")
Back to Journals » Drug Design, Development and Therapy » Volume 17
An Integrative Approach to Study the Inhibition of Providencia vermicola FabD Using C2-Quaternary Indolinones
Authors Khataniar A, Das A, Baruah MJ, Bania KK, Rajkhowa S , Al-Hussain SA , Zaki ME
Received 22 June 2023
Accepted for publication 25 October 2023
Published 14 November 2023 Volume 2023:17 Pages 3325—3347
DOI https://doi.org/10.2147/DDDT.S427193
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Ankita Khataniar,1,* Abhichandan Das,1,* Manash J Baruah,2 Kusum K Bania,2 Sanchaita Rajkhowa,1,* Sami A Al-Hussain,3 Magdi EA Zaki3
1Centre for Biotechnology and Bioinformatics, Dibrugarh University, Dibrugarh, As-786004, India; 2Department of Chemical Sciences, Tezpur University, Tezpur, As-784028, India; 3Department of Chemistry, Imam Mohammad Ibn Saud Islamic University (IMSIU), Riyadh, Saudi Arabia
*These authors contributed equally to this work
Correspondence: Sanchaita Rajkhowa; Magdi EA Zaki, Email [email protected]; [email protected]
Background: The present study investigates the potential bioactivity of twelve experimentally designed C-2 quaternary indolinones against Providencia spp., a bacterial group of the Enterobacteriaceae family known to cause urinary tract infections. The study aims to provide insights into the bioactive properties of the investigated compounds and their potential use in developing novel treatments against Providencia spp. The experimental design of indolinones, combined with their unique chemical structure, makes them attractive candidates for further investigation. The results of this research may contribute to the development of novel therapeutic agents to combat Providencia spp. infections.
Methods: The synthesized indolinones (moL1-moL12) are evaluated to identify any superior activity, particularly focusing on moL12, which possesses aza functionality. The antimicrobial activities of all twelve compounds are tested in triplicates against six different Gram-positive and Gram-negative organisms, including P. vermicola (P< 0.05). Computational methods have been employed to assess the pharmacokinetic properties of the compounds.
Results: Among the synthesized indolinones, moL12 exhibits superior activity compared to the other compounds with similar skeleton but different functional moieties. All six strains tested, including P. vermicola, demonstrated sensitivity to moL12. Computational studies support the pharmacokinetic properties of moL12, indicating acceptable absorption, distribution, metabolism, excretion, and toxicity characteristics.
Conclusion: Utilizing the PPI approach, we have identified a promising target, FabD, in Gram-negative bacteria. Our analysis has shown that moL12 exhibits significant potential in binding with FabD, thereby, might inhibit cell wall formation, and display superior antimicrobial activity compared to other compounds. Consequently, moL12 may be a potential therapeutic agent that could be used to combat urinary tract infections caused by Providencia spp. The findings of this research hold significant promise for the development of new and effective treatments for bacterial infections.
Keywords: C-2 quaternary indolinones, Providencia vermicola, Providencia stuartii, multi-drug resistance, molecular docking, molecular dynamics simulation
Graphical Abstract:
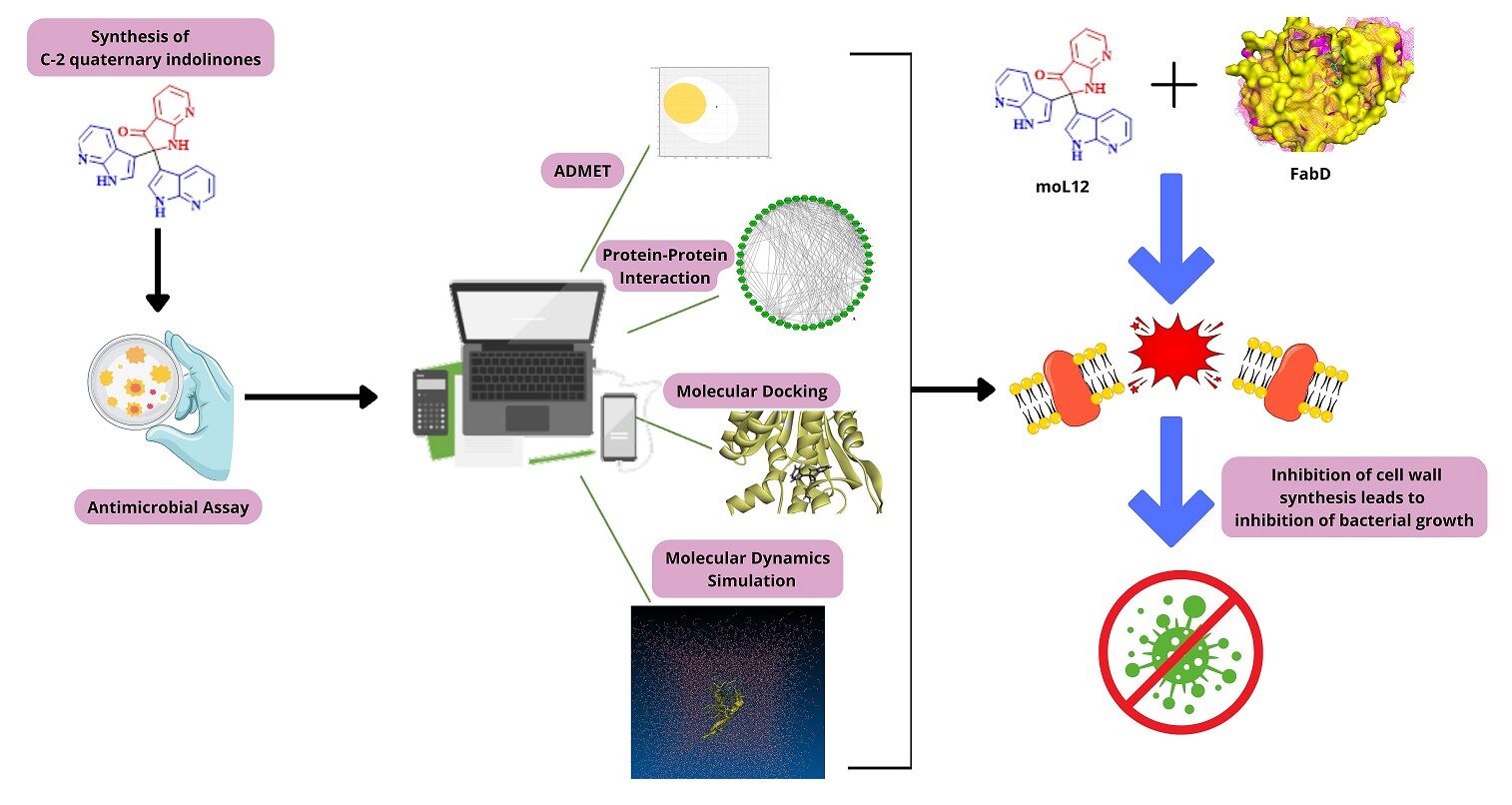
Introduction
Microbial resistance has become a problem in the contemporary medical environment due to the inadequate development of new antibiotics and the asinine prescription of antibiotics in both developed and developing nations. Antimicrobial resistance (AMR), which affects wealthy and developing nations, is a global concern.1 AMR is a problem for public health that has broad societal, economic, and health repercussions. Multi-drug resistance (MDR) is defined as non-sensitivity to at least one agent in all but two or fewer antimicrobial groups (i.e., bacterial isolates remain susceptible to only one or two categories), extensively-drug resistance (XDR) is defined as non-sensitivity to at least one agent in all but one or two antimicrobial groups and pan-drug resistance (PDR) is defined as resistance to all drugs in all antimicrobial classifications.2 Providencia vermicola and Providencia stuartii are two pathogenic species of Enterobacteriaceae, a family already classified by the WHO as a Critical level Priority 1 of AMR.3 In this regard, N-bonded heterocycles, 2,2-Di(3-indolyl)-3-indolones, i.e., C2-quaternary indolinones having three-dimensional acyclic chemical architecture with quaternary sp3-carbon are of significant importance.4 The C2-quaternary indolinone compounds are known to occur naturally in marine sponges, the poisonous mucus of the boxfish “Ostracion cubicus”, and a marine-derived bacterial strain in seawater.5,6 The tris-indole components are used in the treatment of various infectious disorders, including antibacterial activity against Gram-positive bacteria.7 They possess antiviral bioactivities and show activity towards other infectious illnesses such as hepatitis, mumps, pneumonia, and influenza.8 They are also identified as potential drugs for the therapeutic targets of SARS-CoV-2 using a computational technique.9 Because of the diverse applications of the indolinone framework, a number of approaches for their synthesis have been established. Generally, they are manufactured in the laboratory involving free radical pathways or metal-catalyzed, oxidant-driven, non-free radical approaches.7–15 Bio-catalytically they are also synthesized from indoles using laccase catalysis, laccase-surfactant catalysis, biocatalytic process, and combinatorial biocatalytic approaches.8,16,17 Likewise, these indolinone derivatives have been synthesized from indoles using a variety of reagents and conditions via some metal-free or metal-catalyzed routes.4,7,13,14,18,19 Metals like palladium (Pd), gold (Au), iridium (Ir), ruthenium (Ru), copper (Cu), and iron (Fe) are used more for such synthesis. Despite these developments, an easy and mild approach using fewer chemicals is required to efficiently synthesise these tris-indoles and supply the resources for diverse bioactivity research. Recently, Cheng et al designed a Ru-based photocatalyst to efficiently facilitate the quaternization of N-H and N-alkyl indoles under mild reaction conditions.7 They also found that the synthesized C2-quaternary indolinones exhibit antiproliferative activity against some human cancer cell lines like breast adenocarcinoma, lung adenocarcinoma, and cervix adenocarcinoma.
Very recently, we have also developed a cost-effective, environment-friendly, as well as recyclable zeolite-Y supported iron oxide (Fe2O3-Y), which was found as an excellent heterogeneous nanocatalyst for the synthesis of the C2-quaternary indolinones from various substituted and un-substituted indoles under ambient reaction conditions (Figure 1).20 In the previous work, we reported the molecules 1–11 as represented in Figure 1. It is known that indolinone derivatives exhibit biological activity; however, there is no record of these compounds being tested for their antimicrobial properties against pathogenic bacteria. Therefore, to further extend the study and to bring out the biological importance of the synthesized indolinones derivatives herein, we have investigated the bioactivity of the previously reported C2-quaternary indolinones (moL1-moL11, Scheme 1) and the newly synthesized one, ie, the molecule 12 (moL12) as shown in Scheme 1.
 |
Figure 1 Isolated derivatives (moL1 to moL12) of C2–quaternary indolinones by quaternization of indoles using Fe2O3–Y as catalyst. |
The rationale for this study is rooted in our recent achievement of developing a cost-effective, environmentally friendly, and recyclable zeolite-Y supported iron oxide (Fe2O3-Y). This innovative catalyst has demonstrated remarkable effectiveness in facilitating the synthesis of C2-quaternary indolinones from both substituted and unsubstituted indoles, all achieved under ambient reaction conditions. Building on our prior research efforts, where we successfully synthesized and characterized compounds 1–11, we found that some compounds displayed activity against certain pathogenic bacterial strains. Driven by these preliminary outcomes, we initiated a fresh undertaking, synthesising and evaluating a novel C2-quaternary indolinone, moL12. This compound was rigorously tested against a panel of six bacterial strains and exhibited sensitivity towards all of them. Encouraged by these promising experimental findings, we deemed it essential to subject moL12 to further computational analyses. The study’s rationale emerges from our prior successes with the catalyst and the observed activity of previously synthesized compounds against pathogenic bacteria. This led us to synthesize moL12 and investigate its antimicrobial potential, a process complemented by computational assessments aimed to advance our understanding of these C2-quaternary indolinones in combating antimicrobial resistance.
Materials and Methods
Sources of the Materials Used in the Study
Bacterial media for inoculation of test organisms were obtained from Himedia. Barium chloride and sulfuric acid for preparing McFarland standards were obtained from Merck. Standard antibiotics were obtained from Himedia. Test organisms were obtained from ATCC.
Determination of Antimicrobial Activity
Antimicrobial activity of the 12 test samples (moL1 to moL12) was assessed against a total of six (6) different bacterial strains, both Gram-positive and Gram-negative, viz Bacillus subtilis (6051), Staphylococcus aureus (25923), Pseudomonas aeruginosa (27853), Enterococcus faecium (51559), Salmonella typhimurium (49416), and Providencia vermicola (KP21).
Well Diffusion Method
Test organisms were inoculated in Nutrient broth and cultured overnight at 37 °C. The freshly prepared broth was diluted in normal saline in order to adjust the turbidity to 0.5 McFarland standard, thus yielding a final inoculum of 1×108 CFU/mL. A sterile petri dish prepped with solidified Muller-Hinton agar media was inoculated with 50 µL of diluted bacterial culture. The inoculum was spread evenly on the surface of the agar plate with a sterilized glass spreader. The wells were drilled into the agar plates containing inoculums using a sterile borer (6 mm in diameter). 50 µL of each sample (20 mg/mL) was then applied to the appropriate well. The plates were placed in a refrigerator to allow the compounds to diffuse properly into the agar after being kept at room temperature for 30 minutes. The plates were then incubated for 24 hours at 37 °C. Antimicrobial activity was detected by measuring the zone of inhibition, which included the well diameter, following the incubation period.21,22
Determination of Minimum Inhibitory Concentration (MIC)
The compounds showing significant inhibition against the test organisms, specifically P. vermicola, were selected for the determination of MIC by performing the rapid INT colorimetric assay. The compounds were diluted into separate concentrations, which was achieved by performing two-fold dilutions placed directly on a microtitre plate, pre-added with 100 µL of Mueller Hinton (MH) Broth. 50 µL of freshly prepared bacterial inoculum of P. vermicola with a concentration of 1.5×108 CFU/mL was added to each well. Commercially available Imipenem was used as the positive control. Wells containing 100 µL MH broth, 50 μL of bacterial inoculum, and DMSO served as the negative control. After resting the plates at room temperature for 30 minutes, the plates were treated to an incubation period of 18 hours at 37 °C. OD was taken at 600 nm.
50 μL p-iodonitrotetrazolium chloride or INT (0.5% w/v) was added post-OD measurement, followed by incubation at 37 °C for 30 minutes. The plates were observed visually. The MIC was regarded as the lowest concentration of sample that prevented the bacteria from growing as viable bacteria reduce the yellow INT dye to a pinkish color.23
Statistical Analysis
Statistical analyses in the study have been carried out in OriginPro, Version 2022.24 OriginLab Corporation, Northampton, MA, USA. All obtained values are expressed as the mean of triplicates ± SD (Standard Deviation). One-way ANOVA has been carried out for each raw data set.
ADMET Analysis of the Indolones
When determining drug exposure at the site of action, it is essential to precisely account for drug absorption, distribution, and metabolism inside a specific organ. It is crucial to understand the pharmacokinetic properties of these chemicals, particularly their absorption, distribution, metabolism, excretion, and toxicity (ADMET), in order to develop medicines with desirable qualities and appropriate dosing regimens.25
For the current study, both SwissADME web tool (http://www.swissadme.ch) and ADMETlab 2.0 (https://admetmesh.scbdd.com) have been used to predict the drug-like properties and pharmacokinetics of the compounds.
Target Fishing Using Protein–Protein Interaction Network
The uprising of resistance to most existing antibiotics highlights the need for novel approaches to bacterial pathogen management. Within the bacteria, a sequence of events occurs that affect the host. Enzymes control these reactions, which result in the production of the required products. Some pathogen metabolic pathways have been shown to be critical for the organism’s survival within the host. The analysis of such reactions is critical in identifying enzymes that might be potential therapeutic targets.
From the experimental analysis, it was found that the indolones showed the highest activity against Providencia vermicola. Thus, P. vermicola was undertaken for further exploration. The leading cause of resistance in Gram-negative bacteria to various antibiotics, such as β-lactams, quinilons, colistins, and other antibiotics, is their outer membrane. Majority of antibiotics require passage through the outer membrane in order to reach their targets; for instance, hydrophobic medications can do so via a diffusion pathway, whereas hydrophilic drugs, such as β-lactams, can do so via porins. Any modification to the outer membrane caused by Gram-negative bacteria, such as a change in the hydrophobic characteristics or a mutation in the porin protein, can result in resistance. Because Gram-positive bacteria lack this crucial layer, Gram-negative bacteria are more resistant to antibiotics than Gram-positive bacteria.26 Thus, targeting the formation and stability of the outer membrane may provide sensitivity to the newly developed antibacterial drugs. Fatty acid biosynthesis (FAB) is reported to be essential to the survival of Gram-negative P. vermicola as it is related to the formation of its membranes.27 However, the lack of available theoretical data for P. vermicola has encouraged the use of other species related closely to P. vermicola. Therefore, in this study, the FAB pathway of Providencia stuartii has been used to analyze and understand the importance of the proteins.28
The crucial enzymes involved in the Protein–Protein Interaction (PPI) network of FAB pathway were checked using topological analysis. A PPI reflects the interaction of the protein with their neighbouring protein in a static manner. They are mathematically constructed networks where a protein is denoted as node and the interaction between them is denoted by edge. They provide an excellent way of studying functional relationships, finding crucial modules and ranking targets. The proteins associated with FAB for P. stuartii were derived from STRING (version 11.5) database.29 The collected data were subjected to an open-source software “Cytoscape”,30 visualizing the PPI of P. stuartii cluster of proteins in a PPI.
MCODE, a clustering plugin was utilized to find out the cluster of the proteins.31,32 The proteins in a cluster tend to have highly interconnecting regions. The MCODE algorithm employs three steps: vertex weighting, complex prediction, and possible postprocessing to remove or add proteins in the generated complexes according to specific connection criteria. The vertex-weighting strategy is based on the clustering coefficient, Ci, which assesses the “cliquishness” of a vertex’s surroundings.
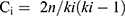
where n is the number of edges in the neighbourhood and ki is the vertex size.
Density of Graph G is defined as G = (V, E), where |V| is the no. of vertices, |E| is the number of edges and the maximum number of edge possible is defined as  . For a graph containing loops
. For a graph containing loops  and for no loops
and for no loops  Thus, density of G is
Thus, density of G is  , value ranging between 0.0 and 1.0.33
, value ranging between 0.0 and 1.0.33
Further, topological analysis was performed to analyze the network’s attributes along with it, Cytohubba,34 a plugin of “Cytoscape” was used to analyze and rank the central nodes present in the PPI network. These central nodes tend to have more connections with their neighbouring nodes. Removal of any one of these central nodes may result in the loss of connectivity of the protein in a network, altering the overall topology.
The ranking methods are divided into two categories based on algorithms: the local-based methods and the global-based methods. Here, Maximum Clique Centrality (MCC) method is used as a local-based method. It is defined as
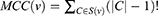
where S(v) represents the collection of maximal cliques containing v (|C|-1)! represents the product of all positive values less than |C|.34
Similarly, for global-based method, “closeness (Clo)” was implemented. It is defined as
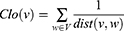
where the length of the shortest path between v and w nodes is represented by dist (v,w).35
After finding out the essential protein, phylogenetic trees were constructed to verify the close relation between P. vermicola and P. stuartii.
Phylogenetic Tree Construction
Due to unavailability of relevant data on P. vermicola, P. stuartii was taken as an alternate organism, as according to a previous study, P. stuartii showed close relation with P. vermicola.32 Therefore, to verify the close relationship, a phylogenetic tree was constructed to estimate the relationship among them and among the common organisms showing homology to the protein. For this study, two proteins were considered which were found to be crucial in the FAB pathway. Both these proteins were used as inputs to search for common ancestors using BLASTp. The top 10 hits were considered for the tree construction using the MEGA-X36 software. The alignment of the sequences was done using the inbuild ClustalW37 alignment tool in MEGA-X. Bootstrap test of phylogeny uses the bootstrap resampling approach,38,39 is one of the most frequently applied tests for evaluating the accuracy of a reconstructed tree. Using either nucleotides, codons or amino acids from the sequence, a phylogenetic tree is constructed. For every sequence, the nucleotides, codons, or amino acids are chosen at random with substitutions, resulting in a new set of sequences. Using the same approach and new set of sequences, a tree (bootstrap tree) is reconstructed, which is then compared with the original one.
The original tree’s each inner branch, which differs from the bootstrap tree, is assigned a 0 score, while the other branches are assigned a 1 score. In general, the best bootstrap value for inner branch is considered to be 95% or higher.40 Using the maximum likelihood method, the phylogenetic tree was constructed.
Protein Structure Prediction and Validation
The unavailability of target proteins’ 3D structure has led to prediction and modeling using computational approaches. The UniProtKB database41 has been used to retrieve primary sequence along with various primary data of P. stuartii.
Using the target sequence of P. stuartii in BLASTp, known protein template structure was found out from protein data bank (PDB),42 and top 5 hits were taken under consideration for further study. The proteins are subjected to SWISS-MODEL43 for model prediction, freely available at https://swissmodel.expasy.org.
The modeled structures were validated using various tools available. The QMEAN scoring algorithm was also used to measure the global and per-residue model quality. Ramachandran plot and Verify 3D were used to validate the three-dimensional modeled structure by examining the amino acid residue using the SWISS-MODEL server and UCLA-DOE LAB server (https://saves.mbi.ucla.edu/). Additionally, MD simulations were performed to evaluate the stability and conformational changes of the proposed models at different time scales. Furthermore, the binding interaction between the indolone compounds and the modeled targets was achieved for molecular docking.
Binding Site Prediction and Comparative Molecular Docking
The binding and active sites of proteins are often associated with structural pockets and cavities of the protein. In this study, the binding site prediction of the model protein was performed using the FTMap server44 and the CASTp server (http://sts.bioe.uic.edu/castp)45 along with a comprehensive evaluation of the literature.
The docking method is used to anticipate the putative binding characteristics of the ligand–receptor complex. Here, the compound showing the best ADMET results was docked onto the modeled FabD and FabG proteins using the CHARMM-based docking software (CDOCKER) of the BIOVIA Discovery Studio v4.5 (DS v4.5).46 The grid-based molecular dynamics simulated annealing method used in CDOCKER is based on the CHARMM force field. By using this algorithm, ligands were kept flexible, whereas non-bonded interactions were softened during the docking simulation. Throughout the entire docking procedure, the protein structure was kept rigid.47 The modeled protein structures were optimized and cleaned using the protein preparation module of DS v4.5. CHARMM-based force field (steepest descent method followed by conjugate gradient) at default mode was employed to optimize the protein models. The active site of proteins was manually predicted and identified using Edit binding site module of DS v4.5. The docked structure with the lowest energy was further analyzed using the Discovery Studio Visualizer.
The best docked protein–ligand complex has been further considered as the initial structure for performing the molecular dynamics simulations for a time interval of 100 ns.
Molecular Dynamics Simulations
MD simulations were carried out to assess the postulated model’s stability and conformational changes as well as to look into the protein–ligand connections at various timeframes. A total of 300 ns of simulation time have been performed for this purpose using the GROMACS 2019.1 package to simulate the modeled FabD and FabG proteins as well as the protein–ligand complex.48 A methodology similar to the work reported previously has been applied to calculate the force field parameters of the ligand, as well as setting up of the MD simulation calculation.49 The XMgrace tool was used to construct the graphs.50–52 Various parameters were used to analyze the results of molecular dynamics simulations generating plots that include root mean square deviation (RMSD), radius of gyration (Rg), and root mean square fluctuation (RMSF).
Results
Determination of Antimicrobial Activity
Out of the twelve compounds considered, the molecule with aza functionality, i.e., moL12 showed the best activity against both Gram-negative and Gram-positive bacterial strains, followed by the compounds moL1, moL10 and moL11. The zones of inhibition of these compounds were considered significant when compared to the standard antibiotic tested against the strains. The results are presented as resistant, intermediate, or sensitive to the tested compounds as shown in Table 1.
 |
Table 1 Antimicrobial Activity of the Tested Compounds (moL1 to moL12) and Standard Antibiotics |
R, Resistant (ZOI<10); I, Intermediate (10mm<ZOI<25mm); S, Sensitive (ZOI>30mm); *IMP, Imipenem; **DMSO, Dimethyl sulfoxide.
Minimum Inhibitory Concentration
MIC is defined as the lowest concentration of an antimicrobial agent that visibly inhibits microorganisms after a certain incubation period. MIC is used to determine the lowest possible concentration of an agent that is being tested as a potential candidate for an antibiotic. Thus, one can avoid using higher concentrations of a tested compound, which might become toxic inside a living body at those levels. In this study, moL12 was selected as it showed significantly better results than the other compounds. For this study, P. vermicola was selected as the test organism. The unique ability of Providencia species is to deaminate particular amino acids by oxidation into their respective keto and ammonia acids, which sets them apart from other Enterobacteriaceae family members. More than 80% of human clinical infections, primarily urinary tract infections, are caused by two species of Providencia, P. stuartii and P. rettgeri, which are naturally immune to numerous antibiotics, including colistin and tigecycline. In a more recent study, it was found that P. vermicola also exhibits multi-drug resistance, which is a cause of major concern in terms of clinical significance.53 The lowest concentration or the MIC for moL12 is 10 mg/mL (as shown in Figure 2).
 |
Figure 2 The above graph depicts the lowest concentration of moL12 that inhibits the growth of P. vermicola. Here 0 mg/mL represents the dilution well that contained no traces of the antimicrobial agent and thus can be used as a standard to measure the growth in the other wells. |
In silico Pharmacokinetic Analysis of the Compounds
ADMET properties of the designed molecules have been evaluated and depicted in Table S1. The molecule 12 (moL12) shows the best ADMET results. The yellow region of the boiled egg graph depicts whether molecules get through the blood–brain barrier. The area of the graph that is white represents molecules that are absorbed by the digestive system. The graph showing the boiled egg indicates that the gastrointestinal tract absorbs moL12 as well as the spider web graph where the physiochemical space that is best for oral bioavailability is indicated by the colored zone shown in Figure S1(a and b), respectively, are analyzed from swissADME server.
Target Fishing Using PPI Network
Fatty Acid Biosynthesis Pathway
The survivability of bacteria is dependent on the membrane bound lipid and the adaptive response of the cell’s lipid composition which can survive a varied range of environment. The membranes resemble the phospholipid bilayer, usually there is a wide variety of membrane structures but significantly the number of glycerolipids with two fatty acid chains is in majority. The genes involved in fatty acid chain formation are essential for the growth of bacteria. Thus, fatty acid biosynthesis (FAB) target pathway is taken under consideration for further exploration.54,55
The fatty acid chains are generated by the type II FAB pathway. This pathway consists of a series of distinct enzymes in comparison to that of type I synthase seen in mammals. The enzymes associated with the pathway are homologous to various model organisms of which crystal structures are available, making it easily accessible bioinformatically as they share the same biochemical activities. Thus, FAB can be a potent target pathway for antibiotic drugs.56 The package Cell Designer57 has been used to create the schematic representation of the pathway. Both reversible and irreversible reactions were included in the model.
Protein–Protein Interaction Network
For analyzing the importance of FAB pathway, a static network was created using the accessible protein–protein interaction network data from STRING database for P. stuartii. The network consisted of a total number of 48 nodes and 245 interactions (as shown in Figure 3a). Using the clustering plugin MCODE taking the degree cut off as 10, four different clusters were found. A comparison of the clusters showed that cluster 1 (Figure 3b) is having the highest number of interactions with respect to other 3 clusters. Proteins engaged in the interactions lead to the formation of dense network, which in turn creates functional systems that aid in cellular activities. Furthermore, removal of these proteins from the network will lower the interaction along with inhibiting the normal functioning of cellular activities.58
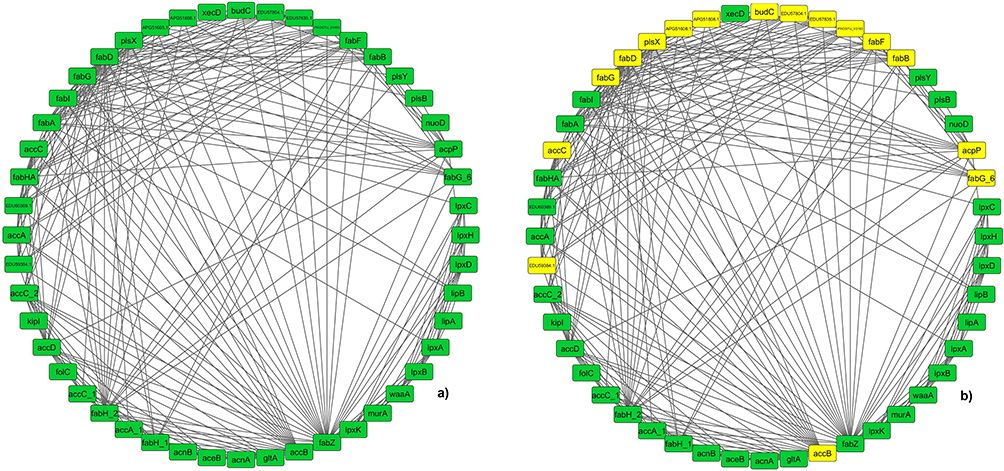 |
Figure 3 (a) The complete PPI network of FAB pathway proteins interacting with each other. The green hexagon denotes the nodes and black lines are the edges. (b) The MCODE results of cluster 1 are shown in PPI network marked in yellow. |
Network analysis gave a distinct understanding of the interactions between two nodes and their neighbouring nodes. The crucial proteins in the interaction network were identified by analyzing the topological parameters. Using the Cytohubba plugin, the best central nodes were identified, and MCC method is used as local centrality test as it is said to be one of the best method available for figuring out the central node in a network. Similarly, for global centrality test Clo method was implemented.31 This resulted in three proteins in top 3, FabD, FabG, and FabZ, as shown in Figure S2 a and b. The top 10 proteins of both the tests are mentioned in Tables S2 and S3. The rank results of both tests are compared with MCODE cluster. The comparison revealed that except FabZ, both FabD and FabG are common in MCODE results, as well as in cytohubba results. Thus, both are considered as an essential node in the overall FAB network.
Upon further literature survey it was found that FabG, a NADPH-dependent ketoacyl-ACP reductase catalyzes the first reductase step in each fatty acid production cycle. This FabG gene is usually transcribed with that of upstream genes. When C-Cm cassette (a transcription terminator) was inserted between FabD and FabG genes, the FabG gene growth and transcription were inhibited. Thus, showing that FabG is an essential enzyme for cell growth along with FabD acting as a promoter.55 The FabD protein provides a remarkable catalytic efficiency and transfer a variety of acyl substituents, which ultimately serve as to fatty acid synthesis a unique motif forming the ω terminus. The active side of FabD provides specificity towards malonyl-CoA leading to the backbone of amino acid chain.59
Evolutionary Relationship
For both the proteins (FabD & FabG) the top 10 hits from BLASTp were extracted, separately. MEGA-X was used for alignment as well as phylogenetic tree construction. Multiple sequence alignment was performed using the CLUSTALW algorithm within the MEGA-X package. For bootstrap, the best model sections for both the proteins FabD & FabG were calculated. Implementing the maximum likelihood method, the best phylogenetic tree was constructed.
FabD Protein
The model selection analysis was performed by considering statistical method as maximum likelihood and gaps/missing data treatment as partial deletion with 95% site coverage cut off. This resulted in WAG+G, ie, Whelan and Goldman’ matrix60 + Gamma distribution with lowest BIC scores (Bayesian Information Criterion) of 3936.387.61 Meanwhile, the non-uniformity of evolutionary rates among sites was modeled by using a discrete Gamma distribution (+G).
Using the maximum likelihood tree method, the phylogenetic tree was constructed. The bootstrap method was used with 1000 as no. of bootstrap replications and WAG+G as the model. The output result showed that the branch P. stuartii and P. vermicola have the most recent common ancestor with 95 as bootstrap support (as shown in Figure 4), which is equal to that of the minimum bootstrap support value. Besides this, Providencia rustigianii and Providencia alcalifaciens also showed 95 bootstrap supports.
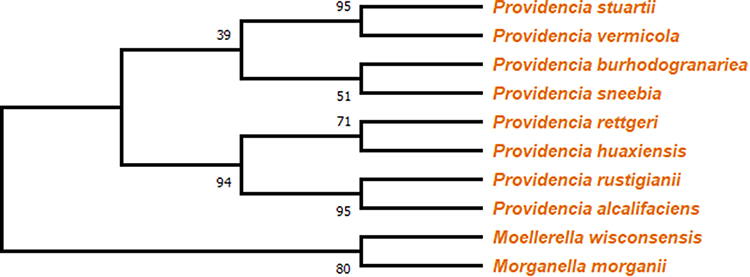 |
Figure 4 Cladogram representing a branched diagram of top 10 BLASTp hits of different species, where P. stuartii, P. vermicola, P. rustigianii and P. alcalifaciens having 95 as bootstrap support. |
FabG Protein
The model selection analysis was done by considering the same parameters as that of FabD protein. The result gave JTT+G, ie, Jones-Taylor-Thornton62 + Gamma distribution with lowest BIC scores (Bayesian Information Criterion) of 2566.021. Meanwhile, the non-uniformity of evolutionary rates among sites was modeled by using a discrete Gamma distribution (+G).
Using the same method, i.e., maximum likelihood tree, the phylogenetic tree was constructed. The bootstrap method was used with 1000 as no. of bootstrap replications and JTT+G as the model. The output result showed that the branch P. stuartii and P. vermicola have the most recent common ancestor with 97 as bootstrap support (as shown in Figure 5) which is above that of the minimum bootstrap support value. Besides this, Morganella morganii and Morganella psychrotolerans showed bootstrap support of 100.
 |
Figure 5 Cladogram representing a branched diagram of top 10 BLASTp hits of different species, where P. stuartii, and P. vermicola have bootstrap support of 97, with Morganella morganii and Morganella psychrotolerans having the highest bootstrap value of 100. |
Analyzing both the cladograms it can be concluded that both protein FabD and FabG of P. stuartii are closely related with that of P. vermicola and hence can be used as a surrogate against P. vermicola. Our results are further supported by the fact that genome comparison of both P. vermicola and P. stuartii revealed a proteome similarity of 90.79%.53 Similar evidence was also observed in another work where P. vermicola type strain in a clade showed 100 bootstrap support in the strains of both the species P. stuartii and P. thailandensis.28
Protein Structure Prediction and Validation
In the absence of crystal structure of both the essential proteins FabD & FabG, they were subjected to homology modeling. The primary sequence of both the proteins were derived. FabD consists of 310 amino acid residues, while FabG consists of 244 amino acid residues. The primary sequence was used for finding the best template. The top few results of BLASTp for both the proteins were taken under consideration for modeling. Table 2 shows the top five hits for both the proteins that have been retrieved by the BLASTp program. A set of 5 structures for both the proteins were modeled. The best models were further evaluated.
 |
Table 2 Showing Various Parameters Associated with the Top-Scoring Matches Acquired Through BLASTp (a) FabD & (b) FabG |
FabD Model Validation
The global model quality estimation (GMQE) is used to predict the local distance difference test (lDDT) score. Estimation of the score is done by integrating target-template alignment and template structural properties. The model reliability is expressed between the range of 0 and 1, the higher the value, the more reliable the model. From Table S4, it was found that model 1 has the highest GMQE score of 0.91. The QMEAN score evaluates the model by comparing the values with high-resolution X-ray crystallography structure. Here, model 1 scored the best out of the other 4 with a value of 0.66.
The Ramachandran plot value showed that model 1 has more amino acid residues in the allowed region of 5.3% and less outlier amino acid residues of 0.4%, compared to other models.
Using verify3D, the models were validated by profile score which are based on the atomic coordinates of the structure. The result almost showed a 100% score for most of the models with ≥0.2 average 3D-1D score. Models having positive profile scores are acceptable.
Comparing the results of GMQE, QMEAN, Ramachandran plot, and Verify3D it was found that Model 1 is best fit model and chosen for further analysis.
FabG Model Validation
Similarly, GMQE is used to predict the local distance difference test (lDDT) score. From Table S5, it was found that models 1, 2 and 3 have the highest GMQE score of 0.90. The QMEAN score evaluates the model by comparing the values with high-resolution X-ray crystallography structure. Here, model 2 scored the best out of the other 4 with a value of 1.
The Ramachandran plot value showed that model 2 has more amino acid residues in the favoured region of 93.4% and both Models 2 and 5 have less outlier amino acid residues of 0.0%. Using verify3D, the models were validated by profile score. The result showed Model 2 has a 90.74% score ≥0.2 average 3D-1D score. Models having positive profile scores are acceptable. Comparing the results of GMQE, QMEAN, Ramachandran plot, and Verify3D it was found that Model 2 is best fit model and chosen for further analysis. For the above data SWISSMODEL & UCLA-DOE LAB servers have been utilized.
Binding Site Prediction and Molecular Docking
The lack of crystal structures for both the FabD and FabG proteins in P. vermicola led to the need for modeling these proteins. The FabD protein in P. vermicola shares its highest similarity, approximately 76.62%, with the crystal structure of malonyl-CoA acyl carrier protein transacylase (FabD) and Acyl Carrier Protein (AcpP) (PDB ID: 6U0J). On the other hand, the P. vermicola FabG protein exhibits the highest identity, approximately 84.02%, with the crystal structure of Salmonella typhimurium FabG bound to NADPH (PDB ID: 6T5X). Although a structural similarity of 76.30% and 84.02%, along with query coverage of 99% and 100%, respectively, suggests a notable resemblance in their overall structure, it does not guarantee that their binding sites will always be identical or highly similar. Hence, to assess the similarity of the binding sites more precisely, we conducted more specific analyses using the FTMap server44 and the CASTp server (http://sts.bioe.uic.edu/castp).45 Additionally, we conducted a thorough review of the existing literature. Furthermore, we performed structural superimposition of the modeled structures with their respective template proteins.
The FTMap server predicted that the most active residues (Figure 6) in FabD, Gly 10, Gln 11, Arg 75, His 91, Asn 160, Asn 162, Ser 200, and His 201 are likely to be involved in ligand binding through hydrogen bonds, as indicated in Figure 6(a). Additionally, the following residues Pro 9, Gly 10, Gln 11, His 91, Met 132, Leu 194, Val 196, etc (in modeled protein) are expected to participate in non-bonded interactions, as shown in Figure 6(b). Furthermore, on comparison of these predicted active sites with P. vermicola FabD (UniProt ID: A0A8E3VSV0), it was found that Ser 92 and His 201 were reported to be documented as active sites in the UniProt database.
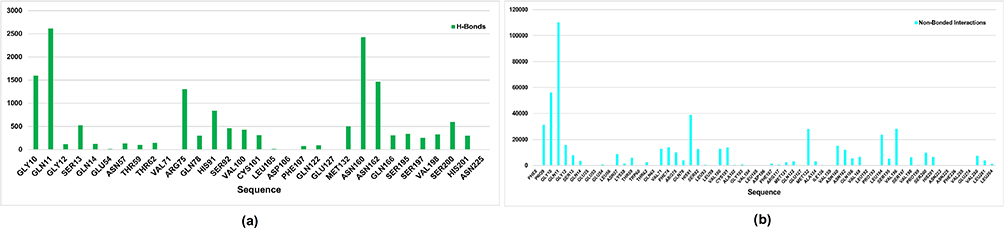 |
Figure 6 Binding site of the FabD modeled protein as predicted using the FTMap server. (a) Bar graph shows the contact frequency of H-bonds per residue; (b) bar graph shows the contact frequency of non-bonded interactions per residue. |
Similarly, for FabG, as depicted (Figure 7), the residues Arg 15, Ile 17, Gly 18, Asn 86, Gly 88, Thr 90, Ser 138, Lyd 155, Gly 182, and Ile 184 are predicted to form hydrogen bonds, while the following residues Gly 12, Arg 15, Gly 16, Ile 17, Gly 18, Asn 86, Ala87, Gly88, Ile 89, Thr 90, Ser 138, Tyr 151, Pro 181, Gly 182, Phe 183, Met 188, etc are anticipated to engage in non-bonded interactions, as illustrated in Figure 7(a and b), respectively. Likewise, on comparison of these predicted active sites with P. vermicola FabG (UniProt ID: A0A8E3VST2), it was found that Tyr 151 was reported to be documented as an active site in the UniProt database.
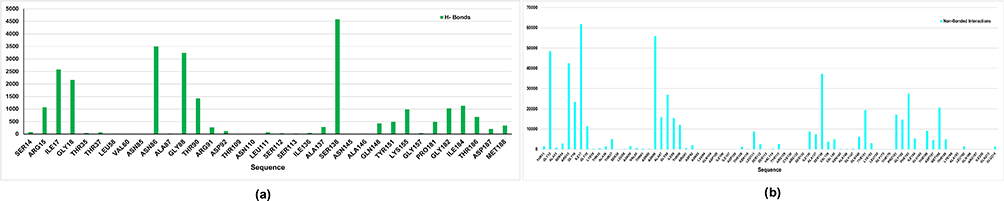 |
Figure 7 Binding site of the FabG modeled protein as predicted using the FTMap server. (a) Bar graph shows the contact frequency of H-bonds per residue; (b) bar graph shows the contact frequency of non-bonded interactions per residue. |
We also conducted an analysis of the CastP calculations, which revealed that the most probable binding cavities in FabD (as shown in Figure 8(a)) have an area of 631.854 Ų and a volume of 677.983 ų. Likewise, for FabG (Figure 8(b)), the cavity has an area of 157.399 Ų and a volume of 38.861 ų. The respective residues for both modeled proteins are listed in Table 3.
 |
Table 3 Residues Involved in Ligand Binding for FabD and FabG. This Table Lists the Residues Within the Binding Cavities of FabD and FabG Modeled Proteins |
 |
Figure 8 CastP calculations revealed the Binding Cavities in (a) FabD and (b) FabG. The yellow color denoting the binding cavities of the modeled proteins. |
The results of structural superimposition revealed that, in the case of FabD and 6U0J, the binding site of the modeled protein largely retained its original configuration (as illustrated in Figure S3). In contrast, for FabG, it appeared that the binding site had shifted somewhat and had notably reduced in size compared to 6T5X, as depicted in Figure S4. Nevertheless, these results are consistent with the findings from the FTMap server and CastP server analyses. Consequently, we selected these cavities as active sites for further docking investigations of the FabD and FabG proteins in P. vermicola.
The molecular docking of the ligand moL12 with both the modeled proteins FabD and FabG was implemented. The results showed that moL12 was able to dock onto both the modeled proteins as shown in Figure 9(a and b), with surrounding residues in Table 4, which were found to be stabilized by forming various non-covalent interactions such as hydrogen bonds (green), Pi-sigma (lavender), Pi-alkyl (pink), Pi-Sulphur (honey) Pi-anion and Pi-cation (orange) and van der Waals interactions (mint) from Figure 9(c and d).
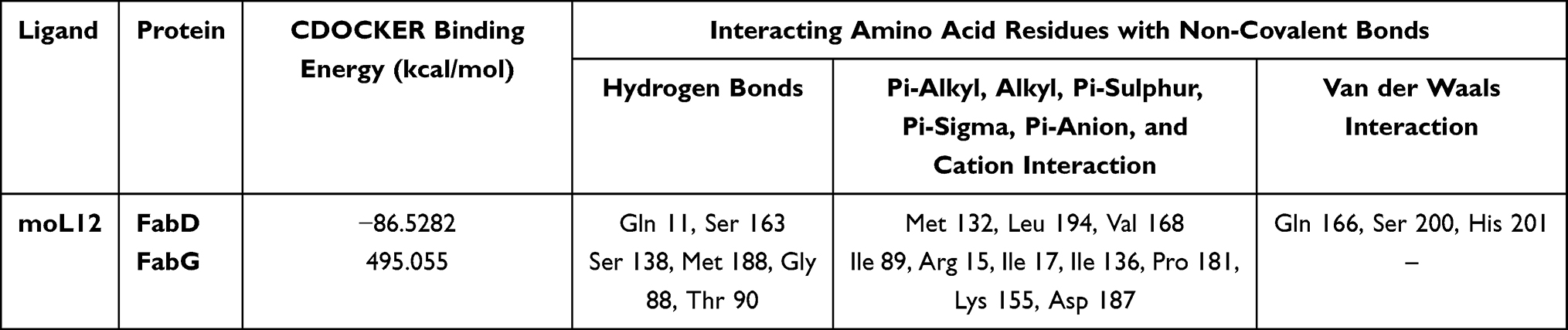 |
Table 4 Binding Energy and Binding Site Residues Forming Non-Covalent Interactions |
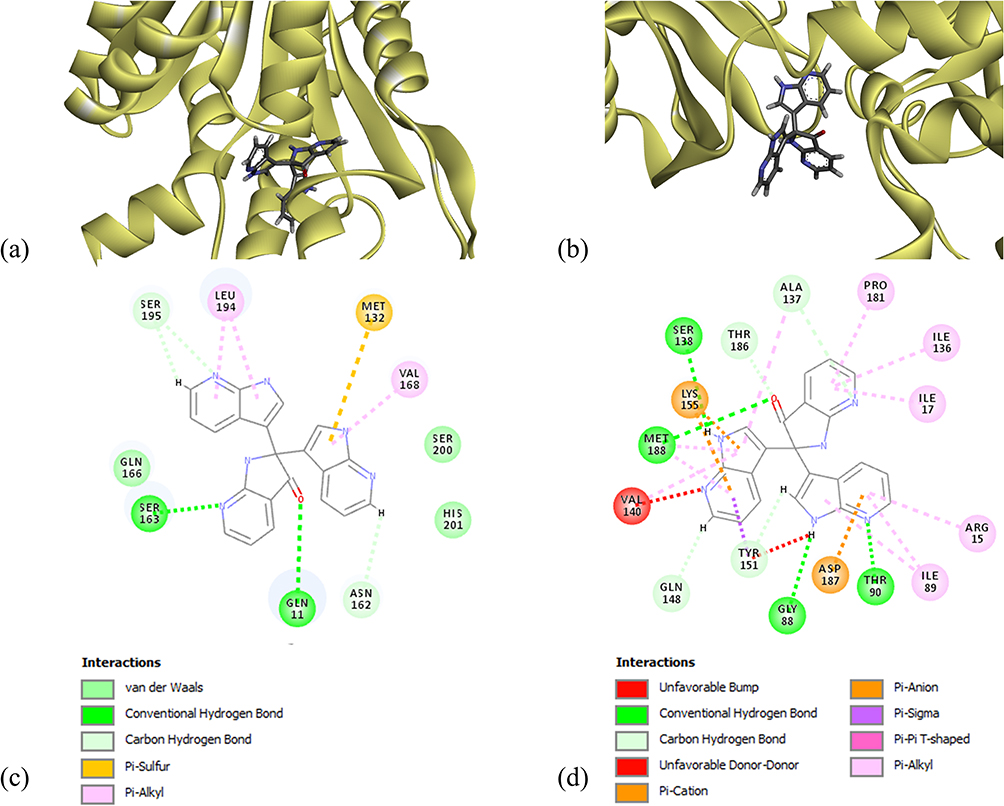 |
Figure 9 Minimum energy docked pose of moL12 with (a) FabD, (b) FabG and 2D diagram of moL12 with (c) FabD, (d) FabG. |
Hydrogen bonding interactions and hydrophobic connections mediated by the residue were the primary factors in stabilizing the complexes. However, from Figure 9(d) apart from the various bonded and non-bonded interactions, one unfavourable bump and one unfavourable donor–donor interaction are found in the case of moL12 and FabG complex. The binding energy was also determined from the docked structure, and it was found that the FabD protein docked with the ligand moL12 showed the best binding energy as shown in Table 4.
It is a fundamental premise in the complex domain of biological systems that molecules cannot occupy within the same spatial coordinates without experiencing repulsive forces. Because of the electron clouds that surround atoms, unfavorable steric conflicts or bumps are suggestive of implausible and unrealistic ligand conformations within a binding site. These repulsive interactions have led us to a discriminating conclusion in the context of our study: it is extremely likely that the complex containing FabG-moL12, the target protein and ligand, will not retain the stability or feasibility necessary for a functioning biological interaction. Moreover, both the complexes have shown different binding energy scores.
From Figure 9 and Table 4, it can be concluded that although moL12 was found to interact with both the modeled proteins; however, its interaction with FabD is much better as compared to FabG. Certain studies that were previously conducted have employed docking interactions and binding energy as a screening process for the evaluation of compounds.63,64 Therefore, for the MD simulations, only moL12 in complex with FabD was selected. The docked structure was taken as the starting conformation for performing the MD simulations.
Molecular Dynamics Simulations
Depending on a broad model of the physics driving neighbouring atoms interactions, molecular dynamics (MD) simulations forecast how each atom in a protein or other molecular structure will interact through time.65
Three plots of MD simulations with a total duration of 100 ns each were employed to determine the stability and fluctuation of the protein and the complex. The two native proteins were used as controls, and the best docked position was chosen for the MD simulation. Separate water simulations were run for each protein. Table 5 lists the many non-covalent interactions for the docked compound at 100 ns of MD simulations. Upon evaluating the table below, it becomes obvious that residues Ser 92 and His 201, which are actively participating in non-bonded interactions, showed an identical match with the relevant information obtained from UniProt data during cross-referencing.
 |
Table 5 Different Non-Covalent Interaction Between Binding Site Residues and Protein–Ligand (FabD-moL12) Complex Obtained from MD Simulation at 100 Ns |
The system’s stability was examined using a variety of other properties.49,66
RMSD
The RMSD (Root Mean Square Deviation) plot comprises of numerous plots that were generated as a result of the MD simulation, which is a key parameter to consider when analyzing the equilibration of MD trajectories. Protein stability is determined utilizing this RMSD trajectory. For both the protein structures, together with the docked complex, the RMSD backbone value is computed using a time simulation between 0 and 100 ns. A higher RMSD value suggests that the protein structure is less stable.
From Figure 10, it can be observed that the RMSD plot of FabG shows a much higher rise as compared to that of FabD. A sharp peak at the RMSD can be observed for FabG at 0 ns to 25 ns, However, post-30 ns, the protein seems stable with deviations ranging from 0.40 to 0.49 nm.
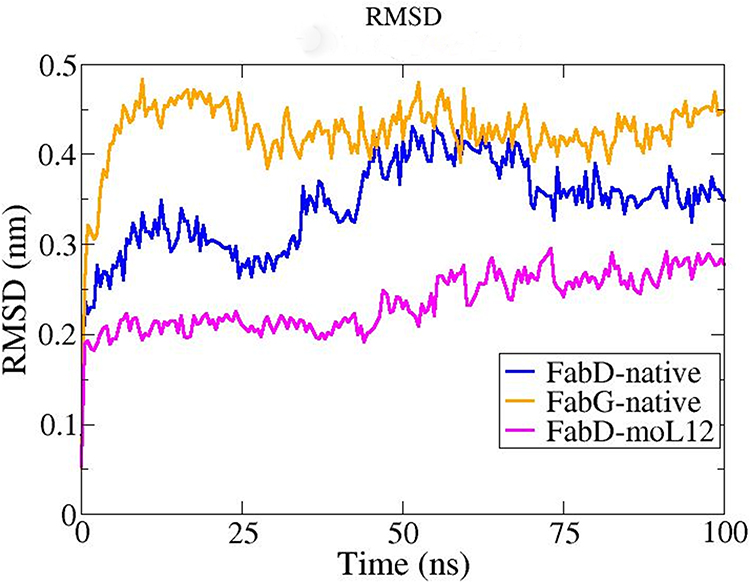 |
Figure 10 RMSD plot obtained from MD Simulation at a total time interval of 100 ns for the FabG and FabD proteins in their native forms (modeled) and FabD-moL12 complex. Plots as obtained from MD simulations performed in Gromacs 2019.1 package and analysed using XMgrace software. |
For the native FabD protein, the RMSD seems to fluctuate to 70 ns but tends to attain stability post-75 ns with fluctuations ranging from approx. 0.32 nm to 0.38 nm. However, when moL12 is bound to FabD, the RMSD seems to be much more stable comparatively, indicating that moL12 provides extra stability to the FabD protein. The protein FabD seems to attain stability in the presence of moL12 with very little fluctuations ranging from 0.21 to 0.29 nm post-50 ns. Hence, the RMSD value for the native protein FabG seems much higher than the RMSD value of the native protein FabD and FabD when in complex with the ligand moL12, which implies that the stability follows the trend of FabD-moL12 > Native FabD > Native FabG.
Rg (Radius of Gyration)
To comprehend the degrees of compactness of the modeled proteins and the docked complex, the Rg plot was analyzed. The root mean square distance of a group of atoms from their shared center of mass is recognized as the Rg. As a result, this report includes with the protein’s total proportions.
The Rg value of native FabG protein was found to be much lower comparatively and is almost constant throughout the 100 ns simulation with very less deviations as compared to native FabD and the FabD-moL12 complex as shown in Figure 8. However, in the case of FabD, the Rg in general shows a much higher value as compared to the FabG. This is probably due to the larger size of FabD protein as compared to FabG. Also, a slight increase in the Rg value from about 45 ns in the FabD native protein can be seen in Figure 11, which tends to attain stability post-60 ns. However, a decrease in the Rg value can be clearly seen in FabD-moL12 as compared to native FabD till the end of 100 ns and is almost constant throughout the 100 ns simulation time.
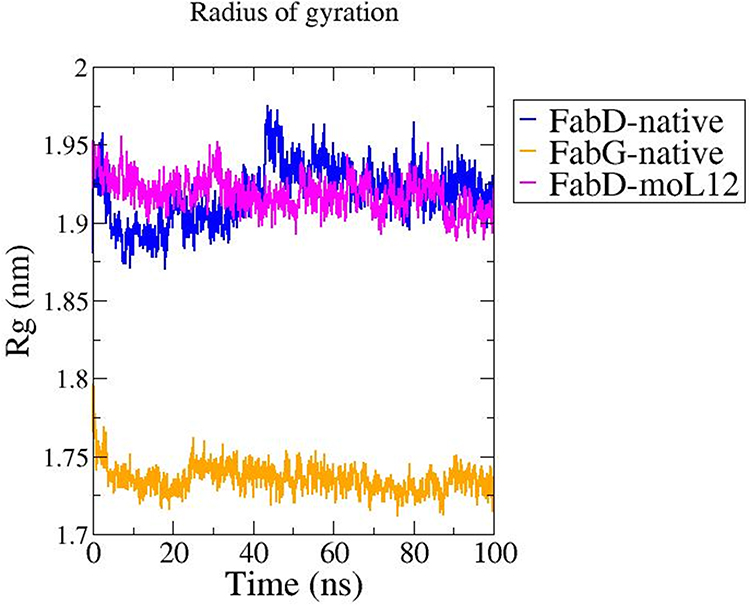 |
Figure 11 Rg plot obtained from MD Simulation at a total time interval of 100 ns for the FabG and FabD proteins in their native forms (modeled) and FabD-moL12 complex. Plots as obtained from MD simulations performed in Gromacs 2019.1 package and analysed using XMgrace software. |
From this plot, it can be inferred that the native FabD protein structure has a higher Rg value with more deviations than the FabD-moL12 complex resulting in the following trend in compactness of native FabG > FabD-moL12 > native FabD.
RMSF
As a means of representing the variations in flexibility between residues, the RMSF (Root Mean Square Fluctuation) with regard to the typical MD simulation conformation is used. For the purpose of examining the flexibility of the backbone structure, the backbone RMSF of each residue in both the proteins and FabD-moL12 complex was determined. When compared to its average position, simulations with increased RMSF values reveal more flexible motions, while simulations with lower RMSF values show more constrained displacements.
Root-mean-square fluctuations (RMSF) were computed to comprehend the residue-wise fluctuations of the proteins across the simulation period (as shown in Figure 12a). Because any significant variations in the flexibility of the functionally significant residues will affect the operation of FabG and FabD, the RMSF analysis clarifies the fluctuations of residues and demonstrates their significance. Figure 12b depicts the overall variation of the stretch that contains crucial FabD residues. Figure 12b shows that the overall variations of the complex’s functionally significant residues in FabD-moL12 are comparable to those of the control. However, the highest fluctuation observed at the functionally important stretch in FabD at 127 position is seen to be much lower when in complex with moL12.
 |
Figure 12 RMSF plots obtained from MD Simulation at a total time interval of 100 ns. (a) RMSF plot of FabG and FabD proteins in their native forms (modeled) and FabD-moL12 complex. (b) RMSF plot of the binding region of FabD protein in its native form (modeled) and when in complex with moL12. Plots as obtained from MD simulations performed in Gromacs 2019.1 package and analysed using XMgrace software. |
Non-Covalent Interactions
The physical organization of life, from small molecules in chemistry to enormous assemblies in living organisms and crystals, is stabilized in significant part by non-covalent interactions. In addition, ions as well as water-mediated solvation both contribute significantly to the operation and control of biological systems. Accordingly, one of the key areas of study in the natural sciences is the intricacies of non-covalent interactions and the function of solvation. Electrostatics, one of these non-covalent interactions, is particularly important in hydrogen bonding, which is well understood. Pi-anion interactions are less well understood than the other traditional non-covalent forces like Pi-cation and Pi-Pi interactions.67–70
From Figure 13, where hydrogen and hydrophobic interactions were seen to be prevalent from the preliminary complex, it is feasible to determine the number of non-bonded contacts between the ligand moL12 and the protein FabD at intervals of 20 nanoseconds. This suggests that the ligand moL12 continue to achieve stability in the binding cavity. The amount of hydrogen bonds and hydrophobic contacts of the FabD-moL12 complex thus demonstrated a clear and significant presence throughout time, contributing to the stability of the complex.
 |
Figure 13 Graph showing the comparison of time evolution in number (at every 20 ns) of Hydrogen bonds and Hydrophobic interactions of the FabD-moL12 complex. |
Discussion
The study details a comprehensive research endeavor that delves into the molecular and functional aspects of FabD and FabG proteins within the Fatty Acid Biosynthesis (FAB) pathway. This study takes a systematic approach, ranging from assessing antimicrobial activity to molecular dynamics simulations and phylogenetic analyses. The wealth of information generated through this investigation carries substantial significance in the context of drug development, particularly for combating multi-drug-resistant pathogens.
The investigation begins with the assessment of twelve compounds, revealing moL12 as the most potent antimicrobial agent against both Gram-negative and Gram-positive bacterial strains. This is a crucial finding, given the alarming rise in antibiotic resistance, especially among Providencia species responsible for clinical infections, including urinary tract infections. The Minimum Inhibitory Concentration (MIC) determination demonstrates that moL12 exhibits considerable potential as an effective antimicrobial agent, thus, a promising candidate in the battle against challenging pathogens.
Furthermore, the study extends to assess the Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties of the compounds, emphasizing moL12ʹs favorability for oral bioavailability and non-toxicity. These properties are essential considerations for drug development as they affect a compound’s effectiveness and safety.
The identification of FabD and FabG as pivotal proteins within the FAB pathway is a pivotal revelation. This pathway, critical for bacterial survival, has long been recognized as a potent target for antibiotic drug development. The construction of a protein–protein interaction network highlights the interconnectedness of these proteins and their roles in cellular function, further underscoring their significance as potential targets for drug development.
The subsequent phylogenetic analysis provides essential insights into the evolutionary relationships between FabD and FabG proteins across various species. The close evolutionary relationship between FabD and FabG in Providencia species, particularly P. stuartii and P. vermicola, makes these proteins suitable surrogates for further study. This has implications not only for understanding the biology of these proteins but also for potential applications in drug discovery.
The study’s core section involves homology modeling, a powerful technique in the absence of crystal structures. Validation through various parameters demonstrates the selection of the best-fit models. These models are subsequently employed in binding site prediction and molecular docking studies. The assessment of active residues and binding interactions provides valuable insights into the ligand–protein relationships. The superior binding energy of moL12 with FabD compared to FabG hints at moL12ʹs potential as a drug candidate against the FabD protein.
The Molecular Dynamics (MD) simulations offer a dynamic view of the proteins’ stability and flexibility. RMSD, Rg, and RMSF plots reveal that the FabD-moL12 complex exhibits enhanced stability and flexibility, essential attributes for drug–protein interactions. The evaluation of non-covalent interactions, including hydrogen bonds and hydrophobic contacts, adds another layer of insight into the complex dynamics of the FabD–moL12 interaction. The favorable profile of moL12 in these assessments further supports its candidacy as a promising antimicrobial agent.
FabD is an essential enzyme involved in the process of fatty acid synthesis, specifically in bacterial cells.71 It plays a crucial role in the elongation of fatty acid chains, a fundamental step in the biosynthesis of fatty acids that are essential for the cell’s structure and various physiological processes. In fatty acid synthesis, FabD serves as an acyl–acyl carrier protein (ACP) synthetase, which catalyzes the activation of a fatty acid molecule by attaching it to an ACP, an important carrier protein in the process.72 This activated fatty acid is then ready for further elongation, modification, and eventual incorporation into complex lipids. The importance of FabD in fatty acid synthesis can be linked to its role in providing the building blocks for membrane lipids, energy storage, and various cellular functions. Fatty acids are critical components of phospholipids, which form the lipid bilayer of cell membranes, ensuring their integrity and function.71 In summary, FabD is an enzyme involved in fatty acid synthesis, particularly in bacterial cells, and plays a crucial role in the generation of fatty acids used for membrane lipids. Therefore, when an inhibitor (moL12) binds to the active site of FabD, it may inhibit this crucial protein, leading to disruption or inhibition of bacterial cell wall of the bacteria.
Thus, this multi-faceted study encompassing antimicrobial activity evaluation, ADMET analysis, protein interaction network construction, phylogenetic analysis, homology modeling, molecular docking, and molecular dynamics simulations represents a holistic approach to studying FabD protein. The findings collectively advance our understanding of this protein and offer valuable prospects in drug development. Furthermore, the significance of this work is magnified by its potential applications in addressing the urgent clinical challenge of antibiotic resistance, particularly in Providencia species. The identified candidate moL12 stands as a promising option for future drug development efforts, bolstered by its structural and functional characteristics outlined in this study.
Conclusion
In conclusion, this study has successfully explored the potential antimicrobial properties of a set of synthesized C2 quaternary indolinones. Through a series of in vivo antimicrobial experiments, the compound moL12 exhibited promising inhibitory effects against a range of bacterial strains, with particular potency observed against the multidrug-resistant P. vermicola. This finding is especially significant, given the escalating global health crisis posed by multidrug-resistant bacteria, emphasizing the need for novel antimicrobial agents.
Furthermore, the ADMET studies provided valuable insights into the pharmacokinetic properties of moL12, highlighting its suitability for further development as a potential therapeutic agent. This supports its potential as a viable antimicrobial compound for future drug development.
The identification of FabD and FabG as critical protein targets for both P. vermicola and P. stuartii is a noteworthy revelation, particularly because crystal structures for these proteins are currently unavailable. The subsequent molecular modeling and stabilization of these proteins through molecular dynamics simulations enabled the investigation of moL12ʹs interactions with FabD, revealing its remarkable stability with this target. This suggests that moL12 has the capacity to disrupt bacterial cell wall formation, given FabD plays a crucial role in the elongation of fatty acid chains, a fundamental step in the biosynthesis of fatty acids that are essential for the cell’s structure and various physiological processes. This finding is significant in the context of combating bacterial growth and infection.
The study’s implications extend beyond laboratory research, as it addresses real-world concerns. The fact that P. vermicola is known to infect fish and insects raises concerns about potential bacterial transmission to humans through the consumption of these organisms, particularly in regions where they are part of the local diet. Therefore, moL12ʹs efficacy against this pathogen could have far-reaching implications for food safety and public health.
In summary, this research represents a promising step towards the development of new antimicrobial agents to combat multidrug-resistant pathogens like P. vermicola and potentially P. stuartii, both of which have clinical and environmental significance. The discovery of moL12ʹs antimicrobial properties and its interactions with essential bacterial proteins may pave the way for innovative strategies to address the global threat of antimicrobial resistance and bacterial transmission through the food chain.
Acknowledgment
Manash J Baruah and Kusum K Bania contributed towards the synthesis of the molecules. The authors would like to acknowledge Dibrugarh University for providing the lab facilities to perform antimicrobial and computational studies. The authors also extend their appreciation to DBT e-Library Consortium (DeLCON) for providing access to some of the important journals used while writing this manuscript. KKB thanks DST-SERB (CRG/2019/000962) and CSIR (NO. 80(0094)/20/EMR-II) for the research grant. MJB acknowledge the Department of Science and Technology (DST), Govt. of India for the DST-INSPIRE Fellowship (No. DST/INSPIRE Fellowship/2018/IF180217). Additionally, the authors extend their appreciation to the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia for supporting and supervising this project.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Chinemerem Nwobodo D, Ugwu MC, Oliseloke Anie C, et al. Antibiotic resistance: the challenges and some emerging strategies for tackling a global menace. J Clin Lab Anal. 2022;36(9):e24655. doi:10.1002/jcla.24655
2. Magiorakos AP, Srinivasan A, Carey RB, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–281. doi:10.1111/j.1469-0691.2011.03570.x
3. Dhingra S, Rahman NAA, Peile E, et al. Microbial resistance movements: an overview of global public health threats posed by antimicrobial resistance, and how best to counter. Front Public Health. 2020;8:535668. doi:10.3389/fpubh.2020.535668
4. Guchhait SK, Chaudhary V, Rana VA, Priyadarshani G, Kandekar S, Kashyap M. Oxidative dearomatization of Indoles via Pd-catalyzed C–H oxygenation: an entry to C2-quaternary indolin-3-ones. Org Lett. 2016;18(7):1534–1537. doi:10.1021/acs.orglett.6b00244
5. Bell R, Carmeli S, Sar N. Vibrindole A, a metabolite of the marine bacterium, Vibrio parahaemolyticus, isolated from the toxic mucus of the boxfish Ostracion cubicus. J Nat Prod. 1994;57(11):1587–1590. doi:10.1021/np50113a022
6. Cai SX, Li DH, Zhu TJ, Wang FP, Xiao X, Gu QQ. Two new Indole alkaloids from the marine-derived bacterium Aeromonas sp. CB101. Helv Chim Acta. 2010;93(4):791–795. doi:10.1002/hlca.200900360
7. Cheng LT, Luo SQ, Hong BC, Chen CL, Li WS, Lee GH. Oxidative trimerization of indoles via water-assisted visible-light photoredox catalysis and the study of their anti-cancer activities. Org Biomol Chem. 2020;18(32):6247–6252. doi:10.1039/D0OB01298J
8. McClay K, Mehboob S, Yu J, et al. Indole trimers with antibacterial activity against Gram-positive organisms produced using combinatorial biocatalysis. AMB Express. 2015;5(1):125. doi:10.1186/s13568-015-0125-4
9. Wu C, Liu Y, Yang Y, et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm Sin B. 2020;10(5):766–788. doi:10.1016/j.apsb.2020.02.008
10. Jiang X, Zhu B, Lin K, Wang G, Su WK, Yu C. Metal-free synthesis of 2,2-disubstituted indolin-3-ones. Org Biomol Chem. 2019;17(8):2199–2203. doi:10.1039/C8OB03057J
11. Chakrabarty M, Sarkar S, Basak R. Reaction of indole and alkylindoles with ceric ammonium nitrate on silica gel. J Chem Res. 2003;2003(10):664–665. doi:10.3184/030823403322656012
12. Rogers JL, MacMillan JB. A labeled substrate approach to discovery of biocatalytic reactions: a proof of concept transformation with N- methylindole. J Am Chem Soc. 2012;134(30):12378–12381. doi:10.1021/ja304767m
13. Xue J, Bao Y, Qin W, et al. Metal-Free–catalyzed oxidative trimerization of indoles using NaNO 2 to construct quaternary carbon centers: synthesis of 2-(1 H -Indol-3-yl)-2,3′-biindolin-3-ones. Synth Commun. 2014;44(15):2215–2221. doi:10.1080/00397911.2014.891743
14. Shukla G, Dahiya A, Alam T, Patel BK. Visible light‐mediated C2‐Quaternarization of N‐alkyl indoles through oxidative dearomatization using Ir(III) catalyst. Asian J Org Chem. 2019;8(12):2243–2248. doi:10.1002/ajoc.201900604
15. Takeshige Y, Egami Y, Wakimoto T, Abe I. Production of indole antibiotics induced by exogenous gene derived from sponge metagenomes. Mol Biosyst. 2015;11(5):1290–1294. doi:10.1039/C5MB00131E
16. Ganachaud C, Garfagnoli V, Tron T, Iacazio G. Trimerisation of indole through laccase catalysis. Tetrahedron Lett. 2008;49(15):2476–2478. doi:10.1016/j.tetlet.2008.02.021
17. Azimi M, Nafissi-Varcheh N, Mogharabi M, Faramarzi MA, Aboofazeli R. Study of laccase activity and stability in the presence of ionic and non-ionic surfactants and the bioconversion of indole in laccase-TX-100 system. J Mol Catal B Enzym. 2016;126:69–75. doi:10.1016/j.molcatb.2016.02.001
18. Gohain SB, Basumatary M, Boruah PK, Das MR, Thakur AJ. Nano Au/Pd-catalysed ‘on-water’ synthesis of C3–C3′ diaryl-oxindole scaffolds via N 2 -selective dearomatization of indole. Green Chem. 2020;22(1):170–179. doi:10.1039/C9GC02370D
19. Deng Z, Peng X, Huang P, Jiang L, Ye D, Liu L. A multifunctionalized strategy of indoles to C2-quaternary indolin-3-ones via a TEMPO/Pd-catalyzed cascade process. Org Biomol Chem. 2017;15(2):442–448. doi:10.1039/C6OB02285E
20. Baruah MJ, Dutta A, Biswas S, et al. Fe 2 O 3 Nanocatalysts Supported on Zeolite-Y for the Selective Synthesis of C2 Di-Indolyl Indolones and Isatins. ACS Appl Nano Mater. 2022;5(1):1446–1459. doi:10.1021/acsanm.1c03987
21. Gonelimali FD, Lin J, Miao W, et al. Antimicrobial properties and mechanism of action of some plant extracts against food pathogens and spoilage microorganisms. Front Microbiol. 2018;9:1639. doi:10.3389/fmicb.2018.01639
22. Manandhar S, Luitel S, Dahal RK. In vitro antimicrobial activity of some medicinal plants against human pathogenic bacteria. J Trop Med. 2019;2019:1–5. doi:10.1155/2019/1895340
23. Kuete V, Betrandteponno R, Mbaveng AT, et al. Antibacterial activities of the extracts, fractions and compounds from Dioscorea bulbifera. BMC Complement Altern Med. 2012;12:228. doi:10.1186/1472-6882-12-228
24. Origin: data analysis and graphing software. Available from: https://www.originlab.com/index.aspx?go=Products/Origin.
25. van de Waterbeemd H, Gifford E. ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Discov. 2003;2(3):192–204. doi:10.1038/nrd1032
26. Breijyeh Z, Jubeh B, Karaman R. Resistance of gram-negative bacteria to current antibacterial agents and approaches to resolve it. Mol Basel Switz. 2020;25(6):E1340. doi:10.3390/molecules25061340
27. Rock CO, Cronan JE. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim Biophys Acta. 1996;1302(1):1–16. doi:10.1016/0005-2760(96)00056-2
28. Andolfo G, Schuster C, Gharsa HB, Ruocco M, Leclerque A. Genomic analysis of the nomenclatural type strain of the nematode-associated entomopathogenic bacterium Providencia vermicola. BMC Genom. 2021;22(1):708. doi:10.1186/s12864-021-08027-w
29. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
30. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi:10.1101/gr.1239303
31. Bora N, Jha AN. In silico metabolic pathway analysis identifying target against leishmaniasis - A kinetic modeling approach. Front Genet. 2020;11:179. doi:10.3389/fgene.2020.00179
32. Udhaya Kumar S, Thirumal Kumar D, Siva R, et al. Dysregulation of signaling pathways due to differentially expressed genes from the B-cell transcriptomes of systemic lupus erythematosus patients – a bioinformatics approach. Front Bioeng Biotechnol. 2020;8:276. doi:10.3389/fbioe.2020.00276
33. Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2. doi:10.1186/1471-2105-4-2
34. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11. doi:10.1186/1752-0509-8-S4-S11
35. Sabidussi G. The centrality index of a graph. Psychometrika. 1966;31(4):581–603. doi:10.1007/BF02289527
36. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi:10.1093/molbev/msy096
37. Li KB. ClustalW-MPI: clustalW analysis using distributed and parallel computing. Bioinforma Oxf Engl. 2003;19(12):1585–1586. doi:10.1093/bioinformatics/btg192
38. Efron B. The Jackknife, the Bootstrap and Other Resampling Plans. Society for Industrial and Applied Mathematics; 1982. doi:10.1137/1.9781611970319
39. Felsenstein J. Confidence limits on Phylogenies: an approach using the Bootstrap. Evol Int J Org Evol. 1985;39(4):783–791. doi:10.1111/j.1558-5646.1985.tb00420.x
40. Chakraborty R. Molecular evolution and phylogenetics, Masatoshi Nei and Sudhir Kumar. Oxford University Press, Oxford, England/New York, USA, 2000, xiv+333 pages (hardback) $75; (paperback) $35.00. Mol Phylogenet Evol. 2002;25(3):569–570. doi:10.1016/S1055-7903(02)00245-2
41. Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot. Methods Mol Biol. 2007;406:89–112. doi:10.1007/978-1-59745-535-0_4
42. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
43. Waterhouse A, Bertoni M, Bienert S, et al. Swiss-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi:10.1093/nar/gky427
44. Kozakov D, Grove LE, Hall DR, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10(5):733–755. doi:10.1038/nprot.2015.043
45. Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46(W1):W363–W367. doi:10.1093/nar/gky473
46. BIOVIA Discovery Studio - BIOVIA - Dassault Systèmes®. Available from: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-discovery-studio/.
47. Wu G, Robertson DH, Brooks CL, Vieth M. Detailed analysis of grid-based molecular docking: a case study of CDOCKER?A CHARMm-based MD docking algorithm. J Comput Chem. 2003;24(13):1549–1562. doi:10.1002/jcc.10306
48. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi:10.1002/jcc.20291
49. Rajkhowa S, Pathak U, Patgiri H. Elucidating the interaction and stability of withanone and withaferin‐a with human serum albumin, lysozyme and hemoglobin using computational biophysical modeling. ChemistrySelect. 2022;7(12):12. doi:10.1002/slct.202103938
50. Kemmerer S, Voss JC, Faller R. Molecular dynamics simulation of dipalmitoylphosphatidylcholine modified with a MTSL nitroxide spin label in a lipid membrane. Biochim Biophys Acta BBA Biomembr. 2013;1828(11):2770–2777. doi:10.1016/j.bbamem.2013.07.030
51. Hünenberger PH. Thermostat algorithms for molecular dynamics simulations. In: Holm C, Kremer K, editors. Advanced Computer Simulation. Vol. 173. Springer Berlin Heidelberg; 2005:105–149. doi: 10.1007/b99427
52. Turner P XMGRACE, Version 5.1. 19. Cent Coast Land-Margin Res Or Grad Inst Sci Technol Beaverton OR; 2005:2.
53. Lupande-Mwenebitu D, Khedher MB, Khabthani S, et al. First genome description of providencia vermicola isolate bearing NDM-1 from blood culture. Microorganisms. 2021;9(8):1751. doi:10.3390/microorganisms9081751
54. Egan AF, Russell RR. Conditional mutations affecting the cell envelope of Escherichia coli K-12. Genet Res. 1973;21(2):139–152. doi:10.1017/s001667230001332x
55. Zhang Y, Cronan JE. Transcriptional analysis of essential genes of the Escherichia coli fatty acid biosynthesis gene cluster by functional replacement with the analogous Salmonella typhimurium gene cluster. J Bacteriol. 1998;180(13):3295–3303. doi:10.1128/JB.180.13.3295-3303.1998
56. Heath RJ, Rock CO. Fatty acid biosynthesis as a target for novel antibacterials. Curr Opin Investig Drugs. 2004;5(2):146–153.
57. Funahashi A, Morohashi M, Kitano H, Tanimura N. CellDesigner: a process diagram editor for gene-regulatory and biochemical networks. BIOSILICO. 2003;1(5):159–162. doi:10.1016/S1478-5382(03)02370-9
58. Rasti S, Vogiatzis C. A survey of computational methods in protein–protein interaction networks. Ann Oper Res. 2019;276(1–2):35–87. doi:10.1007/s10479-018-2956-2
59. Marcella AM, Barb AW. The R117A variant of the Escherichia coli transacylase FabD synthesizes novel acyl-(acyl carrier proteins). Appl Microbiol Biotechnol. 2017;101(23–24):8431–8441. doi:10.1007/s00253-017-8586-9
60. Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18(5):691–699. doi:10.1093/oxfordjournals.molbev.a003851
61. Bauldry S. Structural equation modeling. In: International Encyclopedia of the Social & Behavioral Sciences. Elsevier; 2015:615–620. doi:10.1016/B978-0-08-097086-8.44055-9
62. Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 1992;8(3):275–282. doi:10.1093/bioinformatics/8.3.275
63. Gogoi D, Chaliha AK, Sarma D, Kakoti BB, Buragohain AK. Identification of potential type 4 cAMP phosphodiesterase inhibitors via 3D pharmacophore modeling, virtual screening, DFT and structural bioisostere design. Med Chem Res. 2017;26(11):3000–3014. doi:10.1007/s00044-017-1998-3
64. Gogoi D, Baruah VJ, Chaliha AK, Kakoti BB, Sarma D, Buragohain AK. 3D pharmacophore-based virtual screening, docking and density functional theory approach towards the discovery of novel human epidermal growth factor receptor-2 (HER2) inhibitors. J Theor Biol. 2016;411:68–80. doi:10.1016/j.jtbi.2016.09.016
65. Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9(9):646–652. doi:10.1038/nsb0902-646
66. Kumar CV, Swetha RG, Anbarasu A, Ramaiah S. Computational analysis reveals the association of Threonine 118 methionine mutation in PMP22 resulting in CMT-1A. Adv Bioinforma. 2014;2014:1–10. doi:10.1155/2014/502618
67. Mahadevi AS, Sastry GN. Cation−π interaction: its role and relevance in chemistry, biology, and material science. Chem Rev. 2013;113(3):2100–2138. doi:10.1021/cr300222d
68. Kollman PA, Allen LC. Theory of the hydrogen bond. Chem Rev. 1972;72(3):283–303. doi:10.1021/cr60277a004
69. Jungwirth P. Biological water or rather water in biology? J Phys Chem Lett. 2015;6(13):2449–2451. doi:10.1021/acs.jpclett.5b01143
70. Balamurugan K, Pisabarro MT. Stabilizing role of water solvation on anion−π interactions in proteins. ACS Omega. 2021;6(39):25350–25360. doi:10.1021/acsomega.1c03264
71. Cronan JE, Thomas J. Chapter 17 Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. In: Methods in Enzymology. Vol. 459. Elsevier; 2009:395–433. doi:10.1016/S0076-6879(09)04617-5
72. Gago G, Diacovich L, Arabolaza A, Tsai SC, Gramajo H. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol Rev. 2011;35(3):475–497. doi:10.1111/j.1574-6976.2010.00259.x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.