14 (100%) received infusions of agalsidase alfa on all scheduled dates and completed the study. The median duration of agalsidase alfa exposure was 54.5 (range 54.0, 59.0) weeks. Five (35.7%) of the patients were male, 13 (92.9%) were white, and one (7.1%) was Hispanic. The median age overall at baseline was 11.8 (range 6.7, 15.9) years: for males it was 10.2 (6.7, 14.4) years and for females 14.8 (10.1, 15.9) years. The median height and height Z-score at baseline were 150 (range 122, 167) cm and −0.04 (−1.10, 2.36), respectively. The median BMI and BMI Z-score at baseline were 20.8 (range 14.0, 33.3) and 1.06 (−1.82, 3.14), respectively.
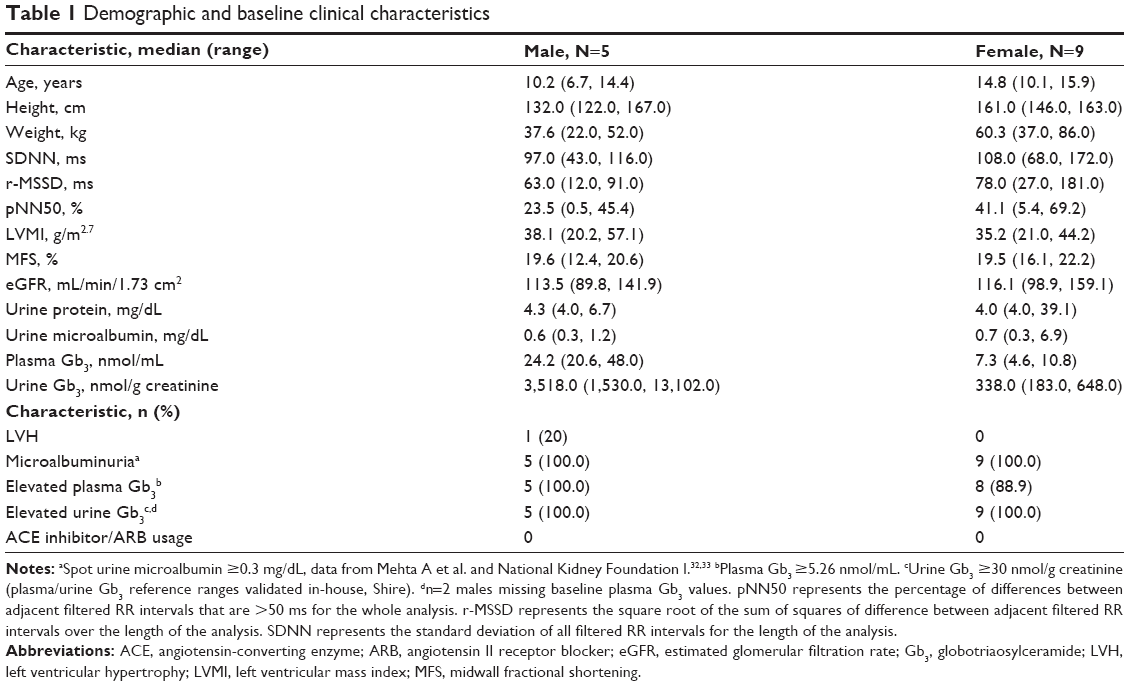 | Table 1 Demographic and baseline clinical characteristics |
Safety of agalsidase alfa
Clinical laboratory values, vital signs, and results from physical examinations and ECGs showed no change during the study. The overall mean change from baseline in height Z-score by week 55 was −0.18 (SD 0.31; median −0.13 [range −0.76, 0.25]) and for BMI it was 0.06 (SD 0.63; median −0.09 [range −0.75, 1.50]), indicating stable growth during the treatment period.
Adverse events
At least one treatment-emergent AE was reported by all 14 patients (100%) during the study, most being mild (5/14; 35.7%) or moderate (6/14; 42.9%) in severity. The most common treatment-emergent AEs (occurring in ≥25%) were pyrexia (n=6; 42.9%), cough, diarrhea, headache, nausea, pain in the extremity, upper respiratory tract infection, and vomiting (each n=4; 28.6%). Three patients (21.4%) experienced severe treatment-emergent AEs of mouth ulceration, tooth malformation, chest discomfort, alveolar ostitis, facial bone fracture, dyspnea, and angioedema (each reported by one patient only).
Eight patients (57.1%) experienced at least one infusion-related AE. The most common were vomiting (n=4; 28.6%); nausea and dyspnea (each n=3; 21.4%); chest discomfort, chills, dizziness, and headache (each n=2; 14.3%). One patient had a likely allergic response with a number of severe infusion-related reactions at week 49. These included angioedema, dyspnea, and chest tightness, all of which resolved following emergency treatment with 2.5 mg salbutamol inhaler and 20 mg intravenous methylprednisolone. The infusion was later completed at a slower rate. Of note, this patient had a prior medical history of reactive airway disease, seasonal allergies, and eczema on the arms and thighs. At week 47, the same patient received a partial dose after the infusion was interrupted due to infusion-related AEs of nausea and vomiting. Both events resolved, but vein access could not be regained. Full doses were received for all subsequent infusions through week 55, although all were extended to over 1 hour. The patient was negative for IgE at all assessments.
Overall, infusion-related AEs were more likely to occur within the first 6 months of treatment with agalsidase alfa (21 patients with infusion-related AEs) than during the last 6 months (ten patients). No deaths or serious AEs occurred during the study and there were no discontinuations due to an AE.
Anti-agalsidase alfa antibodies
One patient tested positive for both IgG and neutralizing antibodies at weeks 49 and 55 (a summary of efficacy results for this patient is shown in Table 2). This patient reported 26 mild or moderate infusion-related AEs and subsequently tested positive for anti-agalsidase alfa antibodies at week 49.
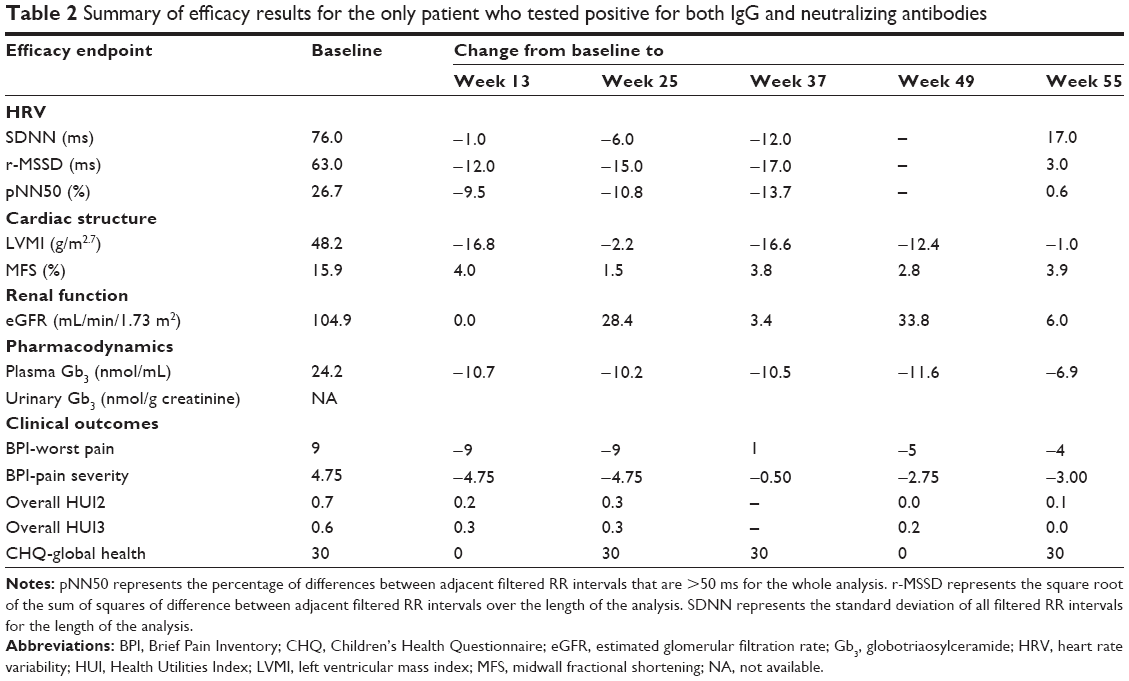 | Table 2 Summary of efficacy results for the only patient who tested positive for both IgG and neutralizing antibodies |
Efficacy and pharmacodynamic parameters
Baseline Z-scores indicated that the safety population had low to abnormal values of SDNN (Figure 1) and normal values of pNN50 and r-MSSD for age. Mean (SD) baseline values were: SDNN 103.46 (32.93) ms; pNN50 32.79% (20%); and r-MSSD 75.92 (45.75) ms. Median baseline values were: SDNN 100.00 (range 43.0, 172.0) ms; pNN50 26.74% (0.5, 69.2%); r-MSSD 67.00 (12.0, 181.0) ms. A subset of patients with mean baseline HRV values below the population median was evaluated to further assess treatment effects. In seven patients with SDNN ≤100 ms, a slight but statistically nonsignificant improvement or no change in SDNN was observed over the course of the study (mean [SD] value at baseline: 81.6 [20.9] ms, mean change [95% confidence interval, CI] from baseline to week 55: 17.4 [2.9, 31.9] ms; median value at baseline: 88.0 [range 43.0, 100.0] ms, median change [95% CI] from baseline to week 55: 19.0 [−2.0, 45.0] ms). For the six patients who had baseline pNN50 ≤26.7%, no change was observed overall as the mean (SD) value at baseline was 16.1% (10.5%; median 20.4% [range 0.5%, 26.2%]) and the mean change (95% CI) at week 55 was 7.7% (−6.5, 21.8%; median change [95% CI] 12.3% [−16.1%, 19.7%]). Similarly, no difference overall was observed in seven patients who had baseline r-MSSD ≤67 ms as the mean (SD) at baseline was 44.6 (19.4) ms (median 48.0 [range 12.0, 67.0] ms) and the mean change (95% CI) from baseline at week 55 was 27.0 (−21.3, 75.3) ms (median change [95% CI] 18.0 [−41.0, 124.0] ms).
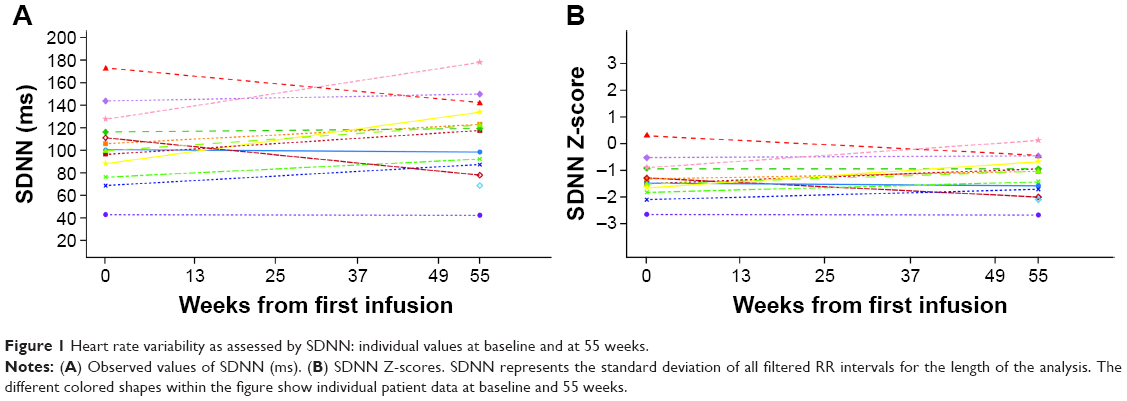 | Figure 1 Heart rate variability as assessed by SDNN: individual values at baseline and at 55 weeks. |
When evaluated by sex, changes from baseline over the course of the study in all three HRV indices were not statistically significant overall (mean [95% CI] change from baseline by week 55 for SDNN: males 7.60 [−5.56, 20.76] ms, females 12.25 [−13.28, 37.78] ms; pNN50: males −1.72% [−14.03%, 10.59%], females −5.64% [−26.7%, 15.4%]; r-MSSD: males 3.00 [−41.67, 47.67] ms, females 0.50 [−53.45, 54.45] ms. Median values for change from baseline by week 55 for SDNN: males 2.00 [range −2.0, 21.0] ms, females 18.00 [−33.0, 51.0] ms; pNN50: males 0.11% [−16.1%, 11.3%], females −5.12% [−58.3%, 19.7%]; r-MSSD: males 1.00 [−41.0, 59.0] ms, females −0.50 [−98.0, 124.0] ms). Males generally had numerically lower values of each HRV parameter than females.
For structural cardiac parameters, the mean [SD] values of LVMI were within the normal range (<51 g/m2.7 for males; <48 g/m2.7 for females) at baseline for the majority of both males (38.75 [14.53] g/m2.7) and females (33.50 [7.08] g/m2.7) and remained so throughout the study (mean [95% CI] change from baseline at week 55 −0.74 [−6.01, 4.53] g/m2.7 for males and 0.66 [−4.77, 6.08] g/m2.7 for females). One male patient had LVH at baseline, with an LVMI value of 57.1 g/m2.7. At week 55 his LVMI value was 59.9 g/m2.7; the interim values both increased and decreased, although all were below his baseline value (Figure 2). Median baseline LVMI was 38.07 (range 20.2, 57.1) g/m2.7 for males and 35.22 (21.0, 44.2) g/m2.7 for females; median change from baseline at week 55 was −1.00 (−6.3, 4.0) g/m2.7 for males and 3.23 (−8.5, 8.8) g/m2.7 for females.
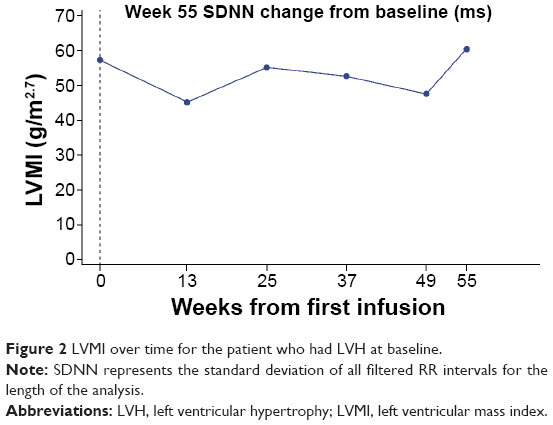 | Figure 2 LVMI over time for the patient who had LVH at baseline. |
There was a strong correlation between increases in SDNN and decreases in LVMI (r=−0.975; Figure 3) in patients with low to abnormal HRV (SDNN <100 ms) and no LVH at baseline. However, a weaker correlation was found between these parameters for patients with SDNN ≥100 ms at baseline (r=0.229).
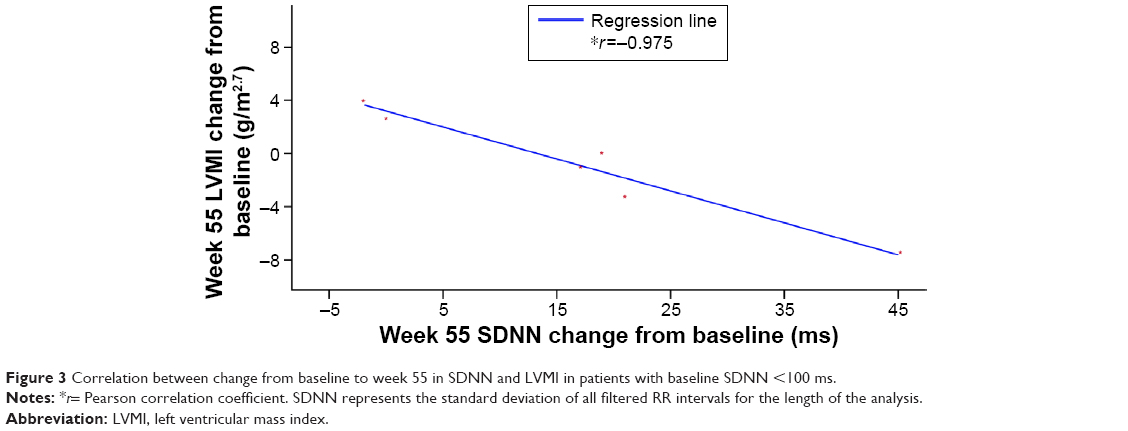 | Figure 3 Correlation between change from baseline to week 55 in SDNN and LVMI in patients with baseline SDNN <100 ms. |
MFS values at baseline were in agreement with MFS results previously reported in normal children28 (mean [SD] 17.75% [3.57%] for males and 19.11% [2.55%] for females; median 19.60% [range 12.4%, 20.6%] for males and 19.52% [16.1%, 22.2%] for females). Changes from baseline were small and remained within the normal range (overall mean change [95% CI] from baseline by week 55: −0.62% [−2.69%, 1.46%]; overall median change from baseline by week 55: −1.27 [range −6.1, 4.6]), as also found when evaluated by sex.
The mean (SD) eGFR at baseline was 117.5 (18.53) mL/min/1.73 m2 (median 115.7 [range 89.8, 159.1] mL/min/1.73 m2), with a mean (95% CI) change from baseline at week 55 of 0.15 (−11.39, 11.70) mL/min/1.73 m2 (median 0.72 [−27.5, 35.9] mL/min/1.73 m2). When examined by sex, slightly lower values for all renal function parameters were found in males compared with females (although the differences were not statistically significant). The mean (SD) eGFR at baseline was 115.3 (19.96) mL/min/1.73 m2 for males and 118.7 (18.81) mL/min/1.73 m2 for females. Median baseline values were 113.5 (range 89.8, 141.9) mL/min/1.73 m2 for males and 116.1 (98.9, 159.1) mL/min/1.73 m2 for females. Mean eGFR values did not change over the course of the study; for males, the mean (95% CI) change from baseline by week 55 was 4.26 (−21.20, 29.71) mL/min/1.73 m2 and for females it was −2.41 (−18.39, 13.57) mL/min/1.73 m2 (Figure 4). The median change from baseline by week 55 was 3.44 (range −24.1, 33.8) mL/min/1.73 m2 for males and 0.31 (−27.5, 35.9) mL/min/1.73 m2 for females.
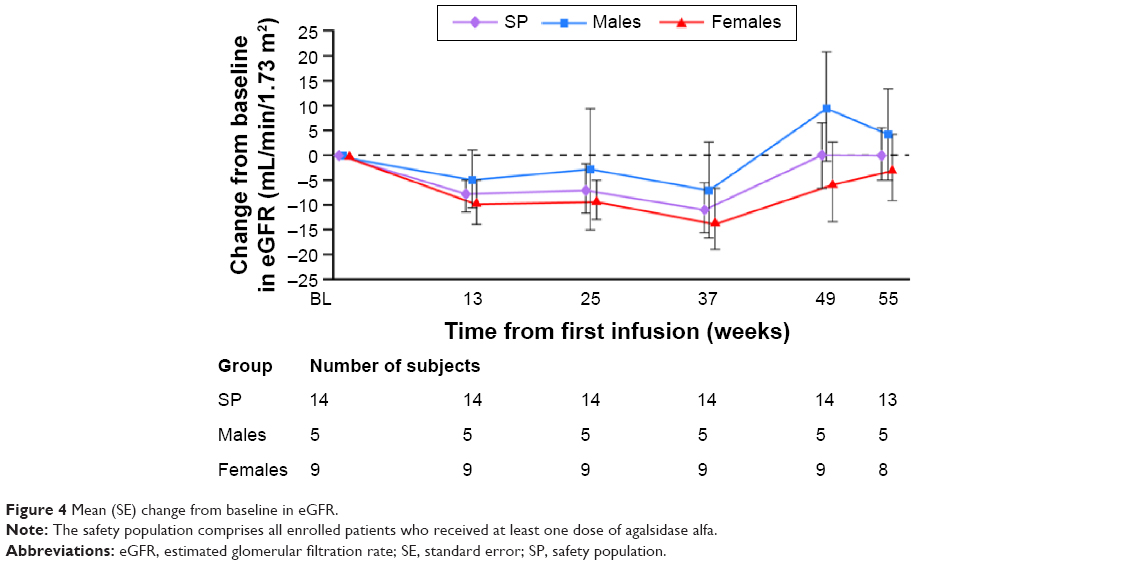 | Figure 4 Mean (SE) change from baseline in eGFR. |
The results for urine protein and microalbumin show no difference over the course of the study. Overall mean (SD) baseline values and mean (95% CI) changes from baseline by week 55 were: urine protein, 7.43 (9.26) and −1.79 (−5.95, 2.37) mg/dL, and urine microalbumin, 1.16 (1.70) and 0.61 (−0.53, 1.75) mg/dL, respectively. Overall median baseline values and changes from baseline at week 55 were 4.05 (range 4.0, 39.1) and 0.00 (−26.2, 3.5) mg/dL for urine protein, and 0.65 (0.3, 6.9) and 0.00 (−0.7, 7.1) mg/dL for urine microalbumin, respectively.
The mean plasma Gb3 level in males showed a considerable decrease from baseline by week 13, with a steady reduction maintained over the remainder of the study (Figure 5); this reduction was lower in females, who had lower baseline levels, and occurred from week 25 onward. Mean (SD) plasma Gb3 levels at baseline were 28.20 (11.36) nmol/mL in males and 7.34 (1.92) nmol/mL in females (median values were 24.2 [range 20.6, 48.0] nmol/mL in males and 7.3 [4.6, 10.8] nmol/mL in females). Mean (95% CI) changes from baseline by week 55 were −15.18 (−25.9, −4.5) nmol/mL and −0.46 (−1.39, 0.47) nmol/mL, respectively (median changes from baseline were −12.9 [range −29.8, −6.9] nmol/mL in males and −0.9 [−2.2, 1.4] nmol/mL in females).
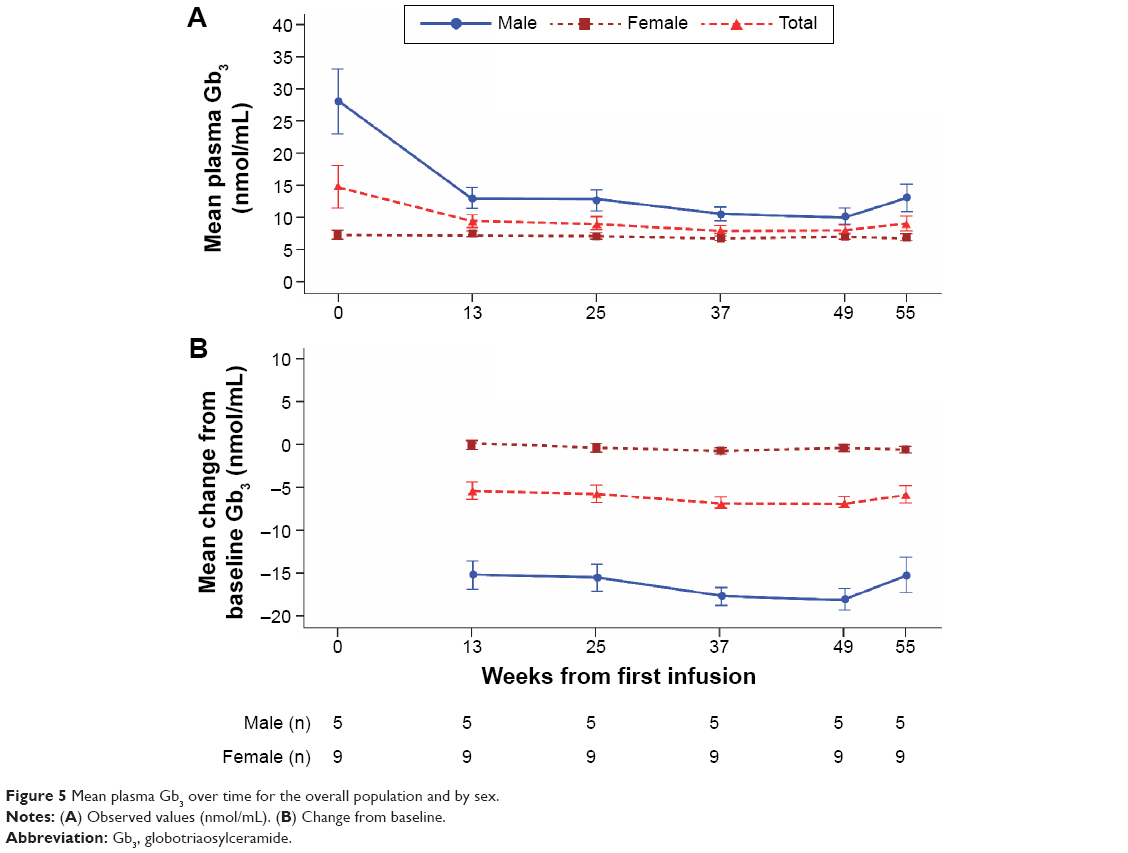 | Figure 5 Mean plasma Gb3 over time for the overall population and by sex. |
Mean (SD) urinary Gb3 levels at baseline were 6,050.0 (6,187.57) nmol/g creatinine in males and 350.11 (155.56) nmol/g creatinine in females; they decreased from baseline at each time point in both (mean change [95% CI] from baseline by week 55: −4,952.0 [−22,067.84, 12,163.84] and −220.33 [−329.78, −110.89] nmol/g creatinine, respectively). Median baseline values and change from baseline by week 55 were 3,518.0 (range 1,530.0, 13,102.0) and −3,160.0 (−12,561.0, 865.0) nmol/g creatinine in males, and 338.0 (183.0, 648.0) and −206.0 (−501.0, −28.0) nmol/g creatinine in females.
Among the patient-reported outcomes for quality-of-life, the BPI variable “worst pain last week”, primarily used to assess changes in neuropathic pain, showed a reduction in pain with a median (range) change from baseline at week 55 of −3.0 (−7, 0) points. The other patient-reported outcome assessments, BPI, HUI, and CHQ, showed little variation over the course of the 55-week study. Overall health-related quality-of-life as assessed by the HUI2 showed a median (range) change from baseline at week 55 of 0.06 (−0.1, 0.4) points, and for the HUI3 it was 0.05 (−0.2, 0.6) points. The CHQ-PF50 raw scores were essentially unchanged from baseline. The median (range) change was −0.6 (−12.9, 31.4) for CHQ-PF50 PhS and 2.16 (−10.7, 16.6) for PsS.
Discussion
While safety and efficacy of ERT with agalsidase alfa have previously been studied in children with FD,14,15 the present study evaluated agalsidase alfa in ERT-naïve children after a change in the manufacturing process of the drug. In agreement with earlier studies,14,15 agalsidase alfa was well tolerated overall, with high rates of study completion and treatment adherence (100% infusion attendance rate). Of note, the majority of patients in this study were females, who typically show later symptom onset than males.7
In general, treatment-emergent AEs were consistent with the known safety profile of agalsidase alfa and typical FD sequelae. Similar to previous pediatric studies, no life-threatening treatment-emergent AEs or deaths were reported,14,15 and the majority of patients reported treatment-emergent AEs that were mild or moderate in intensity. All treatment-emergent AEs resolved and no new safety concerns were identified.
Despite protocol differences between studies in the reporting of infusion-related AEs (unlike in previous studies, in this study, all adverse drug reactions that occurred within 12 hours were considered infusion-related AEs), the overall type and rate of adverse drug reactions reported by patients in this study are similar to those found in earlier studies.14,15 The incidence of infusion-related AEs decreased after the first 6 months of ERT with agalsidase alfa.
The overall incidence of IgG antibodies is similar to previous experience,14,15 and in the majority, the formation of anti-agalsidase alfa antibodies had no apparent short-term impact on the safety or efficacy of agalsidase alfa. The male patient who tested positive for both IgG and neutralizing antibodies had stable efficacy parameters throughout the study.
An impaired HRV is considered to be an early sign of FD, earlier than that of increasing LVM, and can be present by adolescence in both sexes despite the lack of major organ dysfunction.2 Previous HRV analyses in children with FD revealed that boys, but not girls, had significantly reduced HRV, reflecting a reduction in parasympathetic stimulation of the heart.13 In boys, HRV indices, particularly SDNN, were shown to improve after treatment with agalsidase alfa for 6 months.16 In long-term studies, there was a sustained improvement in time-domain indices of HRV in boys on agalsidase alfa ERT over a period of 4 years.15,17 In the current study, most of the HRV indices were normal at baseline. This study showed no deterioration in cardiac function over the 55 weeks of treatment, including children with low or abnormal HRV (SDNN) but without evidence of LVH. The fact that a reduction in LVM strongly correlated with an increase in SDNN in patients with low to abnormal SDNN at baseline indicates that early intervention could be beneficial in preventing LVH. This correlation was not shown in patients with normal SDNN. A longer study of agalsidase alfa treatment in such patients would be needed to evaluate whether improvements in cardiac function could occur, but it could be possible that starting agalsidase alfa ERT in childhood, before major organ dysfunction has occurred, can help prevent the longer-term complications of FD.
Biopsy data in patients with FD show that glycosphingolipid deposits are present in cardiac myocytes and conduction tissue.29 Therefore, the possibility of a concurrent treatment effect in patients with advanced conduction abnormalities was examined. A subset of patients with the lowest SDNN at baseline was selected; in these patients, LVMI was generally within the normal range (all except for one patient, whose LVMI values varied throughout and are difficult to interpret) while SDNN was low or abnormal for age, as assessed by Z-scores. The change in SDNN for patients with low or abnormal SDNN was plotted against the change in LVMI. The selected patients had a baseline SDNN that was lower than the median of the study population (ie, SDNN <100 ms). The cutoff point of 100 ms also represents an important prognostic cutoff point for HRV.30,31 In fact, there is general consensus that HRV is considered normal (confers good prognosis) if SDNN >100 ms. The corollary is not true, as there is no clear consensus about which SDNN values are associated with a poor prognosis. A strong correlation was observed between increasing SDNN and decreasing LVMI after treatment began in patients with abnormal baseline SDNN. For completeness, changes in SDNN and LVMI were also plotted for patients with preserved HRV, as defined by an SDNN ≥100 ms (or at or above the median of this study). As expected, in the latter group, no correlation was observed. However, almost all patients had normal baseline values of LVMI and MFS,28 and it was not possible to assess whether a similar correlation would hold true in patients with more advanced disease, such as patients with low or abnormal HRV and LVH. Overall, these results indicate a potential stabilization or trend toward improvement in cardiac function when ERT is started as soon as children show low HRV and before they develop LVH, and are consistent with previous findings that showed increases in HRV and stable cardiac function and structure over time.15,16
Renal parameters showed no change during treatment in the majority of patients. Changes in levels of urinary or plasma Gb3 have not been correlated with efficacy of ERT; however, they can demonstrate the pharmacological effect of ERT. In this study, a desired pharmacological effect of treatment with agalsidase alfa was reflected by a general decrease in plasma and urinary Gb3, more so in males than females, as found previously.14 The female patients in this study had notably lower baseline plasma Gb3 levels compared with males.
The patient-reported quality-of-life outcome measures generally showed average to good results at baseline and did not change throughout the duration of the study. A benefit of ERT with agalsidase alfa was reflected by a reduction from baseline in the BPI “worst pain last week” subscale.
Some limitations of this current study include the following. It was primarily designed to assess safety and thus was an open-label clinical trial with no placebo control for comparison. The number of patients in the total safety group and thus subgroup analyses was small; only descriptive statistics were used. Furthermore, patients with FD in this study were young, with no overt clinical manifestations of the disease to reverse. Finally, for this study, comparisons with similar studies on agalsidase alfa only focus on general trends because of the differences in protocol design,14,15 and comparisons with natural history cohorts were not possible due to the lack of data on the natural course of FD in children.
In addition to assessing the safety and efficacy of agalsidase alfa and despite the small sample size, this study represents a valuable addition to the body of evidence of ERT treatment effects in children, since very few such studies have been conducted in children to date.
Conclusion
The safety profile of agalsidase alfa in this study was consistent with that found in previous studies on children with FD. Agalsidase alfa was generally well tolerated in both males and females and no apparent short-term impact on safety or efficacy from IgG or neutralizing antibodies was found. Treatment with agalsidase alfa may slow the progression of cardiac, renal, and autonomic nervous system parameters. Long-term studies are needed to confirm whether later-onset complications of FD could be prevented with timely agalsidase alfa therapy in the pediatric age group.
Acknowledgments
Medical writing support was provided by Tina Rose, of Excel Scientific Solutions, and was funded by Shire.
Author contributions
All authors conducted the study and collected the data. PC performed statistical analyses. All authors participated fully in data analysis and interpretation, and helped draft the manuscript. All authors read and approved the final manuscript. Providing patient anonymity can be assured, patient-level data will be made available upon reasonable request.
Disclosure
AP is now an employee of Dicerna Pharmaceuticals. OG-A has received research support from Actelion, Amicus, Genzyme Corp, Pfizer-Protalix Biotherapeutics, and Shire, as well as payments for consultancy from Actelion, Pfizer-Protalix Biotherapeutics, and Shire, and payments for speaker bureaus from Actelion, Genzyme, and Shire. NL has received payments for consultancy from BioMarin and Shire, and research support from Amicus, BioMarin, Genzyme, Hyperion, Pfizer-Protalix Biotherapeutics, and Shire. MM has received training grant funding from Genzyme. SPS has been site Primary Investigator in clinical trials and received research support and educational grants from Actelion, Amicus, Biomarin, Genzyme, Protalix, and Shire, and has received honoraria for speaker bureaus from Genzyme and Shire. RS has received honoraria, travel reimbursement, and research support from Amicus, Genzyme, and Shire. PC and YS are employees of Shire. At the time of writing, AP was an employee of Shire. The authors report no other conflicts of interest in this work.
References
Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337(2):319–335. | ||
Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. | ||
Schiffmann R, Kopp JB, Austin HA 3rd, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–2749. | ||
Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105(8):2812–2817. | ||
Desnick RJ, Ioannou YA, Eng CM. α-Galactosidase A deficiency: Fabry disease. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001:3733–3774. | ||
Wang RY, Lelis A, Mirocha J, Wilcox WR. Heterozygous Fabry women are not just carriers, but have a significant burden of disease and impaired quality of life. Genet Med. 2007;9(1):34–45. | ||
Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(2):112–128. | ||
Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M; European FOS Investigators. Natural history of Fabry disease in females in the Fabry Outcome Survey. J Med Genet. 2006;43(4):347–352. | ||
MacDermot KD, Holmes A, Miners AH. Natural history of Fabry disease in affected males and obligate carrier females. J Inherit Metab Dis. 2001;24:13–14, discussion 11–12. | ||
Sestito S, Ceravolo F, Concolino D. Anderson-Fabry disease in children. Curr Pharm Des. 2013;19(33):6037–6045. | ||
Ries M, Gupta S, Moore DF, et al. Pediatric Fabry disease. Pediatrics. 2005;115(3):e344–e355. | ||
Ramaswami U, Whybra C, Parini R, et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta Paediatr. 2006;95(1):86–92. | ||
Kampmann C, Wiethoff CM, Whybra C, Baehner FA, Mengel E, Beck M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008;97(4):463–469. | ||
Ramaswami U, Wendt S, Pintos-Morell G, et al. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta Paediatr. 2007;96(1):122–127. | ||
Schiffmann R, Martin RA, Reimschisel T, et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr. 2010;156(5):832–837, e831. | ||
Ries M, Clarke JT, Whybra C, et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics. 2006;118(3):924–932. | ||
Schiffmann R, Pastores GM, Lien YH, et al. Agalsidase alfa in pediatric patients with Fabry disease: a 6.5-year open-label follow-up study. Orphanet J Rare Dis. 2014;9(1):169. | ||
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline: Comparability of Biotechnological/Biological Products Subject to Changes in their Manufacturing Process; 2004. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q5E/Step4/Q5E_Guideline.pdf. Accessed March 15, 2016. | ||
European Medicines Agency. Replagal, agalsidase alfa; 2015. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000369/human_med_001029.jsp&mid=WC0b01ac058001d124. Accessed February 22, 2016. | ||
World Health Organization. Growth reference data for 5–19 years; 2007. Available from: http://www.who.int/growthref/en/. Accessed January 8, 2014. | ||
Massin M, von Bernuth G. Normal ranges of heart rate variability during infancy and childhood. Pediatr Cardiol. 1997;18(4):297–302. | ||
Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology. J Am Soc Echocardiogr. 2005;18(12):1440–1463. | ||
National High Blood Pressure Education Program Working Group on High Blood Pressure in Children and Adolescents. The fourth report on the diagnosis, evaluation, and treatment of high blood pressure in children and adolescents. Pediatrics. 2004;114(2 Suppl 4th Report):555–576. | ||
Kampmann C, Linhart A, Baehner F, et al. Onset and progression of the Anderson-Fabry disease related cardiomyopathy. Int J Cardiol. 2008;130(3):367–373. | ||
Charles S. Cleeland. The Brief Pain Inventory User Guide; 1991. Available from: http://www.mdanderson.org/education-and-research/departments-programs-and-labs/departments-and-divisions/symptom-research/symptom-assessment-tools/BPI_UserGuide.pdf. Accessed October 14, 2014. | ||
Furlong WJ, Feeny DH, Torrance GW, Health Utilities Inc. Self-Complete Questionnaire Manual; 2012. Available from: http://www.healthutilities.com/manual.htm. Accessed October 14, 2014. | ||
Healthactchq. CHQ: Child Health Questionnaire™; 2014. Available from: https://www.healthactchq.com/chq.php. Accessed October 14, 2014. | ||
Crepaz R, Cemin R, Pedron C, Gentili L, Trevisan D, Pitscheider W. Age-related variations of left ventricular endocardial and midwall function in healthy infants, children, and adolescents. Ital Heart J. 2005;6(8):634–639. | ||
Morrissey RP, Philip KJ, Schwarz ER. Cardiac abnormalities in Anderson-Fabry disease and Fabry’s cardiomyopathy. Cardiovasc J Afr. 2011;22(1):38–44. | ||
Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol. 1987;59(4):256–262. | ||
Zuanetti G, Neilson JM, Latini R, Santoro E, Maggioni AP, Ewing DJ. Prognostic significance of heart rate variability in post-myocardial infarction patients in the fibrinolytic era. The GISSI-2 results. Gruppo Italiano per lo Studio della Sopravvivenza nell’ Infarto Miocardico. Circulation. 1996;94(3):432–436. | ||
Mehta A, West ML, Pintos-Morell G, et al. Therapeutic goals in the treatment of Fabry disease. Genet Med. 2010;12(11):713–720. | ||
National Kidney Foundation I. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease; 2007. Available from: http://www2.kidney.org/professionals/KDOQI/guideline_diabetes/guide1.htm. Accessed September 30, 2015. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.