")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
An antagonist of the platelet-activating factor receptor inhibits adherence of both nontypeable Haemophilus influenzae and Streptococcus pneumoniae to cultured human bronchial epithelial cells exposed to cigarette smoke
Authors Dhar Shukla S , Fairbairn R, Gell D, Latham R, Sohal SS, Walters EH, O'Toole R
Received 17 March 2016
Accepted for publication 21 May 2016
Published 25 July 2016 Volume 2016:11(1) Pages 1647—1655
DOI https://doi.org/10.2147/COPD.S108698
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Russell
Shakti D Shukla,1,* Rory L Fairbairn,1,* David A Gell,1 Roger D Latham,1 Sukhwinder S Sohal,1,2 Eugene H Walters,1 Ronan F O’Toole1
1Breathe Well Centre, School of Medicine, Faculty of Health, University of Tasmania, Hobart, TAS, Australia; 2School of Health Sciences, Faculty of Health, University of Tasmania, Launceston, TAS, Australia
*These authors contributed equally to this work
Background: COPD is emerging as the third largest cause of human mortality worldwide after heart disease and stroke. Tobacco smoking, the primary risk factor for the development of COPD, induces increased expression of platelet-activating factor receptor (PAFr) in the lung epithelium. Nontypeable Haemophilus influenzae (NTHi) and Streptococcus pneumoniae adhere to PAFr on the luminal surface of human respiratory tract epithelial cells.
Objective: To investigate PAFr as a potential drug target for the prevention of infections caused by the main bacterial drivers of acute exacerbations in COPD patients, NTHi and S. pneumoniae.
Methods: Human bronchial epithelial BEAS-2B cells were exposed to cigarette smoke extract (CSE). PAFr expression levels were determined using immunocytochemistry and quantitative polymerase chain reaction. The epithelial cells were challenged with either NTHi or S. pneumoniae labeled with fluorescein isothiocyanate, and bacterial adhesion was measured using immunofluorescence. The effect of a well-evaluated antagonist of PAFr, WEB-2086, on binding of the bacterial pathogens to BEAS-2B cells was then assessed. In silico studies of the tertiary structure of PAFr and the binding pocket for PAF and its antagonist WEB-2086 were undertaken.
Results: PAFr expression by bronchial epithelial cells was upregulated by CSE, and significantly associated with increased bacterial adhesion. WEB-2086 reduced the epithelial adhesion by both NTHi and S. pneumoniae to levels observed for non-CSE-exposed cells. Furthermore, it was nontoxic toward the bronchial epithelial cells. In silico analyses identified a binding pocket for PAF/WEB-2086 in the predicted PAFr structure.
Conclusion: WEB-2086 represents an innovative class of candidate drugs for inhibiting PAFr-dependent lung infections caused by the main bacterial drivers of smoking-related COPD.
Keywords: airway epithelium, NTHi, pneumococci, WEB-2086, platelet-activating factor receptor, PAFr antagonist
Introduction
COPD is a progressive and poorly reversible airflow obstructive lung illness manifested as chronic cough and shortness of breath. It is emerging as the third largest cause of human mortality after heart disease and stroke, killing over 3 million people worldwide each year.1 Tobacco smoking is the most important causative factor of COPD.2–4 Exposure to cigarette smoke has been shown to significantly increase invasive respiratory pneumococcal infections.5 Moreover, chronic exposure of mice to cigarette smoke resulted in significantly higher loads of nontypeable Haemophilus influenzae (NTHi) and Streptococcus pneumoniae in the lungs.6 NTHi and S. pneumoniae are the foremost respiratory bacterial pathogens involved in both chronic airway infection in COPD and acute exacerbations (AE) of COPD,7–9 but the reason for their prevalence in these lung conditions is poorly understood.
One potential epithelial adhesion site for chronic bacterial colonization of the lower respiratory tract airway is the platelet-activating factor receptor (PAFr), a G-protein-coupled receptor (GPCR).10–12 Cundell et al13 provided the first potential link between PAFr expression on lung structural cells and S. pneumoniae infection. Similarly, NTHi adheres to human bronchial epithelial cells in a PAFr-dependent manner.14 The importance of PAFr in respiratory bacterial infections has been reviewed previously.15
We have recently shown that PAFr expression is significantly upregulated in the airway epithelium of both the large and small airways in smokers and COPD patients.16,17 NTHi and S. pneumoniae adhere to PAFr expressed on the luminal surface of human respiratory tract epithelial cells through physicochemical interactions with phosphorylcholine (ChoP) (a molecular mimic of PAF) present on the cell wall surface of these bacteria.13,14,16 We have previously determined that cigarette smoke, the primary risk factor for development of COPD, induces increased expression of PAFr in lower airway epithelial cells, which correlates with enhanced adhesion of S. pneumoniae.18 In this work, we further investigate PAFr as an anti-infective therapeutic target and test the potential of a well-characterized PAFr antagonist, WEB-2086,19–21 to prevent bacterial adhesion of NTHi and S. pneumoniae to respiratory epithelial cells.
Methods
Bacterial strains and in vitro BEAS-2B cell culture
Clinical isolates of S. pneumoniae (strain 132) and NTHi (strain RHH3) were obtained from the respiratory tracts of patients at the Royal Hobart Hospital, Tasmania. This study was approved by the Tasmania Health & Medical Human Research Ethics Committee (approval number EC00337). The microbial isolates were collected as part of routine hospital laboratory diagnosis and no research participants or patients were recruited for this purpose. NCTC-4560 (ATCC 19418), an NTHi laboratory reference strain, was also used. For each bacterial strain, a multiplicity of infection (MOI) of 10–50 bacterial cells per BEAS-2B cell was used. Fluorescein isothiocyanate (FITC; 1 mg/mL, Sigma-Aldrich, St Louis, MO, USA) was used to tag the bacterial strains as previously described.22,23 We found no significant change in bacterial colony counts following FITC treatment for the experiment duration. A primary immortalized cell line of bronchial epithelial cells, BEAS-2B (Catalogue no 95102433, Sigma-Aldrich), was maintained at 37°C, 5% CO2 in bronchial epithelial cell growth medium (Lonza, Basel, Switzerland) supplemented with the BulletKit provided by the manufacturer (Lonza).13,18,22 BEAS-2B bronchial epithelial cells were selected for use in this work based on their previous application in the study of PAFr expression in response to cigarette smoke exposure.18 Cells were seeded in collagen-coated T75 flasks, sterile clear-flat bottom 24-well plates (Corning Inc., Corning, NY, USA), and sterile Millicell EZ-Slide 8-well glass plates (EMD Millipore, Billerica, MA, USA) at a concentration of 1.6×105 cells/mL (passage number ≤18).
Cigarette smoke extract (CSE) stimulation and bacterial adhesion
In our earlier work in collaboration with Grigg et al,18 exposure to CSE at a concentration of 1.0% (w/v) for 4 hours was chosen as the standard condition for induction of PAFr expression in lung cells, following testing of CSE at a range of concentrations. Bronchial epithelial BEAS-2B cells were exposed to 1% CSE diluted in cell culture medium for 4 hours, at 37°C, 5% CO2. An infection assay was adapted from previous work.18,22,24 Briefly, bacterial adhesion assays were performed on S. pneumoniae 132, and NTHi strains NCTC-4560 and RHH3 using sterile Millicell EZ-Slide 8-well glass plates (EMD Millipore). Cell monolayers were first exposed to 1% CSE followed by inoculation of FITC-labeled bacteria in bronchial epithelial cell growth medium without gentamicin and incubated for 1 hour at 37°C, 5% CO2. All bacterial adhesion assays were run as three biological replicates.
Treatment with PAFr antagonists
A 1 mM stock solution of WEB-2086 was prepared in dimethyl sulfoxide (≤1% concentration) and then diluted in a medium to a final concentration of 10 μM, 1 μM, 100 nM, and 10 nM. After CSE treatment, the cells were exposed to PAFr antagonist for 1 hour, followed by challenge with FITC-tagged bacteria.
Immunocytochemistry
After exposure to FITC-tagged bacteria, the cells were washed in phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 20 minutes at room temperature and rinsed before treatment with ice-cold acetone. Cells were blocked/permeabilized with 1% bovine serum albumin (Oxoid Limited, Basingstoke, Hampshire, UK), 1% Triton-X-100 (Sigma-Aldrich) in PBS for 30 minutes, and incubated with a 1:100 dilution of anti-PAFr monoclonal antibody (Cayman Chemical Company, Ann Arbor, MI, USA) in blocking buffer overnight at 4°C in the dark. After thorough rinsing with PBS, an AlexaFluor 594-conjugated goat anti-mouse secondary antibody (Molecular Probes Inc., Eugene, OR, USA), diluted to 1:500 in blocking buffer, was added, which was followed by incubation at room temperature for 1 hour. The cells were rinsed and then stained with 4′,6-diamidino-2-phenylindole (Life Technologies, Carlsbad, CA, USA), diluted 1:5,000 in PBS, and then incubated in the dark at room temperature for 15 minutes. The cells were washed thrice with PBS before slides were mounted with fluorescent-mounting media (Dako, North Sydney, Australia).
Microscopy and image analyses
Micrographs were analyzed using an Olympus BX50 Fluorescence Microscope (Olympus; Tokyo, Japan) with NIS elements software (Nikon; Tokyo, Japan) and CoolSnap Hq2 CCD camera (Photometrics, Tucson, AZ, USA). Five images were taken per well using each immunofluorescence channel to take pictures of 4′,6-diamidino-2-phenylindole (eukaryotic cell nuclei; 30 ms exposure, 405 nm), PAFr (PAFr expression; 1–3 second exposure, 594 nm), and FITC (bacteria; 1–3 second exposure, 488 nm) signals under ×400 magnification. NIH elements imaging software was used to perform eukaryotic and bacterial cell counts, while image merging was completed using Adobe Photoshop CS6 software (Adobe Systems, San Jose, CA, USA). Cell tracing was achieved using Image-Pro Plus 7.0 (Media Cybernetics, Inc., Rockville, MD, USA).
BEAS-2B cell viability assay
Cells were passaged in sterile clear-flat 96-well plates (Sigma-Aldrich, CLS3340) with PAFr antagonist diluted to concentrations used within bacterial adhesion assays and added as triplicates to incubate for 1 hour. Alamar Blue (Life Technologies) was then added to each well at a final concentration of 10% (w/v) before hourly absorbance readings (585 nm) were taken up to 8 hours and finally, at 24 hours using a Spectromax Spectrophotometer Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). A positive control of 100% (w/v) Alamar Blue reduced by autoclaving was used in experiments. Cell viability data are representative of the three biological replicates and expressed as 50% inhibition (IC50) values as per the manufacturer’s guidelines (Life Technologies).
Quantitative real-time polymerase chain reaction
Cells were seeded into sterile clear-flat bottom 24-well plates (Corning Inc) before stimulation with CSE (1%, 4 hours). RNA was prepared using the Reliaprep™ Mini RNA cell Miniprep system (Promega, Sydney, Australia) with total RNA quantified by Nanodrop 1000 (Thermo Fisher Scientific, Waltham, MA, USA). Complementary DNA was then collected using the Promega complementary DNA synthesis kit. The level of PAFr transcript was determined by quantitative polymerase chain reaction using the Corbett Rotor-Gene 6000 system (Qiagen, Hilden, Germany). Thermocycling controls were run as previously described.25 The relative change of PAFr mRNA expression was normalized to three reference genes (18S rRNA, β-actin, and β2-microglobulin) using the primers and recommended conditions provided by the manufacturer (Qiagen, Catalogue # PPH05820C). Data were derived from two independent experiments performed in duplicate.
In silico analyses of PAFr antagonist compound docking
The software package, MODELLER (Andej Sali, San Francisco, CA, USA),26 was used to create a PAFr protein structure based on the known X-ray crystal structures of other GPCRs, namely, neurotensin receptor type 1 (pdb 4buo, 2.75 Å resolution), bovine rhodopsin (pdb 1u19, 2.2 Å resolution), and an engineered nociception/orphanin FQ receptor (pdb 4ea3, 3.0 Å resolution). A second model was obtained using the Iterative Threading ASSEmbly Refinement web server and a database of structural models including all 1,026 GPCRs in the human genome,27 which identified C–C chemokine receptor 5 (CCR5; pdb 4mbs, 2.7 Å resolution) as a template with significant similarity to PAFr. Ligand docking was performed using AutoDock Vina software (Molecular Graphics Laboratory, La Jolla, CA, USA).28 Atomic coordinate files for WEB-2086 and PAF in Protein Data Bank format were generated using Open Babel29 and input files for docking were prepared using AutoDockTools.30 Docking results were visualized and molecular graphics generated using PYMOL (Schrödinger, New York, NY, USA).
Statistical analysis
Data were expressed as mean ± standard error using the Microsoft Excel Statistics package (Microsoft Corporation, Redmond, WA, USA). Comparisons between groups using one-way analysis of variance and unpaired two-tailed t-tests, and regression analysis, were completed with GraphPad Prism (GraphPad Software Inc., La Jolla, CA, USA). Data were considered statistically significant where P<0.05.
Results
CSE treatment upregulates PAFr expression in bronchial epithelial cells
Epithelial cells treated with 1% CSE exhibited significantly higher PAFr expression per cell compared to untreated cells as measured by immunofluorescence (P<0.0001) (Figure 1A–C). PAFr mRNA expression (relative to three housekeeping genes) was 2.93±0.18 (n=4) in BEAS-2B cells exposed to CSE, and 2.28±0.33 (n=4) in control cells (Figure 1D).
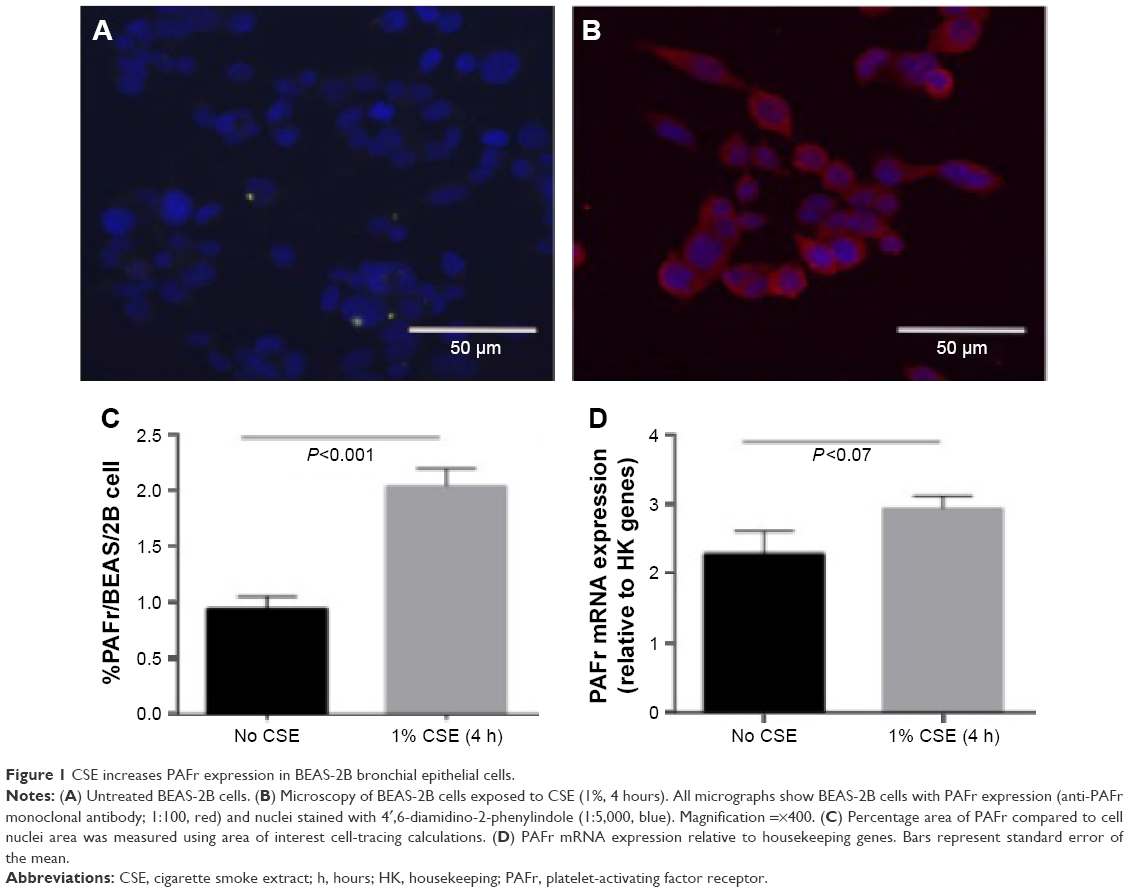 | Figure 1 CSE increases PAFr expression in BEAS-2B bronchial epithelial cells. |
CSE significantly increases NTHi and S. pneumoniae adherence to BEAS-2B cells
Exposure of BEAS-2B cells to 1% CSE for 4 hours significantly enhanced epithelial cell binding of S. pneumoniae (Figure 2A–C) and both NTHi strains (Figure 2D and E). Regression analyses were performed on levels of epithelial PAFr expression against bacterial adhesion. This demonstrated that there was a linear relationship between an increase in PAFr protein levels and elevated levels of adhesion to CSE-pretreated lung cells for all three bacterial strains (r≥0.9; P<0.0001) (Figure 3A–C).
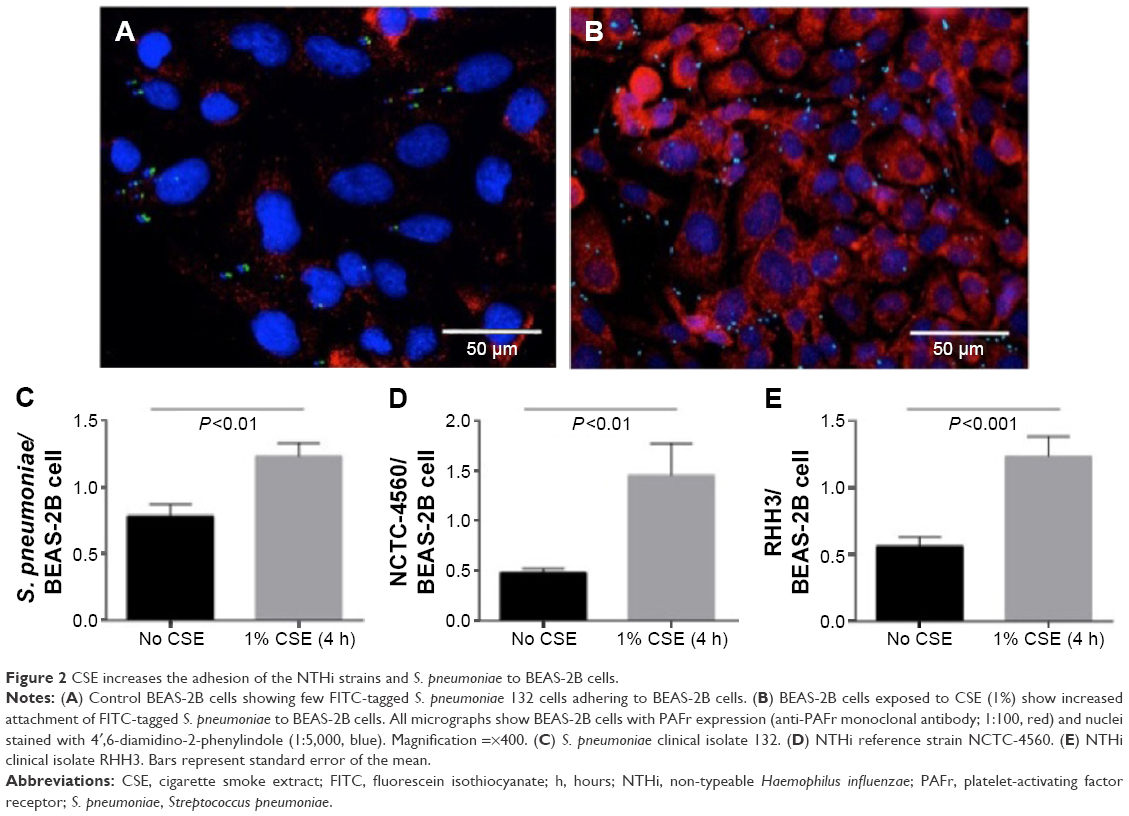 | Figure 2 CSE increases the adhesion of the NTHi strains and S. pneumoniae to BEAS-2B cells. |
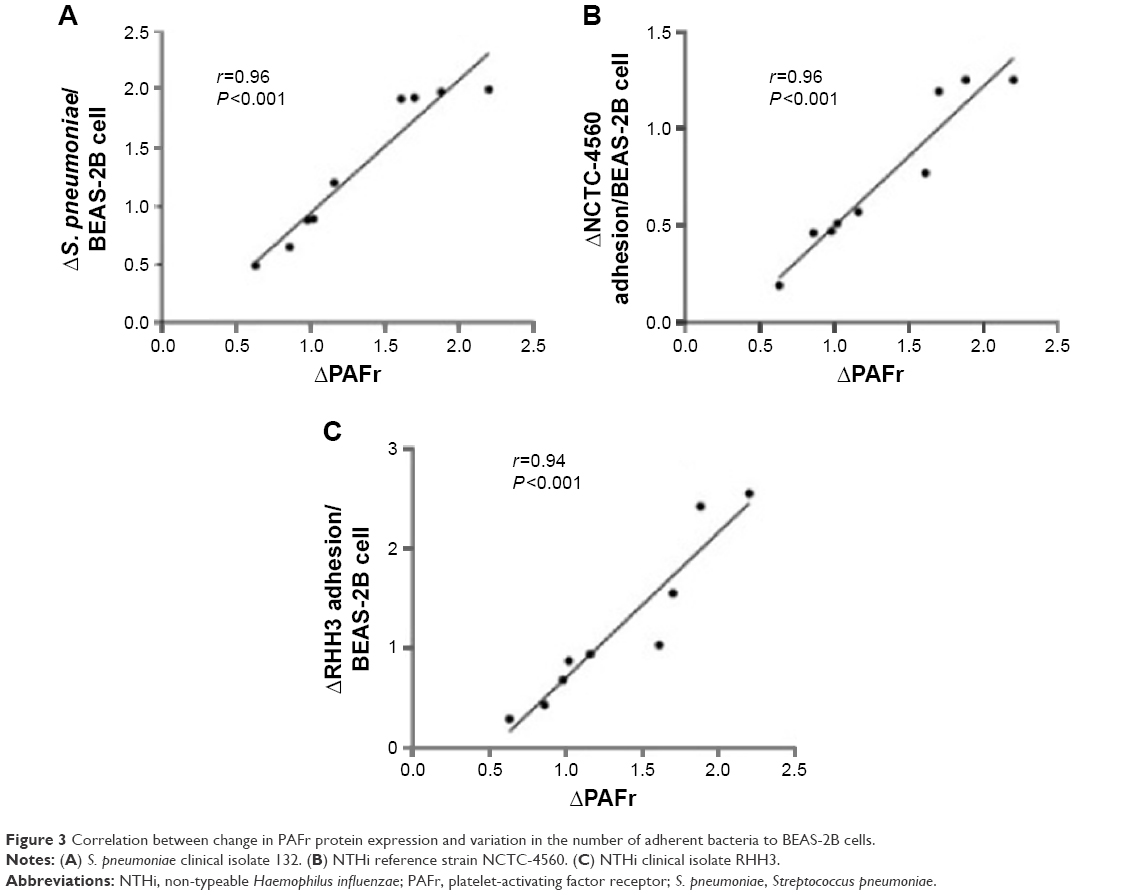 | Figure 3 Correlation between change in PAFr protein expression and variation in the number of adherent bacteria to BEAS-2B cells. |
WEB-2086 reduces the adhesion of S. pneumoniae and NTHi to CSE-treated bronchial epithelial cells
WEB-2086 decreased S. pneumoniae adhesion to CSE-treated BEAS-2B cells back to control levels at a concentration of 100 nM (P=0.0058) (Figure 4A–C). WEB-2086 at 10 μM, but not at lower concentrations, also reduced adhesion of the reference strain of NTHi to CSE-treated cells to control levels (P<0.0001) (Figure 4D). Adhesion of the clinical strain of NTHi to CSE-treated BEAS-2B cells was reduced to control levels by WEB-2086 at a concentration of 1 μM (Figure 4E). WEB-2086 was nontoxic to epithelial cells at the highest concentration tested, that is, IC50 >10 μM.
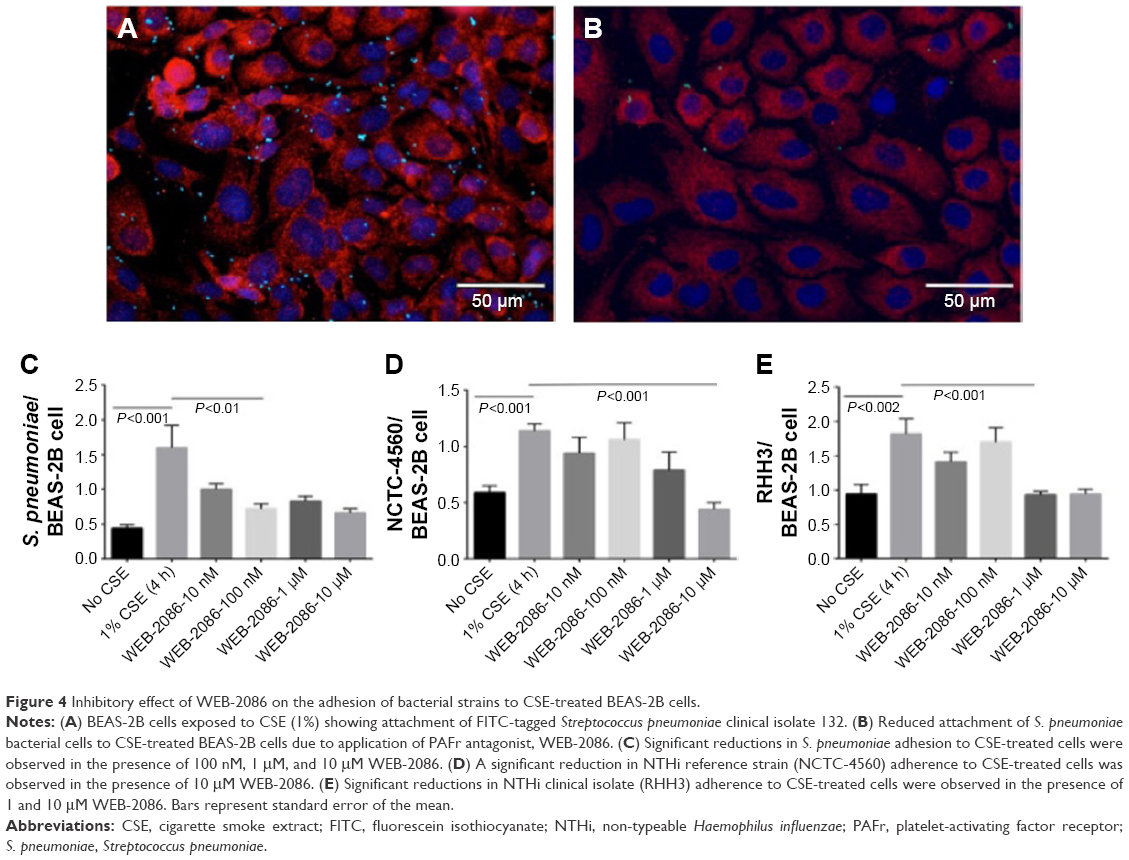 | Figure 4 Inhibitory effect of WEB-2086 on the adhesion of bacterial strains to CSE-treated BEAS-2B cells. |
Docking WEB-2086 to a molecular model of PAFr
Initial models of PAFr produced by MODELLER and Iterative Threading ASSEmbly Refinement were very similar (r.m.s. deviation of 3 Å over 295 Cα atom pairs), and predicted that the same set of amino acids would contribute to the ligand binding site. A final consensus model of PAFr was generated in MODELLER using neurotensin receptor type 1 (NTR1) and C-C chemokine receptor 5 (CCR5) as template structures (Figure 5A), taking advantage of regions of local high sequence/structure similarity to each template. The natural PAF ligand and WEB-2086 were docked to the model using the AutoDock Vina package. In a search of the whole molecular surface, both PAF and WEB-2086 consistently docked into the same deep cavity on the predicted extracellular face of the PAFr model (Figure 5A). The cavity is substantially larger than the WEB-2086 compound (Figure 5B) and a fine-grained docking search of this pocket identified a number of potential docking poses.
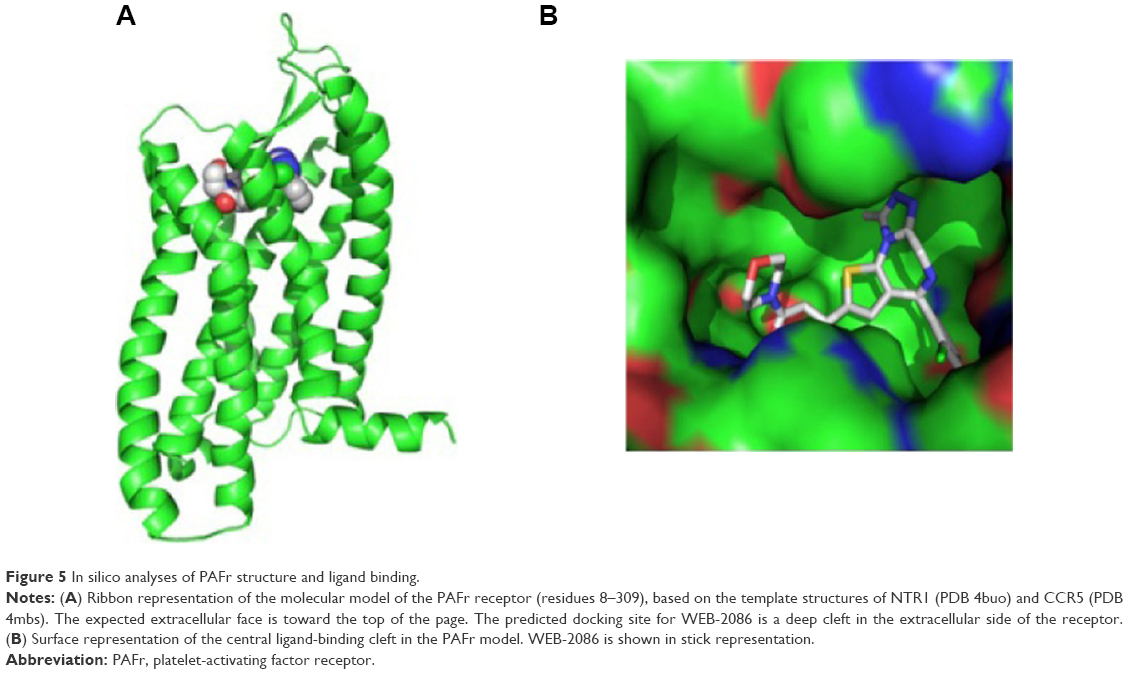 | Figure 5 In silico analyses of PAFr structure and ligand binding. |
Discussion
In this study, we found that in vitro exposure to CSE increases PAFr expression and adhesion of NTHi and S. pneumoniae to bronchial epithelial cells (Figures 1 and 2). Regression analysis identified a correlation between PAFr expression and levels of bacterial adherence. As PAFr expression increased on the surface of the epithelial cells, the number of NTHi and S. pneumoniae cells binding per BEAS-2B cell increased linearly (Figure 3).
Addition of WEB-2086, a PAFr antagonist, reduced the adherence of both respiratory bacterial pathogens down to control levels (Figure 4). Interestingly, the adherence of the S. pneumoniae clinical isolate was inhibited at a lower concentration of PAF antagonist compared to the NTHi strains tested. There were no consistent differences in the slope of the relationship between PAFr receptor upregulation and bacterial attachment for the different strains tested to suggest an interspecies difference in PAFr-dependent bacterial adhesion. A larger number of isolates of both species would need to be investigated to ascertain any species- or strain-specific susceptibility to WEB-2086 but it should be noted that varying levels of adhesion to airway epithelial cells have been observed across different clinical isolates of NTHi.31
This is the first study reporting the efficacy of WEB-2086 in specifically reducing NTHi adherence to CSE-pretreated lung cells. This is also true for pneumococci, although a similar inhibitory effect was observed for WEB-2086 in an epithelial cell culture model of pneumococcal infection, in which PAFr was upregulated by particulate air pollution.24 We have previously shown that airway biopsies taken directly from normal smokers showed significant upregulation of PAFr immunostaining on the surface of their airway epithelium, while normal nonsmokers lacked detectable PAFr expression on their airway epithelial cells.16 Subsequently, we also reported markedly higher PAFr expression in the large and small airway epithelium, and lung parenchyma of COPD patients.17 PAFr is an established receptor for the bacterial wall adhesin ChoP, which is presented by both NTHi and S. pneumoniae.14,32 Variation in ChoP expression leads to NTHi variants with modified functions, including altered NTHi adherence and invasion, and reduced resistance to pulmonary clearance.33–35 ChoP-negative NTHi strains exhibit significantly lower adhesion and invasion of bronchial epithelial cells, suggesting that the ChoP–PAFr interaction is integral for bronchial epithelium colonization by H. influenzae.36–39 Hence, the available data suggest that exposure to cigarette smoke may promote infection of the lung epithelium of COPD patients by the main bacterial drivers of AE of COPD, NTHi, and S. pneumoniae, via upregulated PAFr, though changes in ChoP expression may also play a part.
The potential clinical role of PAFr expression in facilitating pneumococcal disease has been examined using in vivo animal models. PAFr-deficient mice exhibit significantly reduced susceptibility to pneumococcal pneumonia and prolonged survival.40,41 Further evidence of the role of PAFr in mediating bacterial infection of the respiratory tract comes from the use of different chemical antagonists of PAFr, whereby their application has been shown to reduce S. pneumoniae adherence to lung epithelium.13,24,42 Therefore, PAFr represents an innovative anti-infective drug target that could be utilized to prevent common bacteria-mediated respiratory tract infections.
In this work, we tested a PAFr antagonist for its ability to inhibit the binding of NTHi and S. pneumoniae to CSE-pretreated airway epithelial cells. WEB-2086 was effective in significantly reducing the adhesion of both respiratory pathogens to CSE-exposed bronchial epithelial cells with demonstrated PAFr upregulation. Interestingly, the bacterial adhesion seen in the presence of WEB-2086 was comparable to control levels observed for the cultured epithelial cells for which PAFr expression had not been induced through CSE exposure. Furthermore, the level of bacterial adhesion was found to be proportionate to the level of PAFr expression on lung epithelial cells. In terms of drug tolerability, PAF antagonists, including WEB-2086, have been shown to be safe in both animals and humans, even at high doses over several months (49–57). Similarly, we did not find WEB-2086 to be toxic to BEAS-2B cells.
There are a number of limitations in this study. We used a commercial airway epithelial cell line, and further work is now needed on the anti-infective effects of WEB-2086 using primary human cells, especially from smokers. Second, the ligand-docking studies are reliant on an in silico model of PAFr, which is derived from the tertiary structure of related GPCRs. X-ray crystallographic work is required to confirm the specific PAFr–WEB-2086 interactions.
In summary, we investigated the antiadhesion activity of WEB-2086 toward NTHi and S. pneumoniae, two major respiratory pathogens, on airway epithelium primed by CSE to upregulate PAFr expression. We demonstrated an association between PAFr upregulation and bacterial adhesion, the latter of which was reduced to control levels in the presence of the PAFr antagonist WEB-2086. Understanding the events leading to colonization of the lower respiratory tract of COPD patients with NTHi and pneumococcus, and to subsequent increase in local and systemic inflammation, may lead to translation into new management strategies for COPD. Given the apparent tolerance of WEB-2086 in human subjects and its ability to block the adhesion of S. pneumoniae and NTHi to respiratory epithelial cells, we are now in a position to undertake exploratory proof-of-concept studies of PAF antagonists in COPD clinical studies to ascertain if they positively affect day-to-day symptoms as well as AE. We have also begun to explore the tertiary structure of PAFr to design new chemical derivatives of WEB-2086 with enhanced binding affinity for PAFr. This opens up the possibility of a novel nonantibiotic, anti-infective drug for therapeutic interventions in COPD patients, who are prone to frequent bacterial exacerbations, and in smokers, more generally, who are vulnerable to episodes of pneumonia.
Disclosure
EHW and RFO are joint senior authors. The authors report no conflicts of interest in this work.
References
WHO. The Top 10 Causes of Death. Geneva: World Health Organization; 2014. | ||
Lokke A, Lange P, Scharling H, Fabricius P, Vestbo J. Developing COPD: a 25 year follow up study of the general population. Thorax. 2006;61(11):935–939. | ||
Lundback B, Lindberg A, Lindstrom M, et al. Not 15 but 50% of smokers develop COPD? – Report from the obstructive lung disease in northern Sweden studies. Respir Med. 2003;97(2):115–122. | ||
Taylor JD. COPD and the response of the lung to tobacco smoke exposure. Pulm Pharmacol Ther. 2010;23(5):376–383. | ||
Nuorti JP, Butler JC, Farley MM, et al. Cigarette smoking and invasive pneumococcal disease. Active Bacterial Core Surveillance Team. N Engl J Med. 2000;342(10):681–689. | ||
Voss M, Wonnenberg B, Honecker A, et al. Cigarette smoke-promoted acquisition of bacterial pathogens in the upper respiratory tract leads to enhanced inflammation in mice. Respir Res. 2015;16(1):41. | ||
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359(22):2355–2365. | ||
Cabello H, Torres A, Celis R, et al. Bacterial colonization of distal airways in healthy subjects and chronic lung disease: a bronchoscopic study. Eur Respir J. 1997;10(5):1137–1144. | ||
Beasley V, Joshi PV, Singanayagam A, Molyneaux PL, Johnston SL, Mallia P. Lung microbiology and exacerbations in COPD. Int J Chron Obstruct Pulmon Dis. 2012;7:555–569. | ||
Grigg J. The platelet activating factor receptor: a new anti-infective target in respiratory disease? Thorax. 2012;67(9):840–841. | ||
Shukla S, Sohal S, Reid D, Grigg J, Walters E. Platelet activating factor receptor (PAFr) expression is increased in airways of COPD patients but is not attenuated by inhaled corticosteroids. Paper presented at: European Respiratory Society Annual Congress; 2013; Barcelona, Spain. | ||
Shukla SD. Platelet-activating factor receptor and signal transduction mechanisms. FASEB J. 1992;6(6):2296–2301. | ||
Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. 1995;377(6548):435–438. | ||
Swords WE, Buscher BA, Ver Steeg Ii K, et al. Non-typeable Haemophilus influenzae adhere to and invade human bronchial epithelial cells via an interaction of lipooligosaccharide with the PAF receptor. Mol Microbiol. 2000;37(1):13–27. | ||
Shukla SD, Sohal SS, O’Toole RF, Eapen MS, Walters EH. Platelet activating factor receptor: gateway for bacterial chronic airway infection in chronic obstructive pulmonary disease and potential therapeutic target. Expert Rev Respir Med. 2015;9(4):473–485. | ||
Shukla SD, Sohal SS, Mahmood MQ, Reid D, Muller HK, Walters EH. Airway epithelial platelet-activating factor receptor expression is markedly upregulated in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:853–861. | ||
Shukla SD, Muller HK, Latham R, Sohal SS, Walters EH. Platelet-activating factor receptor (PAFr) is upregulated in small airways and alveoli of smokers and COPD patients. Respirology. 2016;21(3):504–510. | ||
Grigg J, Walters H, Sohal SS, et al. Cigarette smoke and platelet-activating factor receptor dependent adhesion of Streptococcus pneumoniae to lower airway cells. Thorax. 2012;67(10):908–913. | ||
Cellai C, Laurenzana A, Vannucchi AM, et al. Growth inhibition and differentiation of human breast cancer cells by the PAFR antagonist WEB-2086. Br J Cancer. 2006;94(11):1637–1642. | ||
Cellai C, Laurenzana A, Vannucchi AM, Della Malva N, Bianchi L, Paoletti F. Specific PAF antagonist WEB-2086 induces terminal differentiation of murine and human leukemia cells. FASEB J. 2002;16(7):733–735. | ||
Kato M, Imoto K, Miyake H, Oda T, Miyaji S, Nakamura M. Apafant, a potent platelet-activating factor antagonist, blocks eosinophil activation and is effective in the chronic phase of experimental allergic conjunctivitis in guinea pigs. J Pharmacol Sci. 2004;95(4):435–442. | ||
Avadhanula V, Rodriguez CA, Ulett GC, Bakaletz LO, Adderson EE. Nontypeable Haemophilus influenzae adheres to intercellular adhesion molecule 1 (ICAM-1) on respiratory epithelial cells and upregulates ICAM-1 expression. Infect Immun. 2006;74(2):830–838. | ||
Adamou JE, Wizemann TM, Barren P, Langermann S. Adherence of Streptococcus pneumoniae to Human Bronchial Epithelial Cells (BEAS-2B). Infection Immun. 1998;66(2):820–822. | ||
Mushtaq N, Ezzati M, Hall L, et al. Adhesion of Streptococcus pneumoniae to human airway epithelial cells exposed to urban particulate matter. J Allergy Clin Immunol. 2011;127(5):1236–1242.e1232. | ||
Latham R, Zhang B, Tristram S. Identifying Haemophilus haemolyticus and Haemophilus influenzae by SYBR Green real-time PCR. J Microbiol Methods. 2015;112:67–69. | ||
Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. | ||
Zhang J, Yang J, Jang R, Zhang Y. GPCR-I-TASSER: A hybrid approach to G protein-coupled receptor structure modeling and the application to the human genome. Structure. 2015;23(8):1538–1549. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33. | ||
Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. 1999;17(1):57–61. | ||
Marti-Lliteras P, Lopez-Gomez A, Mauro S, et al. Nontypable Haemophilus influenzae displays a prevalent surface structure molecular pattern in clinical isolates. PLoS One. 2011;6(6):e21133. | ||
Swords WE, Ketterer MR, Shao J, Campbell CA, Weiser JN, Apicella MA. Binding of the non-typeable Haemophilus influenzae lipooligosaccharide to the PAF receptor initiates host cell signalling. Cell Microbiol. 2001;3(8):525–536. | ||
Clark SE, Snow J, Li J, Zola TA, Weiser JN. Phosphorylcholine allows for evasion of bactericidal antibody by Haemophilus influenzae. PLoS Pathog. 2012;8(3):e1002521. | ||
Weiser JN, Pan N, McGowan KL, Musher D, Martin A, Richards J. Phosphorylcholine on the lipopolysaccharide of Haemophilus influenzae contributes to persistence in the respiratory tract and sensitivity to serum killing mediated by C-reactive protein. J Exp Med. 1998;187(4):631–640. | ||
Weiser JN, Shchepetov M, Chong ST. Decoration of lipopolysaccharide with phosphorylcholine: a phase-variable characteristic of Haemophilus influenzae. Infect Immun. 1997;65(3):943–950. | ||
McCrea KW, Xie J, Marrs CF, Gilsdorf JR. Prevalence of genetic differences in phosphorylcholine expression between nontypeable Haemophilus influenzae and Haemophilus haemolyticus. BMC Microbiol. 2010;10:286. | ||
Weiser JN, Love JM, Moxon ER. The molecular mechanism of phase variation of H. influenzae lipopolysaccharide. Cell. 1989;59(4):657–665. | ||
Michel V, Yuan Z, Ramsubir S, Bakovic M. Choline transport for phospholipid synthesis. Exp Biol Med (Maywood). 2006;231(5):490–504. | ||
Fan X, Pericone CD, Lysenko E, Goldfine H, Weiser JN. Multiple mechanisms for choline transport and utilization in Haemophilus influenzae. Mol Microbiol. 2003;50(2):537–548. | ||
Rijneveld AW, Weijer S, Florquin S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J Infect Dis. 2004;189(4):711–716. | ||
van der Sluijs KF, van Elden LJ, Nijhuis M, et al. Involvement of the platelet-activating factor receptor in host defense against Streptococcus pneumoniae during postinfluenza pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006;290(1):L194–L199. | ||
Ishizuka S, Yamaya M, Suzuki T, et al. Acid exposure stimulates the adherence of Streptococcus pneumoniae to cultured human airway epithelial cells: effects on platelet-activating factor receptor expression. Am J Respir Cell Mol Biol. 2001;24(4):459–468. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.