")
Back to Journals » Journal of Inflammation Research » Volume 10
An in vitro model of renal inflammation after ischemic oxidative stress injury: nephroprotective effects of a hyaluronan ester with butyric acid on mesangial cells
Authors Baraldi O, Bianchi F, Menghi V , Angeletti A , Croci Chiocchini AL, Cappuccilli M, Aiello V, Comai G, La Manna G
Received 31 March 2017
Accepted for publication 2 June 2017
Published 8 September 2017 Volume 2017:10 Pages 135—142
DOI https://doi.org/10.2147/JIR.S138431
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Olga Baraldi,1 Francesca Bianchi,2,3 Viola Menghi,1 Andrea Angeletti,1 Anna Laura Croci Chiocchini,1 Maria Cappuccilli,1 Valeria Aiello,1 Giorgia Comai,1 Gaetano La Manna1
1Department of Experimental, Diagnostic and Specialty Medicine, Nephrology, Dialysis and Renal Transplant Unit, Sant’Orsola-Malpighi Hospital, University of Bologna, Bologna, 2Stem Wave Institute for Tissue Healing, Gruppo Villa Maria Care & Research – Ettore Sansavini Health Science Foundation, Lugo, Ravenna, 3National Institute of Biostructures and Biosystems at the Department of Experimental, Diagnostic and Specialty Medicine, Sant’Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy
Background: Acute kidney injury, known as a major trigger for organ fibrosis and independent predictor of chronic kidney disease, is characterized by mesangial cell proliferation, inflammation and unbalance between biosynthesis and degradation of extracellular matrix. Therapeutic approaches targeting the inhibition of mesangial cell proliferation and matrix expansion may represent a promising opportunity for the treatment of kidney injury. An ester of hyaluronic acid and butyric acid (HB) has shown vasculogenic and regenerative properties in renal ischemic-damaged tissues, resulting in enhanced function recovery and minor degree of inflammation in vivo. This study evaluated the effect of HB treatment in mesangial cell cultures exposed to H2O2-induced oxidative stress.
Materials and methods: Lactate dehydrogenase release and caspase-3 activation were measured using mesangial cells prepared from rat kidneys to assess necrosis and apoptosis. Akt and p38 phosphorylation was analyzed to identify the possible mechanism underlying cell response to HB treatment. The relative expressions of matrix metallopeptidase 9 (MPP-9) and collagen type 1 alpha genes were also analyzed by quantitative real-time polymerase chain reaction. Cell proliferation rate and viability were measured using thiazolyl blue assay and flow cytometry analysis of cell cycle with propidium iodide.
Results: HB treatment promoted apoptosis of mesangial cells after H2O2-induced damage, decreased cellular proliferation and activated p38 pathway, increasing expression of its target gene MPP-9.
Conclusion: This in vitro model shows that HB treatment seems to redirect mesangial cells toward apoptosis after oxidative damage and to reduce cell proliferation through p38 MAPK pathway activation and upregulation of MPP-9 gene expression involved in mesangial matrix remodeling.
Keywords: acute kidney injury, apoptosis, hyaluronan ester of butyric acid, mesangial cells
Introduction
Acute kidney injury (AKI) is a common problem in critically ill patients, and AKI-induced organ inflammation and tubular fibrosis represent a major risk factor for the development of end-stage renal failure.1 Efforts aimed at reducing AKI incidence and improving its clinical management may benefit patients’ survival and delay the initiation of dialysis with all the related complications.2,3 Despite advances in intensive care, renal replacement therapy techniques4 and decrease in in-hospital mortality,5 effective treatments to preserve renal function after AKI have not been developed yet and outcomes have not changed significantly over time.6
During injury, tubular cell death, changes in filtration barrier, glomerular misfiltration, vasoconstriction and tubular obstruction, as well as proliferation of mesangial cells and proliferative/apoptotic damage of podocytes usually occur.7–9 Various models of renal inflammation and ischemia have highlighted a role of reactive oxygen species (ROS) in glomerular injury. During the oxidative damage, mesangial cells induce cell death or hyperproliferation and accumulation of extracellular matrix in the surviving population, thus initiating kidney fibrosis.10,11 Therefore, therapeutic approaches targeting inhibition of mesangial cell proliferation may provide promising opportunities to prevent fibrosis and defend the kidney from concomitant damages during inflammation.
We have recently demonstrated the ability of a differentiating agent, an ester of hyaluronic acid and butyric acid (HB), to enhance the regenerative processes of the kidney after induction of ischemia/reperfusion injury. In our previous ischemic AKI rat model, treatment with HB-preconditioned mesenchymal stem cells accelerated renal function recovery and resulted in a minor degree of inflammation, thus promoting the restoration of the damaged tissue.12
The bioactivity of HB, a four-carbon chain fatty acid (CH3CH2CH2COOH), is related to its ability to modify nuclear architecture, to induce apoptosis and to change the structure of chromatin through modulation of posttranslational modifications involved in metabolic activities and different signal pathways, namely, cell differentiation, proliferation, motility, induction of cell-cycle arrest and apoptosis.13–19 More recent studies have described a further biological role of butyrate as an inhibitor of histone deacetylases (HDACs), a class of enzymes involved in several biological pathways: fibroblast proliferation, chemokine production and regulation of the expression of connective tissue growth factor (CTGF), collagen and α-smooth muscle actin (α-SMA) in interstitial renal fibroblast and tubular epithelial cells.20
Previous findings from our group highlighted the enhanced efficacy of hyaluronan monoesters with butyric acid to promote the recovery of damaged tubular epithelial cells or the differentiation and proliferation of potential residential stem cells.12,21,22 The rationale for the use of hyaluronate, rather than butyrate alone, lies in its efficiency as a targeting system, through its receptor CD44, expressed on several cell types. The ligand–receptor interaction allows the internalization of the transported molecules, including butyric acid.23
Here, we analyzed in vitro the potential protective role of HB on proliferation, death and matrix deposition of mesangial cells exposed to oxidative stress in order to evaluate the possibility of using this molecule to prevent the progression of AKI into organ fibrosis.
Materials and methods
Experimental model
Cell culture
Two-day-old male Wistar rats were provided by Charles River Laboratories, Inc. (Lecco, Italy). The use of animals and all animal procedures used in this study was approved by the local bioethics committee of the University of Bologna. The procedures conformed to the protocols of the local Bioethics Committee of Nephrology Department of the University, number 02/2010.
Kidneys were removed aseptically and homogenized in Hanks’ solution, and mesangial cells were extracted as previously described.24 The small cubes of renal cortex tissue were washed three times with Hanks’ Balanced Salt Solution (HBSS; Invitrogen/Gibco, Carlsbad, CA, USA), minced and passed through sieves to isolate glomeruli. Next, glomeruli were digested by a solution containing 0.1% collagenase IV at 37°C and centrifuged at 400× g for 5 min. The pellets were resuspended in Roswell Park Memorial Institute (RPMI) 1640 with 10% fetal bovine serum. The released cells were then cultured in RPMI 1640 medium (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin (all purchased from EuroClone, Pero, Italy) and 5 μg/mL each of insulin (Sigma-Aldrich Co., St Louis, MO, USA). A total of 50 µM H2O2 was used to simulate in vitro ischemia/reperfusion injury. To evaluate the effect of HB treatment in the prevention of H2O2 damage, cells were divided into three groups:
- HB–H2O2 group: mesangial cells pretreated with HB (1 g/L) for 24 h, before adding H2O2
- H2O2 group: cells untreated with HB before adding H2O2
- Negative control group: cells not treated with HB or H2O2
Lactate dehydrogenase assay
H2O2 toxicity was assessed by measuring the lactate dehydrogenase (LDH) released in culture media at 2, 6, 16 and 24 h after H2O2 treatment using Cell Toxicity Colorimetric Assay Kit (Sigma-Aldrich Co.). The results were expressed as percentage of viable cells versus untreated control cells. In order to distinguish apoptosis from necrosis, 20 µM N-acetyl-Asp-Glu-Val-Asp-aldehyde (DEVD-CHO), a caspase-3-preferred peptide inhibitor, was added to the cells for 1 h prior to H2O2. To evaluate HB toxicity, an aliquot of cell culture was treated only with HB and no addition of H2O2.
Caspase-3 activity assay
The activity of caspase-3 was determined by Caspase Colorimetric Assay Kit (Sigma-Aldrich Co.) according to the manufacturer’s protocol. Briefly, cells were collected and lyzed in the lysis buffer provided with the kit. Caspase-3 substrate was added to each well. The hydrolysis of the peptide substrate acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) by caspase-3 results in the release of the p-nitroaniline (pNA) moiety. The chromophore pNA was detected using a spectrophotometer at a wavelength of 405 nm. The caspase enzymatic activity in cell lysates was directly proportional to the color reaction.
Viability assay
The thiazolyl blue (MTT) assay (Boehringer Mannheim, Indianapolis, IN, USA) was performed to evaluate viability of cells. Cells were cultured for 6, 16, 24, 48 and 72 h and analyzed according to the manufacturer’s instructions. Cytotoxicity was calculated by expressing experimental absorbance values as percentage of control values (untreated cells).
Cell-cycle analysis
Cells were collected after H2O2 treatment, resuspended in phosphate-buffered saline (PBS) at a final concentration of 1 × 106 cells/mL and then fixed with Trisodium Citrate (1 mg/mL), NaCl (0.5 mg/mL), Nonidet P40 (300 µg/L) and 10 µg/mL ribonuclease A (RNase A) for 1 h at 4°C. Next, citric acid (15 mg/mL) and saccarose (85 mg/mL) were added, and the cells were incubated for 45 min. Fixed cells were stained with propidium iodide (PI) for 30 min at room temperature in the dark and analyzed by a FACSAria flow cytometer (BD Biosciences, San Jose, CA, USA).
Western blot
Mesangial cell lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). P-p38, P-Akt and GAPDH were detected by incubation with polyclonal rabbit P-p38 specific antibody (1:500 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA), polyclonal rabbit P-Akt antibody (1:2000 dilution; Cell Signaling Technology, Danvers, MA, USA) and a monoclonal rabbit GAPDH-specific antibody (1:1000 dilution; Cell Signaling Technology), respectively.
Gene expression
Gene expression of collagen 1 alpha and MMP-9 was analyzed at 2, 6, 16 and 24 h after H2O2 treatment. Total RNA was extracted from the cultured mesangial cells using TRIzol Reagent (Invitrogen), according to the manufacturer’s directions. Following reverse transcription, ~20 ng of cDNA was relatively quantified by quantitative polymerase chain reaction (qPCR) using SYBR Green FastStart Kit (Lightcycler FastStart DNA MasterPLUS SYBR Green I; Roche Applied Science, Penzberg, Germany) on a LightCycler System (Roche). The primers were predesigned by QuantiTect Primer Assays (Qiagen, Hilden, Germany), and gene expression from quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) data was normalized using GAPDH as a housekeeping gene.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM). Two-sided Student’s t-test for independent samples was used to evaluate the different effects of the treatment between groups. Comparison of more than two groups was performed by one-way analysis of variance (ANOVA), followed by Bonferroni post hoc test. A p-value <0.05 was assumed as the limit of significance. Data analysis was performed using GraphPad Prism for Windows software (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Effect of HB treatment on cell viability
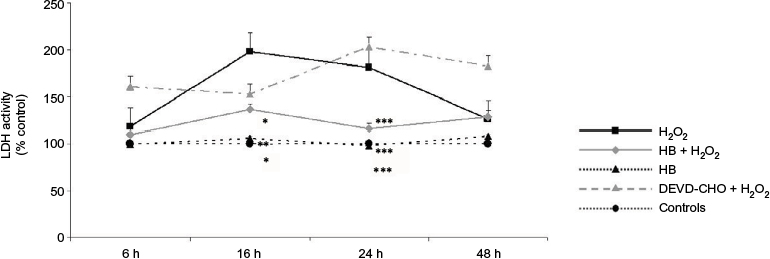
A significant increased release of LDH into the medium was observed in cells added with H2O2 without HB pretreatment (H2O2 group), which at 16 h was two times higher than in untreated negative control cells. At the same time point, the percentage of death cells was significantly lower in the HB–H2O2 group than in the H2O2 group. At 24 h, the level of LDH release found in the H2O2 group was significantly higher in comparison with all the other groups. Later, there was a decrease in the percentage of dead cells until 48 h, probably due to the hyperproliferation of the surviving cells. Pretreatment with DEVD-CHO, an inhibitor of capsase-3, did not reduce cell death rate, suggesting that H2O2 preferentially induced necrosis. Treatment with HB alone was not toxic as it did not affect LDH release (Figure 1A). When analyzing apoptosis through caspase-3 activity, we observed that in the presence of DEVD-CHO, caspase-3 was inhibited, whereas pretreatment with HB induced activation with the highest effect observed at 16 h (Figure 1B).
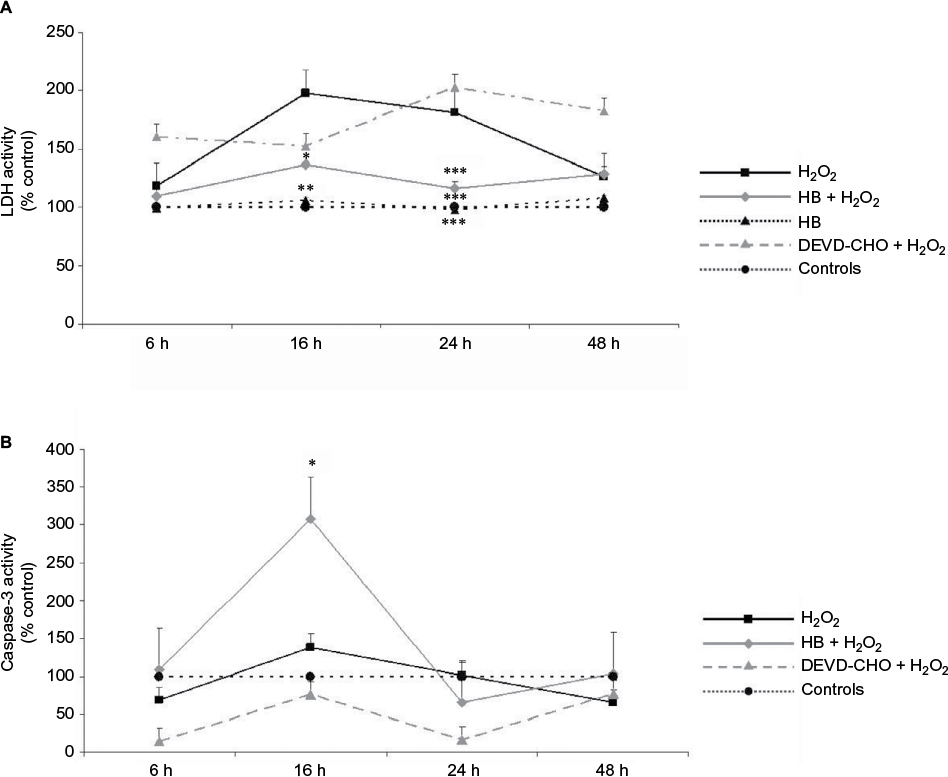 | Figure 1 HB treatment induces apoptosis after H2O2-induced oxidative stress. Notes: (A) LDH release into the medium: H2O2 treatment induced cell death as demonstrated by high LDH release starting from 16 h; HB had a protective effect, and LDH release was reduced. The pretreatment with DEVD-CHO did not affect LDH release in H2O2-treated cells, indicating that H2O2 triggered necrotic cell death. The results are expressed as the percentage of viable cells compared with negative control cells (not treated with HB or H2O2) referred to as 100%. (B) Caspase-3 activity: HB treatment activated caspase-3, while H2O2 alone did not. Negative control cells are referred as 100%. Data are given as mean ± SEM of three independent experiments. Statistical analysis: one-way ANOVA (n = 3). Significantly different from H2O2-treated cells (*p < 0.05, **p < 0.01 and ***p < 0.001). Abbreviations: HB, hyaluronic acid and butyric acid; LDH, lactate dehydrogenase; DEVD-CHO, N-acetyl-Asp-Glu-Val-Asp-aldehyde; SEM, standard error of the mean; ANOVA, analysis of variance. |
Taken together, these results indicate that, in the presence of H2O2, cells died preferentially via necrosis, since cell death was not prevented by the treatment with DEVD-CHO, an inhibitor of caspase-3. Conversely, HB pretreatment reduced cell death rate and activated caspase-3.
Cell cycle was then analyzed to better investigate the doubled release of LDH observed in cells added only with H2O2. At 24 h, 35% of H2O2-treated cells were in the G2/S phase, in comparison with 10% of untreated controls and 23% of HB-pretreated cells (Figure 2).
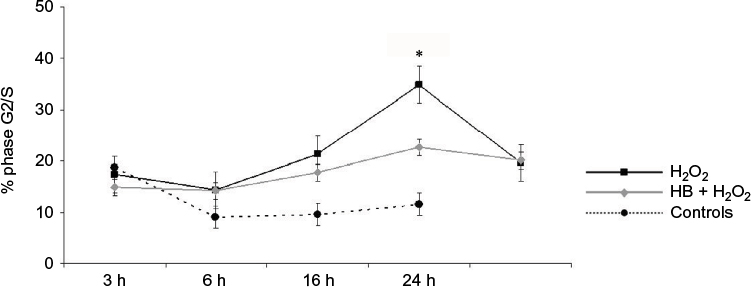 | Figure 2 HB treatment reduces cellular proliferation after H2O2-induced oxidative stress. Notes: Cell-cycle analysis indicated that the percentage of cells in phase G2/S increased after treatment with H2O2, but it was significantly lower when cells had been pretreated with HB. Untreated cells (not treated with HB or H2O2) were considered as negative controls. Data are given as mean ± SEM of three independent experiments. Statistical analysis: one-way ANOVA (n = 3). Significantly different from H2O2 (*p < 0.05). Abbreviations: HB, hyaluronic acid and butyric acid; SEM, standard error of the mean; ANOVA, analysis of variance. |
The results of MTT assay confirmed the protective effect of HB on the death of mesangial cells. H2O2-treated cells immediately lost their metabolic activity, while HB pretreatment preserved a nearly normal activity till 6 h after H2O2 treatment. Then, metabolic activity was greatly reduced, but it remained higher than in non-pretreated cells (Figure 3).
 | Figure 3 HB preserved cellular metabolism after H2O2-induced oxidative stress. Notes: MTT assay revealed that oxidative stress reduced cellular metabolism, but this effect was prevented when cells had been pretreated with HB. Negative control cells (not treated with HB or H2O2) are referred as 100%. Data are given as mean ± SEM of three independent experiments. Statistical analysis: one-way ANOVA (n = 3). Significantly different from H2O2-treated cells (*p < 0.05; **p < 0.01; ***p < 0.001). Abbreviations: HB, hyaluronic acid and butyric acid; SEM, standard error of the mean; ANOVA, analysis of variance; DEVD-CHO, N-acetyl-Asp-Glu-Val-Asp-aldehyde. |
Effect of HB treatment on Akt, p38 phosphorylation and gene expression
The phosphorylation status of Akt and p38 was detected at 2, 6 and 24 h after the treatment with H2O2 using Western blot (Figure 4A). Akt phosphorylation at 2 and 6 h in the H2O2 group was found to be increased in comparison with untreated control cells, but it was significantly reduced by the pretreatment with HB (Figure 4B). The level of p38 phosphorylation at 6 and 24 h was significantly higher in HB-pretreated cells compared to the other groups (Figure 4C).
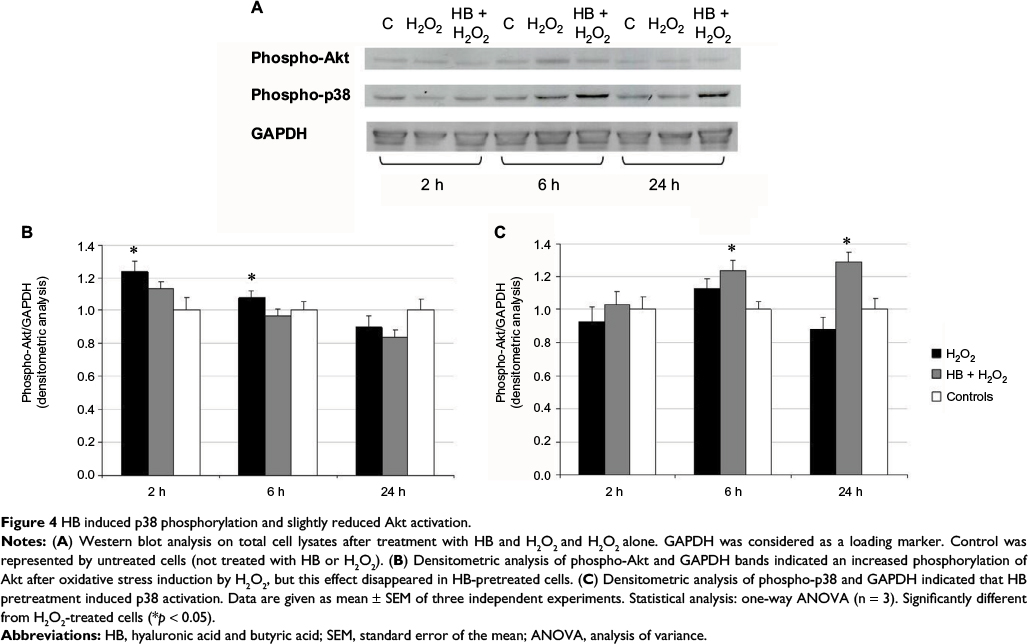 | Figure 4 HB induced p38 phosphorylation and slightly reduced Akt activation. Notes: (A) Western blot analysis on total cell lysates after treatment with HB and H2O2 and H2O2 alone. GAPDH was considered as a loading marker. Control was represented by untreated cells (not treated with HB or H2O2). (B) Densitometric analysis of phospho-Akt and GAPDH bands indicated an increased phosphorylation of Akt after oxidative stress induction by H2O2, but this effect disappeared in HB-pretreated cells. (C) Densitometric analysis of phospho-p38 and GAPDH indicated that HB pretreatment induced p38 activation. Data are given as mean ± SEM of three independent experiments. Statistical analysis: one-way ANOVA (n = 3). Significantly different from H2O2-treated cells (*p < 0.05). Abbreviations: HB, hyaluronic acid and butyric acid; SEM, standard error of the mean; ANOVA, analysis of variance. |
qPCR showed no differences in collagen 1 alpha gene expression between H2O2 group and HB–H2O2 group until 16 h following treatment, and then at 24 h, collagen 1 alpha gene expression was lower in cells that had been pretreated with HB before oxidative stress insult by H2O2 (Figure 5B). Conversely, MMP-9 expression was unaffected by H2O2 alone, while it appeared to be enhanced by HB pretreatment at each time point, even if the difference met statistical significance at 24 h (Figure 5A).
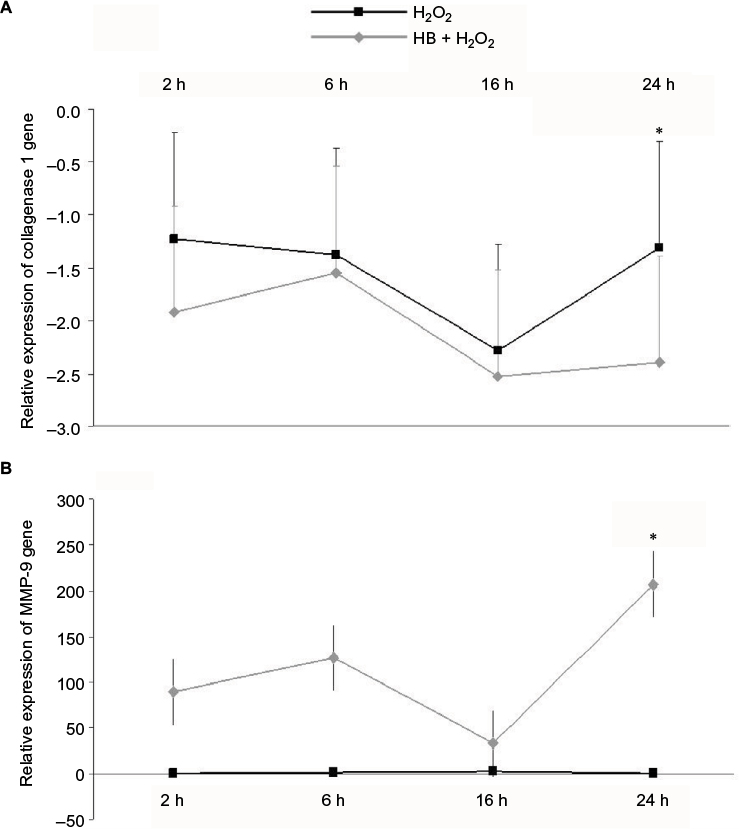 | Figure 5 HB treatment immediately upregulated MMP-9 expression and reduced collagen 1 alpha expression. Notes: (A) Collagen 1 alpha gene expression was downregulated after H2O2 treatment and further decreased in cells that had been pretreated with HB, with a significant difference between the groups at 24 h following H2O2 treatment. (B) Conversely, MMP-9 expression was unaffected by H2O2 alone, but it tended to be increased in the presence of HB (significant at 24 h). Data were normalized on GAPDH expression; untreated cells (not treated with HB or H2O2) were used as a control to determine the relative quantification. Statistical analysis: one-way ANOVA (n = 3). Significantly different from H2O2 (*p < 0.05). Abbreviations: HB, hyaluronic acid and butyric acid; ANOVA, analysis of variance. |
Discussion
In this study, we investigated the mechanistic role of HB on proliferation, death and matrix deposition of mesangial cells in an in vitro model of AKI after oxidative stress induction. We have previously described in two different in vivo models the ability of hyaluronan esters of butyric acid to enhance the rescuing potential of human mesenchymal stem cells.12,21 In spite of the promising results, the need of several weeks to collect and expand cellular cultures leads to a substantial delay for autologous stem cell transplantation and for a timely intervention during the early phases of AKI. We have hypothesized that HB is able to reduce the degree of injury, and it may serve as a first effort to repair a damaged kidney. Here, we designed an in vitro model of oxidative stress to reproduce ischemia/reperfusion injury by exposing mesangial cells to hydrogen peroxide.
In our experimental model, we used a 50 μM concentration of H2O2 to induce ischemic damage, after testing scalar H2O2 concentrations (25, 50, 75 and 100 μM) to assess the mortality rate achieved after 24 h of treatment. We found that 24 h vitality was 95% using the 25 μM concentration, 80% with the 50 μM concentration and 40% with the 75 μM concentration, the latter resulting in noticeable damage. An important point is the discrimination of cellular response to H2O2 insult between necrosis and apoptosis. In cells not previously added with HB, hydrogen peroxide triggered preferentially necrotic cell death, while HB pretreatment reduced LDH release and led to caspase-3 activation, the main effector of apoptosis. Therefore, it seems that HB may predispose mesangial cells to programmed cell death, rather than necrosis, a process commonly associated with inflammation and fibrosis.11 A peak of LDH release was found 16 h after oxidative stress insult; then, the percentage of dead cells showed a progressive reduction. Interestingly, in cell-cycle analysis, we observed a significant increase in cells of the G2/S phase 24 h after H2O2 exposure, thus confirming the hypothesis that mesangial cells initially respond to oxidative damage with necrosis and then start to hyperproliferate, in line with previous evidence.11 HB pretreatment seems to limit the proliferative effect elicited by peroxide on mesangial cells. The role of apoptosis on mesangial biology is still controversial. However, these cells are committed to apoptosis or acquire an activated phenotype in response to metabolic, immunologic or hemodynamic injury and undergo hypertrophy and proliferation with overproduction of matrix proteins, growth factors, chemokines and cytokines. These soluble factors may act through either autocrine or paracrine pathways on mesangial or glomerular cells, respectively.24 Therefore, a treatment able to switch cell commitment toward apoptosis, to reduce proliferation and to control inflammation after injury may exert a beneficial effect on the damaged tissue. In different animal models of renal failure, kidney injury has been associated with activation of apoptotic signaling. Interestingly, the regression of apoptotic features is concomitant with renal function recovery.25,26 Thus, the reversibility of apoptosis may limit inflammation and facilitate tissue restoring.
There is a large body of evidence to indicate that many glomerular diseases are associated with increased activation of PI3-kinase/Akt signaling in mesangial cells. It has been reported that both glomerulosclerosis and albuminuria are worsened in Akt2(–/–) mice as compared to wild-type animals,27 and podocyte survival and foot process effacement are directly regulated by Akt2 pathway.27,28 Diabetic nephropathy is characterized by mesangial cell expansion and extracellular matrix accumulation; in this context, PI3-k/Akt activation via Fox3a phosphorylation induces cellular survival and oxidative stress.29,30 Furthermore, hydrogen peroxide has been proven to induce Akt activation in mesangial cells in vitro.31
Considering that Akt pathway is usually involved in cell survival and proliferation,32 our analysis highlighted a significant increase in Akt phosphorylation levels in H2O2-treated cells compared to controls. However, a similar rise in Akt phosphorylation was found in HB-pretreated cells, suggesting that HB might prevent the activation of survival pathways. Hyperproliferation is often associated with fibrosis, increased matrix deposition and scar formation.33 The p38 signaling has a key role in the regulation of cell proliferation, extracellular matrix gene expression and apoptosis.34 In particular, p38 pathway controls gene expression of MMP-9 and MMP-2,35 matrix metalloproteinases specifically secreted by mesangial cells in normal conditions to control the dynamic turnover of mesangial matrix.36 An intriguing finding emerging from our study was the increased p38 phosphorylation in HB-pretreated cells and a consistent upregulation of MMP-9 transcription revealed by qPCR analysis. Thus, we postulate a possible mechanism by which HB pretreatment could promote a recovery of mesangial function in extracellular matrix regulation. After the oxidative damage, HB might be able to redirect mesangial cells toward apoptosis and to reduce cellular proliferation; concomitantly, it might activate p38 MAPK pathway and enhance MMP-9 expression.
Conclusion
These preliminary findings might provide a basis for further in vivo studies to better elucidate the rationale for the use of HB in injured kidney to prevent and possibly reverse fibrosis and to promote tissue regeneration.
Disclosure
The authors report no conflicts of interest in this work.
References
Lameire NH, Bagga A, Cruz D, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–179. | ||
Cianciolo G, Colí L, La Manna G, et al. Is beta2-microglobulin-related amyloidosis of hemodialysis patients a multifactorial disease? A new pathogenetic approach. Int J Artif Organs. 2007;30(10):864–878. | ||
Cianciolo G, Donati G, La Manna G, et al. The cardiovascular burden of end-stage renal disease patients. Minerva Urol Nefrol. 2010;62(1):51–66. | ||
Colì L, La Manna G, Comai G, et al. Automatic adaptive system dialysis for hemodialysis-associated hypotension and intolerance: a noncontrolled multicenter trial. Am J Kidney Dis. 2011;58(1):93–100. | ||
Waikar SS, Curhan GC, Wald R, McCarthy EP, Chertow GM. Declining mortality in patients with acute renal failure, 1988 to 2002. J Am Soc Nephrol. 2006;17(4):1143–1150. | ||
Kellum JA, Sileanu FE, Bihorac A, Hoste EA, Chawla LS. Recovery after acute kidney injury. Am J Respir Crit Care Med. 2017;195(6):784–791. | ||
Lameire N, Van Biesen W, Vanholder R. Acute kidney injury. Lancet. 2008;372(9653):1863–1865. | ||
Migliorini A, Ebid R, Scherbaum CR, Anders HJ. The danger control concept in kidney disease: mesangial cells. J Nephrol. 2013;26(3):437–449. | ||
Rodriguez F, Bonacasa B, Fenoy FJ, Salom MG. Reactive oxygen and nitrogen species in the renal ischemia/reperfusion injury. Curr Pharm Des. 2013;19(15):2776–2794. | ||
Heyman SN, Rosen S, Rosenberg C. Renal parenchymal hypoxia, hypoxia adaptation, and the pathogenesis of radiocontrast nephropathy. Clin J Am Soc Nephrol. 2008;3(1):288–296. | ||
Lee HS, Song CY. Differential role of mesangial cells and podocytes in TGF-beta-induced mesangial matrix synthesis in chronic glomerular disease. Histol Histopathol. 2009;24(7):901–908. | ||
La Manna G, Bianchi F, Cappuccilli M, et al. Mesenchymal stem cells in renal function recovery after acute kidney injury: use of a differentiating agent in a rat model. Cell Transplant. 2011;20(8):1193–1208. | ||
Kien CL, Chang JC, Cooper JR. Butyric acid is synthesized by piglets. J Nutr. 2000;130(2):234–237. | ||
Smith JG, Yokoyama WH, German JB. Butyric acid from the diet: actions at the level of gene expression. Crit Rev Food Sci Nutr. 1998;38(4):259–297. | ||
Li RW, Li C. Butyrate induces profound changes in gene expression related to multiple signal pathways in bovine kidney epithelial cells. BMC Genomics. 2006;7:234. | ||
Machado RA, Constantino LS, Damiani Tomasi C, et al. Sodium butyrate decreases the activation of NF-kB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol Dial Transplant. 2012;27(8):3136–3140. | ||
Li CJ, Elsasser TH. Butyrate-induced apoptosis and cell cycle arrest in bovine kidney epithelial cells: involvement of caspase and proteasome pathways. J Anim Sci. 2005;83(1):89–97. | ||
Adams JM. Ways of dying: multiple pathways to apoptosis. Genes Dev. 2003;17(20):2481–2495. | ||
Pang M, Ma L, Liu N, et al. Histone deacetylase 1/2 mediates proliferation of renal interstitial fibroblasts and expression of cell cycle proteins. J Cell Biochem. 2011;112(8):2138–2148. | ||
Khan S, Jena G. Sodium butyrate, a HDAC inhibitor ameliorates eNOS, iNOS and TGF-b1-induced fibrogenesis, apoptosis and DNA damage in kidney of juvenile diabetic rats. Food Chem Toxicol. 2014;73:127–139. | ||
Cavallari G, Olivi E, Bianchi F, et al. Mesenchymal stem cells and islet cotransplantation in diabetic rats: improved islet graft revascularization and function by human adipose tissue-derived stem cells preconditioned with natural molecules. Cell Transplant. 2012;21(12):2771–2781. | ||
Bianchi F, Sala E, Donadei C, Capelli I, La Manna G. Potential advantages of acute kidney injury management by mesenchymal stem cells. World J Stem Cells. 2014;6(5):644–650. | ||
Knudson W, Chow G, Knudson CB. CD44-mediated uptake and degradation of hyaluronan. Matrix Biol. 2002;21(1):15–23. | ||
Scivittaro V, Ganz MB, Weiss MF. AGEs induce oxidative stress and activate protein kinase C-beta(II) in neonatal mesangial cells. Am J Physiol Renal Physiol. 2000;278(4):F676–F683. | ||
Shuvy M, Nyska A, Beeri R, et al. Histopathology and apoptosis in an animal model of reversible renal injury. Exp Toxicol Pathol. 2011;63(4):303–306. | ||
La Manna G, Conte D, Cappuccilli ML, et al. An in vivo autotransplant model of renal preservation: cold storage versus machine perfusion in the prevention of ischemia/reperfusion injury. Artif Organs. 2009;33(7):565–570. | ||
Canaud G, Bienaime F, Viau A, et al. Akt2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19(10):1288–1296. | ||
Reiser J. Akt2 relaxes podocytes in chronic kidney disease. Nat Med. 2013;19(10):1212–1213. | ||
Kato M, Yuan H, Xu ZG, et al. Role of the Akt/FoxO3 a pathway in TGFb-1-mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. J Am Soc Nephrol. 2006;17(12):3325–3335. | ||
Chen N, Hao J, Li L, Li F, Liu S, Duan H. Carboxy-terminal modulator protein attenuated extracellular matrix deposit by inhibiting phospho-Akt, TGF-β1 and α-SMA in kidneys of diabetic mice. Biochem Biophys Res Commun. 2016;474(4):753–760. | ||
Venkatesan B, Mahimainathan L, Das F, Ghosh-Choudhury N, Ghosh Choudhury G. Downregulation of catalase by reactive oxygen species via PI 3 kinase/Akt signaling in mesangial cells. J Cell Physiol. 2007;211(2):457–467. | ||
Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. | ||
Cortes P, Riser BL, Yee J, Narins RG. Mechanical strain of glomerular mesangial cells in the pathogenesis of glomerulosclerosis: clinical implications. Nephrol Dial Transplant. 1999;14(6):1351–1354. | ||
Ma FY, Sachchithananthan M, Flanc RS, Nikolic-Paterson DJ. Mitogen activated protein kinases in renal fibrosis. Front Biosci (Schol Ed). 2009;1:171–187. | ||
Itatsu K, Sasaki M, Harada K, et al. Phosphorylation of extracellular signal-regulated kinase 1/2, p38 mitogen-activated protein kinase and nuclear translocation of nuclear factor-kappaB are involved in upregulation of matrix metalloproteinase-9 by tumour necrosis factor-alpha. Liver Int. 2009;29(2):291–298. | ||
Martin J, Eynstone L, Davies M, Steadman R. Induction of metalloproteinases by glomerular mesangial cells stimulated by proteins of the extracellular matrix. J Am Soc Nephrol. 2001;12(1):88–96. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.