Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
An Alpha-1 Antitrypsin Deficiency Screening Study in Patients with Chronic Obstructive Pulmonary Disease, Bronchiectasis, or Asthma in Turkey
Authors Tural Onur S ![]() , Natoli A, Dreger B, Arınç S, Sarıoğlu N
, Natoli A, Dreger B, Arınç S, Sarıoğlu N ![]() , Çörtük M, Karadoğan D
, Çörtük M, Karadoğan D ![]() , Şenyiğit A, Yıldız BP
, Şenyiğit A, Yıldız BP ![]() , Köktürk N
, Köktürk N ![]() , Argun Barıs S, Kodalak Cengiz S
, Argun Barıs S, Kodalak Cengiz S ![]() , Polatli M
, Polatli M ![]()
Received 14 June 2023
Accepted for publication 27 October 2023
Published 28 November 2023 Volume 2023:18 Pages 2785—2794
DOI https://doi.org/10.2147/COPD.S425835
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Seda Tural Onur,1 Antonino Natoli,2 Bettina Dreger,2 Sibel Arınç,3 Nurhan Sarıoğlu,4 Mustafa Çörtük,1 Dilek Karadoğan,5 Abdurrahman Şenyiğit,6 Birsen Pınar Yıldız,1 Nurdan Köktürk,7 Serap Argun Barıs,8 Sümeyye Kodalak Cengiz,7 Mehmet Polatli9
1Department of Pulmonology, Yedikule Chest Diseases and Thoracic Surgery Education and Research Hospital, University of Health Sciences, Istanbul, Türkiye; 2Scientific and Medical Affairs, Scientific Innovation Office, Grifols, Frankfurt, Deutschland; 3Clinic of Chest Diseases, University of Health Sciences Turkey, S.B.Ü. Süreyyapaşa Chest Diseases and Thoracic Surgery Training and Research Hospital, İstanbul, Türkiye; 4Department of Pulmonology, Balıkesir University Faculty of Medicine, Pulmonology Clinic, Balıkesir, Türkiye; 5Department of Chest Diseases, Recep Tayyip Erdoğan University, School of Medicine, Rize, Türkiye; 6Department of Chest Diseases, Dicle University Faculty of Medicine Hospital, Diyarbakır, Türkiye; 7Department of Pulmonary Medicine, Gazi University, School of Medicine, Ankara, Türkiye; 8Department of Pulmonary Diseases, Faculty of Medicine, Kocaeli University, Kocaeli, Türkiye; 9Faculty of Medicine, Aydin Adnan Menderes University, Aydin, Türkiye
Correspondence: Mehmet Polatli, Faculty of Medicine, Aydin Adnan Menderes University, Zafer Mahallesi, Efeler, Aydin, 09100, Türkiye, Email [email protected]
Purpose: Alpha-1 antitrypsin deficiency (AATD) is a rare hereditary condition characterized by decreased serum alpha-1 antitrypsin (AAT) levels. We aim to identify AATD in patients with chronic obstructive pulmonary disease (COPD), bronchiectasis, or asthma and to report the frequency of AAT variants in Turkey.
Patients and Methods: This non-interventional, multicenter, prospective study was conducted between October 2021 and June 2022. Adult patients with COPD, bronchiectasis, asthma, liver symptoms, or family members with AATD were included. Demographic and clinical characteristics, pulmonary diagnosis, respiratory symptoms, and AAT serum levels were assessed. Whole blood samples were collected as dried blood spots, and the most common AATD mutations were simultaneously tested by allele-specific genotyping.
Results: A total of 1088 patients, mainly diagnosed with COPD (92.7%) and shortness of breath (78.7%), were assessed. Fifty-one (5%) were found to have AATD mutations. Fifteen (29.4%) patients had Pi*S or Pi*Z mutations, whereas 36 (70.6%) patients carried rare alleles Pi*M malton (n=18, 35.3% of mutations), Pi*I (n=8, 16%), Pi*P lowell (n=7, 14%), Pi*M heerlen (n=2, 4%), and Pi*S iiyama (n=1, 2%). The most common heterozygous combinations were Pi*M/Z (n=12, 24%), and Pi*M/M malton (n=11, 22%). Ten patients with severe AATD due to two deficiency alleles were identified, two with the Pi*Z/Z genotype, four with the genotype Pi*M malton/M malton, three with Pi*Z/M malton, and one with Pi*Z/M heerlen.
Conclusion: Our results identified AATD mutations as a genetic-based contributor to lung disease in patients with COPD or bronchiectasis and assessed their frequency in a population of Turkish patients.
Keywords: alpha-1 antitrypsin deficiency, genotype, diagnosis, chronic obstructive pulmonary disease
Introduction
Alpha-1 antitrypsin (AAT) is a glycoprotein serine protease inhibitor (coded by the SERPINA1 gene). AAT is mainly synthesized in the liver and released into serum. The main physiological function of AAT is to protect the lung from damage by inhibiting neutrophil elastase but also has anti-inflammatory, immunomodulatory, and anti-infective properties on a wide range of cell types.1,2 The allele coding for the normal AAT protein is designated as M and the homozygous Pi*MM genotype is present in 85–90% of individuals, which leads to the production of serum levels of functional AAT between 90 and 200 mg/dL.3
Alpha-1 antitrypsin deficiency (AATD) is a chronic, autosomal co-dominant hereditary condition characterized by decreased serum AAT levels, which may potentially lead to a loss of pulmonary function, emphysema, and development of liver disease, panniculitis, and vasculitis.4,5 So far, more than 150 mutations of the SERPINA1 gene have been identified, in which 40% of them are responsible for causing AATD.6 The risk for lung disease due to AATD depends on the AAT genotype, the AAT serum levels, and environmental exposures to hazardous agents.4,7 The two most common deficiency mutations are the S and Z alleles, which result in five deficiency genotypes: MS, SS, MZ, SZ, and ZZ.3 The homozygous Pi*ZZ genotype is characterized by a reduction in AAT levels below the protective threshold of 11 µM,8 and predisposes the carriers to develop lung disease (emphysema and premature onset of chronic obstructive pulmonary disease [COPD]) and liver disease (cirrhosis in children and adults, cholestasis, and hepatocarcinoma).9 The remaining deficient AAT genotypes are grouped as rare variants, such as Pi*I and Pi*M malton, and null variants, in which no serum AAT levels are detected.10
Clinical practice guidelines recommend that all symptomatic patients with COPD, emphysema, or asthma with airflow obstruction and relatives of someone with AATD or COPD should undergo specific testing for AATD.4,5,11,12 However, AATD is still a highly underdiagnosed disorder, with less than 10% of patients diagnosed.3 This is often due to the fact that a confirmatory testing needs to be conducted in specialized centers.13,14 Early identification of patients with AATD is recommended because delayed diagnosis worsens clinical status, including COPD-related symptoms.15
Overall, it is estimated that 3.4 million individuals worldwide have one of the deficient allele combinations. When reviewing the epidemiology of the disease, AATD frequency varies markedly among countries worldwide. Specifically, the mean prevalence of Pi*ZZ genotype in Europe is 1 in 35,702, being higher in north-western countries and decreasing gradually from west to east, and in America, the Pi*ZZ prevalence is 1 in 26,002 patients.3 Therefore, there are approximately 74,000 individuals in the European countries and 44,000 in North America with severe AATD of the Pi*ZZ genotype.3 In addition, differences in the risk for developing AATD have been reported depending on the ethnic of the individuals.2 Of note is that mutations rarely described in European patients may be predominantly detected in other countries.16 In some regions of the Mediterranean area, such as southern Italy and central Tunisia, the frequency of rare variant PI*M malton has been described over Pi*Z.16,17
In Turkey, there are limited data on the frequency of AATD and the presence of rare variants.18,19 Considering that prevalence data may differ greatly among countries, regions, and ethnicities, targeted screening programs are needed in countries such as Turkey to identify carriers of AATD variants. Therefore, the aims of the study were to identify AATD as a genetic underlying cause of lung disease in patients with COPD, bronchiectasis, or asthma and to report the frequency of AATD alleles in Turkey.
Materials and Methods
This was a non-interventional, multicenter, prospective study conducted at eight sites in Turkey between October 2021 and June 2022. The study was based on analysis of clinical data obtained under standard clinical practice conditions and enrolled patients with documented COPD or other reasons for AATD testing, such as a relative in whom AATD has been diagnosed.
Patients
Patients were selected on the basis of the following inclusion criteria: male or female adult (≥18) of any ethnic origin with documented respiratory symptoms, ie, COPD, bronchiectasis, or asthma, or another reason for wanting to exclude or confirm AATD (for example, liver symptoms or AATD in family members), and willingness to participate in the study. Patients were excluded if they had been previously tested for AATD. Screening of COPD patients in search of AATD was carried out following the ATS-ERS AATD Guidelines.4,5
The protocol was reviewed and approved by the independent ethics committee from the Aydin Adnan Menderes University Medical Faculty and submitted to the national regulatory health authorities (approval number E-66175679-514.05.04–443436) prior to patient enrollment. All patients signed a written informed consent form before the study was initiated, and all study procedures were compliant with the ICH standards for Good Clinical Practice (GCP). The study was conducted in full conformance with applicable local and national laws and regulations and the Declaration of Helsinki.
Assessments
At screening, demographic and clinical characteristics of patients were recorded (age, sex, ethnicity, and family members with known AATD or COPD), liver disease history (neonatal hepatitis, or a diagnosis with any liver disease), diagnosis of pulmonary disease (COPD, emphysema, bronchiectasis, or asthma), pulmonary function tests (forced expiratory volume in 1 sec % [FEV1], forced vital capacity [FVC], FEV1/FVC), smoking history, respiratory symptoms (chronic cough, sputum production, or shortness of breath), number of COPD exacerbations during the last year, and the number hospitalizations due to exacerbations during the last 2 years. Data from patients were collected on a case report form (CRF).
AAT levels were measured by nephelometry.20 AAT levels should not be measured when patients have an infection since AAT levels would be increased as an acute-phase reaction. Adverse events (AEs) were recorded during the study visit including up to at least 30 minutes after the collection of blood samples. This study was conducted with an in vitro device with no direct contact with the patient except for the blood draw.
Genotyping Test
Whole blood samples were collected as dried blood spots (DBS) by finger stick capillary blood on filter paper for AATD genotyping testing. The diagnostic test also allowed sampling from oral mucosa with a buccal swab. However, that procedure was not approved by health authorities when the study was conducted. Genomic DNA was extracted from whole-blood sample and the allele-specific genotyping was carried out with the validated A1AT Genotyping Test (Progenika, a Grifols company, Derio, Spain). This was a qualitative, PCR, and hybridization-based in vitro diagnostic test (FDA cleared, and CE marked). It relies on allele-specific probes attached to color-coded microspheres, which hybridize specifically to the labelled PCR products. A subsequent fluorescent labelling step allows detection and quantification of the hybridization signal with the Luminex 200TM (Diasorin, Saluggia, Italy) instrument for the simultaneous detection and identification of 14 allelic variants and their associated alleles found in the AAT codifying gene SERPINA1: PI*F, PI*I, PI*S, PI*Z, PI*M procida, PI*M malton, PI*S iiyama, PI*Q0 granite falls, PI*Q0 west, PI*Q0 bellingham, PI*P lowell, PI*Q0 mattawa, PI*Q0 clayton, and PI*M heerlen. The absence of any of the 14 alleles included in the analysis was interpreted, with over 99% of probability, as an M/M genotype.
The AAT allelic variant genotypes and associated allele results, when used in conjunction with clinical findings and other laboratory tests, are intended as an aid in the diagnosis of individuals with AATD.
Statistical Analysis
A sample size of approximately 1000 patients was planned anticipating 10% AATD gene carriers and between three and five cases of severe AATD in that population.3,21 Continuous variables were summarized using the following standard descriptive statistics: number of observations, mean, standard deviation, or median and minimum and maximum ranges, as applicable. Categorical data are described using absolute and relative frequencies. All patients with genotyping analysis results were analyzed. Statistical analysis was descriptive by calculating the different percentages to obtain the frequencies of each AATD genotype. No inferential statistical analysis was conducted.
Results
Study Patients’ Characteristics and Disease Activity
A total of 1090 patients were enrolled during the study period. Two of them (0.18%) did not meet eligibility criteria and were excluded from the analysis. Baseline patient characteristics were available for a total of 1087 patients. The majority were male (85.6%), with a mean age of 61.7 years, and current smokers (32.2%). Patients were diagnosed with COPD (92.7%), bronchiectasis (20.7%) or asthma (19.2%). Some patients had overlapping diagnoses with two or more conditions. The most frequent respiratory symptoms were shortness of breath in 855 (78.7%), sputum production in 683 (68.2%) and chronic cough in 628 (57.8%) patients (Table 1).
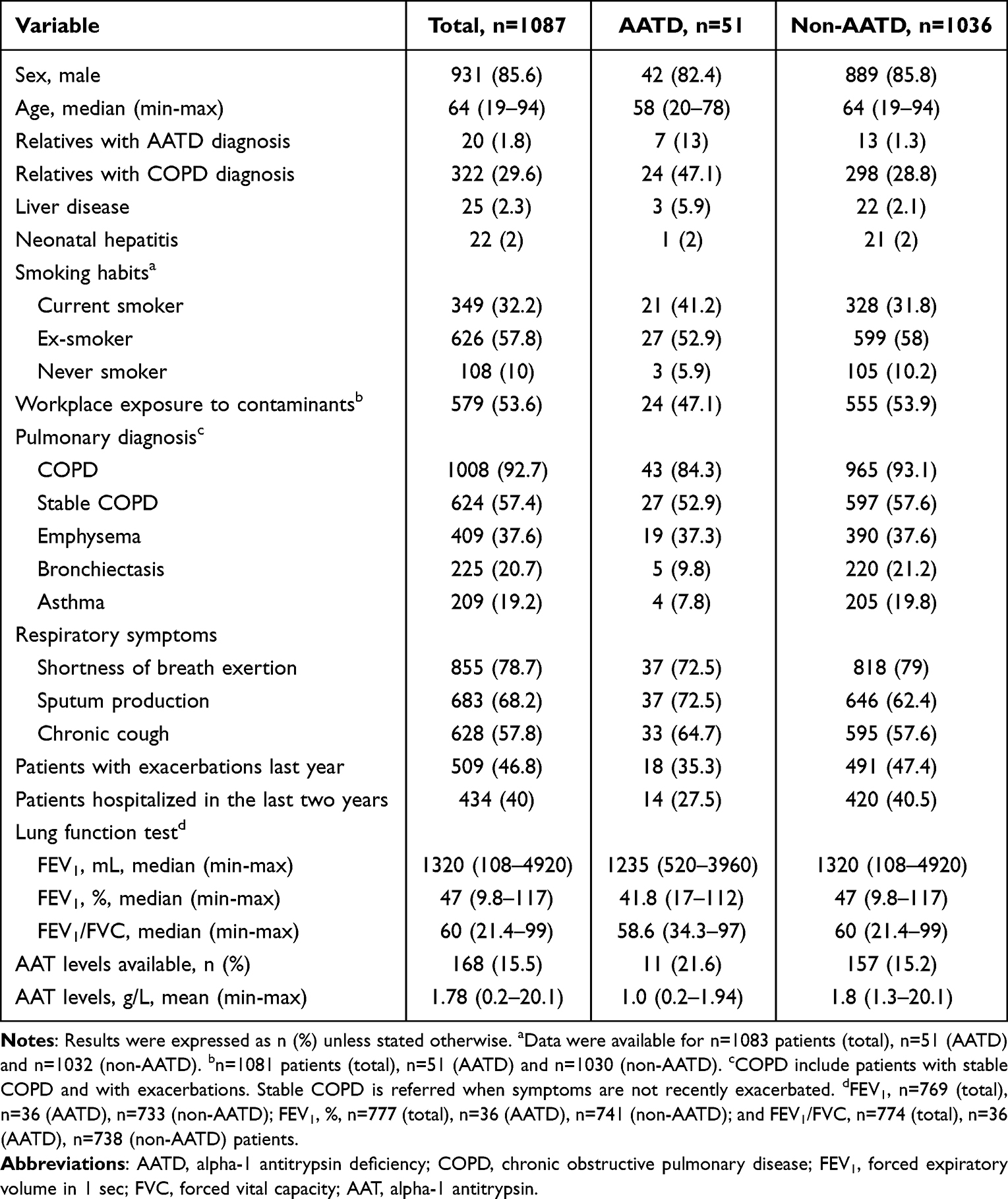 |
Table 1 Demographic and Clinical Data of Assessable Patients Included in the Study. Data Were Available for n=1087 Patients |
When patients were stratified by the presence or absence of AATD mutation, no relevant differences between groups were observed in respirometry, smoking habits, or workplace exposure to dust, fumes, or gases. Likewise, no differences were observed in the percentage of patients diagnosed with COPD or emphysema. Liver disease was slightly more common in the group containing AATD mutation than in the non-AATD group (5.9% vs 2.1%). In the AATD group, 35% of the patients had exacerbations in the last year, compared with the 47% in the non-AATD group, although the mean number of exacerbations was the same (Table 1).
AAT serum levels were available for 168 (15%) of patients, with a mean (range) of 1.78 (0.2–20.1) g/L. As expected, AAT serum levels in AATD patients were lower (1 [0.2–1.94] g/L) than in patients without AATD (1.8 [1.3–20.1] g/L).
No adverse events (AEs) or any other safety signal were reported. This was an in vitro device with no direct contact of the device with the patient and no patient contact except for the blood draw.
AATD Genotyping results
The distribution of mutations is shown in Figure 1. Overall, there were 1037 patients (95%) carrying no mutations and 51 (5%) patients with a AATD mutation of any type. Of the patients with mutations, 15 (29.4%) showed the well-known mutations S or Z, whereas 36 patients (70.6%) carried rare alleles (Pi*M malton, Pi*I, Pi*P lowell, Pi*M heerlen, and Pi*S iiyama). The most frequent combinations reported were Pi*M/Z (n=12, 24%), followed by Pi*M/M malton (n=11, 22%). The percentage of patients carrying two deficiency alleles, was 19.6% (n=10 patients): two of them with Pi*Z/Z genotype, seven had a severe deficiency associated with the M malton allele (Pi*M malton/M malton and Pi*Z/M malton), and one had the genotype Pi*Z/M Heerlen (Figure 1).
 |
Figure 1 Distribution of AATD mutations in the study cohort. Data are expressed as absolute values with percentages in parenthesis: the first percentage is referred to the total number of patients with mutations (n=51 patients); the second percentage is referred to the total number of patients in the study cohort (n=1088 patients). |
Eighteen (35.3%) patients presented with the Pi*Z mutation. Most of them were former or current smokers (n=17) and had COPD (n=16). Moreover, evidence of advanced COPD (post-bronchodilator FEV1 of 60% or lower) was reported in 10 patients, with the following genotypes: Pi*Z/Z (n=2), Pi*Z/M malton (n=2), Pi*Z/M heerlen (n=1), and Pi*M/Z (n=5).
Rare Genotypes
In this study, AATD mutation Pi*M malton was observed in 18 patients (35.3% of the total mutations), achieving the same frequency as the Z allele. Of those patients with one or two M malton alleles, 11 (61%) had the diagnosis COPD, and all of them were former or current smokers. Three patients with genotypes Pi*M malton/M malton (n=2) and Pi*Z/M malton (n=1) had a post-bronchodilator FEV1 below 30%, indicating severe COPD and emphysema. Six patients were diagnosed with the M malton mutation without having major respiratory symptoms, and one patient was diagnosed with the severe genotype Pi*M malton/M malton who suffered from COPD and was a relative of an individual with AATD.
The heterozygous genotypes P*M/I and P*M/P lowell were identified in eight (16%) and seven (14%) patients, respectively. All of them were current or former smokers who suffered from COPD. The Pi*M heerlen mutation was detected in two (4%) patients. One had the combination with the Z allele (Pi*Z/M heerlen) and had severe AATD. The other was heterozygous with one normal allele (Pi*M/M heerlen). Both patients were former smokers and suffered from severe COPD (FEV1 of 36% and 22%, respectively) with AAT serum levels of 0.44 g/L and 1.03 g/L, respectively. The heterozygous genotypes Pi*M/S and Pi*M/S iiyama corresponded to former smokers with advanced COPD (post-bronchodilator FEV1% of 44%) and emphysema.
The geographic distribution of AATD alleles in Turkey is shown in Figure 2. Geographic data were available for 46 out of 51 patients (90.2%). Most of the AATD mutations (n=24, 52%) were documented in the Eastern Black Sea region of Turkey, whereas in the south (Mediterranean region), no AATD mutations were reported. Of note is that many of the patients diagnosed with an M malton allele came from the Black Sea region (Rize, n=8) and Eastern Anatolia (Erzurum, n=2).
 |
Figure 2 Geographic distribution of AATD mutations among the seven geographical regions of Turkey: Black Sea, Marmara, Aegean, Central Anatolia, Eastern Anatolia, Southeastern Anatolia and Mediterranean. Out of n=51 patients with AATD mutations, data about their geographic origin was available for n=46 patients. Map obtained from www.d-maps.com. |
AAT Levels in Patients with AATD Mutation
AAT serum levels were available in 11 (21.1%) patients with AATD mutation and varied according to the mutation. The highest AAT level (1.94 g/L) corresponded to a patient with the Pi*M/I genotype and a FEV1 of 82%, whereas the lowest AAT level (0.21 g/L) was associated with a patient with two deficiency alleles, Pi*Z/M malton and a FEV1 of 60% (Table 2).
 |
Table 2 Alpha-1 Antitrypsin (AAT) Serum Levels (g/L) and Clinical Phenotype Based on Each Genotype in Patients with AATD. AAT Levels Were Available for n=11 (21.6%) Patients with AATD Mutations |
Discussion
This prospective study conducted under standard clinical practice conditions described the frequency of each AATD genotype in a selected sample population of Turkish individuals with pulmonary disease. Specifically, this study was designed to identify patients who were previously diagnosed with COPD, bronchiectasis, or asthma that have a mutation in the SERPINA1 gene, a potential underlying genetic cause of lung disease.
Despite knowing that early diagnosis could help manage patients with AATD, this condition remains widely underdiagnosed.4,11 The present study evidenced that AATD was detected in 5% of patients with COPD, bronchiectasis, and asthma. Interestingly, this percentage was similar to two recent studies, which reported genetic AAT mutations in 7.1% and 3.5% of Turkish patients with COPD.18,19 Similarly, these results were aligned with previous studies that evaluated AATD distribution in patients with other pulmonary diseases,22,23 and reinforced the utility of routine screening for AATD in these patients. The percentage of male patients in our study (85.6%) was unusually high when compared with the COPD and AATD series from other countries, but it was similar to values reported in other Turkish studies (80.6%, 90.5%).18,19 It is possible that cultural reasons in Turkey keep women more hesitant to consult a doctor regarding pulmonary and other diseases.24
Family members of patients with AATD are expected to have more AATD mutations, and a targeted approach for this subgroup usually yields a higher AATD detection rate.25 We identified seven AATD patients with a family history of AATD, six of whom had no respiratory symptoms. This result emphasizes the importance of early testing for AATD in adults with first-degree relatives with severe AATD, regardless of respiratory symptoms, as an important test targeting strategy. Altogether, more extensive screening for AATD could prevent certain clinical consequences by allowing patients to receive early therapeutic intervention or at least smoking prevention or cessation counseling.
The prevalence of AATD mutations in European countries has been extensively evaluated, but there is limited evidence in Turkey. In Finland, the allele frequencies were 19.7 per 1000 habitants for Pi*Z and 10.2 cases per 1000 habitants for Pi*S.26 Similarly, Poland reported a frequency of 17.5 per 1000 for the Pi*S allele and 10.5 per 1000 habitants for the Pi*Z allele.27 The highest frequency of the S allele was observed in the Iberian Peninsula (100–200 cases per 1000 habitants).28
In the present study, we observed a considerably lower frequency for the Pi*S genotype (0.09%) compared with these studies. These differences between geographic areas are to be expected since the distribution of Pi*Z and Pi*S alleles is different among countries and even within different regions of the same country.28 Interestingly, the proportions of the Pi*Z and Pi*S mutations in our Turkish population (35.3% and 2%, respectively) were consistent with the percentages obtained in a feasibility study of the A1AT genotyping test that evaluated the percentage of these mutations in Turkey (36.4% and 5%, respectively).29
The use of AATD genotyping testing is also useful to analyze the most prevalent rare variants in each region. In Turkey, we identified a total of 36 patients who carried rare alleles (Pi*M malton, Pi*I, Pi*P lowell, Pi*M heerlen, and Pi*S iiyama). This high frequency of rare alleles has been previously detected in other countries, evidencing that the so-called rare AATD alleles may not be as rare as expected.10 In Italian patients with AATD, the prevalence of rare AAT genotypes was 11%.17 Similarly, in Switzerland, rare AAT alleles represented 7%,30 and in Japan, the most common deficient variant is the PI*S iiyama, a rare variant present in most patients with AATD.31
Regarding the worldwide geographic distribution of AATD, the highest frequency of Pi*ZZ is located in the Atlantic region of Europe. Then, it decreases gradually from west to east, and in the most remote regions of the south of the continent, until it almost disappears in Asia.28,32 Here, we observed a similar geographic distribution in the frequency of AATD variants since a higher percentage of mutations was observed in the north and decreasing gradually to the south of Turkey.
This study revealed the high frequency of rare AAT variants in Turkey, especially the Pi*M malton, being the most frequent mutation detected. Thirty-five percent of all AATD mutations were associated with the Pi*M malton rare allele, which has the same frequency as the most prevalent mutation (Z allele) in other regions. This finding is in agreement with previous research in which the rare variant Pi*M malton prevails over Z and S alleles in particular regions of the Mediterranean area, such as southern Italy,17 central Tunisia, and North Africa.16,33 Human migrations, commercial purposes, and ethnic relationships between these countries have been proposed to be responsible for spreading rare AATD variants in these regions.16,19,33 The Pi*M malton mutations and other rare AATD variants seem to be widespread in regions in which the frequency of Pi*ZZ is lower.16,17 In the current study, the geographic distribution of patients carrying the rare AATD variant Pi*M malton were mainly located in the Black Sea region in northern Turkey. More importantly, individuals with the Pi*M malton may develop pulmonary emphysema and polymeric intrahepatic inclusions like in AATD patients with Pi*ZZ genotype.10 Indeed, it has been shown that rare AATD mutations could be identified in up to 17% of clinical cases.34 In the present study, most of the reported Pi*M malton cases presented with pulmonary emphysema. Since rare AATD variants remain unexplored in many regions worldwide, targeted screening programs could be recommended even in countries in which a high prevalence of the disease has been reported.
One of the potential benefits of conducting extensive AATD genetic testing is to consider providing early lifestyle interventions, such as smoking prevention, and to prevent delayed introduction of augmentation therapy to preserve lung parenchyma in patients with emphysema. The S iiyama allele is another extremely rare variant mainly identified among Japanese patients that has very rarely been found outside of Japan.31 It was found in the study cohort in a heterozygous Pi*M/S iiyama variant patient who was a former smoker with advanced COPD. Altogether, more than 150 mutations of the SERPINA-1 gene have been described in the literature,34 and new mutations are currently being identified.35 Evidence is emerging that rare AATD mutations may play an important role in Turkey. For example, Pi*M malton was found to have the same frequency as Pi*Z and lead to comparable clinical symptoms.
AATD is characterized by a diverse clinical expression and prognosis. Augmentation therapy with AAT is indicated for patients with severe AATD, in which serum AAT levels are below the protective threshold of 11 µM (or 0.5 g/L),4 and who have evidence of advanced lung disease. Early identification of those patients with extremely low AAT levels is paramount since they are more prone to developing COPD and emphysema.8,36 In our analysis, only three patients were documented to have AAT serum levels below the protective threshold: 0.33 g/L (Pi*ZZ genotype), 0.21 g/L (Pi*Z/M malton) and 0.44 g/L (Pi*Z/M heerlen). AAT serum levels were not routinely available in the study centers. It would be important to better establish AAT serum-level testing in Turkey for the future.
One of the limitations of this study is that the real AATD prevalence was overestimated since this was a highly selected population in which patients with family members known to have AATD disease were included. This could have increased the probability of identifying a patient with AATD. AATD prevalence in a whole-population-based study is usually lower compared to a study conducted in a targeted population.37 Additional limitations are that AAT serum levels were only available for a small number of patients because it was not routinely collected and that female patients were underrepresented. The inclusion of these patients would have strengthened the generalizability of the study results.
Conclusion
In conclusion, our results confirm AATD as a genetic underlying cause of lung disease and determine the frequencies of different AATD alleles in a selected population of Turkish individuals.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
Eugenio Rosado, PhD, and Jordi Bozzo, PhD CMPP (Grifols), are acknowledged for medical writing and editorial support in the preparation of this manuscript. The authors wish to thank all the patients who contributed to this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas and took part in drafting, revising, or critically reviewing the article. MP: conceptualization, study design, methodology, resources, data acquisition, data curation, investigation, writing – review and editing. STO, SA, NS, MÇ, DK, AŞ, BPY, NK, SAB, and SKC: study design, methodology, resources, data acquisition, data curation, validation, investigation, writing – review and editing. AN and BD: conceptualization, formal analysis, project administration, supervision, validation, visualization, writing – review and editing. All authors critically revised, edited, agreed, and approved the final version of the article before submission, and during the revision, of the manuscript. Authors agreed in the journal to which the article is submitted, take responsibility, and are accountable for the contents of the article.
Funding
This study was funded by Grifols, manufacturer of A1AT Genotyping Test and plasma-derived alpha-1 antitrypsin medicinal products.
Disclosure
AN and BD are full-time employees of Grifols. DK reports honoraria paid to her institution, as speaker, from AstraZeneca, Abdi İbrahim, Novartis and Grifols, outside the submitted work. The remaining authors have no conflicts of interest to declare for this work.
References
1. de Serres F, Blanco I. Role of alpha-1 antitrypsin in human health and disease. J Intern Med. 2014;276(4):311–335. doi:10.1111/joim.12239
2. de Serres FJ, Blanco I, Fernández-Bustillo E. Ethnic differences in alpha-1 antitrypsin deficiency in the United States of America. Ther Adv Respir Dis. 2010;4(2):63–70. doi:10.1177/1753465810365158
3. Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017;12:561–569. doi:10.2147/copd.S125389
4. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J. 2017;50(5). doi:10.1183/13993003.00610-2017
5. Stoller JK, American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. doi:10.1164/rccm.168.7.818
6. Silva D, Oliveira MJ, Guimarães M, Lima R, Gomes S, Seixas S. Alpha-1-antitrypsin (SERPINA1) mutation spectrum: three novel variants and haplotype characterization of rare deficiency alleles identified in Portugal. Respir Med. 2016;116:8–18. doi:10.1016/j.rmed.2016.05.002
7. Mayer AS, Stoller JK, Bucher Bartelson B, James Ruttenber A, Sandhaus RA, Newman LS. Occupational exposure risks in individuals with PI*Z alpha(1)-antitrypsin deficiency. Am J Respir Crit Care Med. 2000;162(2 Pt 1):553–558. doi:10.1164/ajrccm.162.2.9907117
8. Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax. 2012;67(8):669–674. doi:10.1136/thoraxjnl-2011-201321
9. Patel D, Teckman JH. Alpha-1-Antitrypsin Deficiency Liver Disease. Clin Liver Dis. 2018;22(4):643–655. doi:10.1016/j.cld.2018.06.010
10. Rodriguez-Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare alpha-1-antitrypsin variants: are they really so rare? Ther Adv Respir Dis. 2012;6(2):79–85. doi:10.1177/1753465811434320
11. Sandhaus RA, Turino G, Brantly ML, et al. The Diagnosis and Management of Alpha-1 Antitrypsin Deficiency in the Adult. Chronic Obstr Pulm Dis. 2016;3(3):668–682. doi:10.15326/jcopdf.3.3.2015.0182
12. Casas F, Blanco I, Martínez MT, et al. Indications for active case searches and intravenous alpha-1 antitrypsin treatment for patients with alpha-1 antitrypsin deficiency chronic pulmonary obstructive disease: an update. Arch Bronconeumol. 2015;51(4):185–192. doi:10.1016/j.arbres.2014.05.008
13. Miravitlles M, Nuñez A, Torres-Durán M, et al. The Importance of Reference Centers and Registries for Rare Diseases: the Example of Alpha-1 Antitrypsin Deficiency. Copd. 2020;17(4):346–354. doi:10.1080/15412555.2020.1795824
14. Gurevich S, Daya A, Da Silva C, Girard C, Rahaghi F. Improving Screening for Alpha-1 Antitrypsin Deficiency with Direct Testing in the Pulmonary Function Testing Laboratory. Chronic Obstr Pulm Dis. 2021;8(2):190–197. doi:10.15326/jcopdf.2020.0179
15. Tejwani V, Nowacki AS, Fye E, Sanders C, Stoller JK. The Impact of Delayed Diagnosis of Alpha-1 Antitrypsin Deficiency: the Association Between Diagnostic Delay and Worsened Clinical Status. Respir Care. 2019;64(8):915–922. doi:10.4187/respcare.06555
16. Denden S, Zorzetto M, Amri F, et al. Screening for Alpha 1 antitrypsin deficiency in Tunisian subjects with obstructive lung disease: a feasibility report. Orphanet J Rare Dis. 2009;4:12. doi:10.1186/1750-1172-4-12
17. Ferrarotti I, Baccheschi J, Zorzetto M, et al. Prevalence and phenotype of subjects carrying rare variants in the Italian registry for alpha1-antitrypsin deficiency. J Med Genet. 2005;42(3):282. doi:10.1136/jmg.2004.023903
18. Çörtük M, Demirkol B, Arslan MA, et al. Frequency of alpha-1 antitrypsin deficiency and unexpected results in COPD patients in Turkey; rare variants are common. Turk J Med Sci. 2022;52(5):1478–1485. doi:10.55730/1300-0144.5486
19. Önür ST. Initial alpha-1 antitrypsin screening in Turkish patients with chronic obstructive pulmonary disease. Turk J Med Sci. 2023;53(4):1012–1018.
20. Ferrarotti I, Scabini R, Campo I, et al. Laboratory diagnosis of alpha-1-antitrypsin deficiency. Transl Res. 2007;150(5):267–274. doi:10.1016/j.trsl.2007.08.001
21. Greulich T, Rodríguez-Frias F, Belmonte I, Klemmer A, Vogelmeier CF, Miravitlles M. Real world evaluation of a novel lateral flow assay (AlphaKit® QuickScreen) for the detection of alpha-1-antitrypsin deficiency. Respir Res. 2018;19(1):151. doi:10.1186/s12931-018-0826-8
22. Carreto L, Morrison M, Donovan J, et al. Utility of routine screening for alpha-1 antitrypsin deficiency in patients with bronchiectasis. Thorax. 2020;75(7):592–593. doi:10.1136/thoraxjnl-2019-214195
23. Veith M, Tüffers J, Peychev E, et al. The Distribution of Alpha-1 Antitrypsin Genotypes Between Patients with COPD/Emphysema, Asthma and Bronchiectasis. Int J Chron Obstruct Pulmon Dis. 2020;15:2827–2836. doi:10.2147/copd.S271810
24. Tackett S, Young JH, Putman S, Wiener C, Deruggiero K, Bayram JD. Barriers to healthcare among Muslim women: a narrative review of the literature. Women’s Studies International Forum. 2018;69:190–194.
25. Brantly M, Campos M, Davis AM, et al. Detection of alpha-1 antitrypsin deficiency: the past, present and future. Orphanet J Rare Dis. 2020;15(1):96. doi:10.1186/s13023-020-01352-5
26. Häggblom J, Kettunen K, Karjalainen J, Heliövaara M, Jousilahti P, Saarelainen S. Prevalence of PI*Z and PI*S alleles of alpha-1-antitrypsin deficiency in Finland. Eur Clin Respir J. 2015;2:28829. doi:10.3402/ecrj.v2.28829
27. Kaczor MP, Sanak M, Libura-Twardowska M, Szczeklik A. The prevalence of alpha(1)-antitrypsin deficiency in a representative population sample from Poland. Respir Med. 2007;101(12):2520–2525. doi:10.1016/j.rmed.2007.06.032
28. Blanco I, de Serres FJ, Fernandez-Bustillo E, Lara B, Miravitlles M. Estimated numbers and prevalence of PI*S and PI*Z alleles of α1-antitrypsin deficiency in European countries. Eur Respir J. 2006;27(1):77–84. doi:10.1183/09031936.06.00062305
29. Lopez-Campos JL, Osaba L, Czischke K, et al. Feasibility of a genotyping system for the diagnosis of alpha1 antitrypsin deficiency: a multinational cross-sectional analysis. Respir Res. 2022;23(1):152. doi:10.1186/s12931-022-02074-x
30. Zorzetto M, Russi E, Senn O, et al. SERPINA1 gene variants in individuals from the general population with reduced α1-antitrypsin concentrations. Clin Chem. 2008;54(8):1331–1338. doi:10.1373/clinchem.2007.102798
31. Seyama K, Nukiwa T, Souma S, Shimizu K, Kira S. Alpha 1-antitrypsin-deficient variant Siiyama (Ser53[TCC] to Phe53[TTC]) is prevalent in Japan. Status of alpha 1-antitrypsin deficiency in Japan. Am J Respir Crit Care Med. 1995;152(6 Pt 1):2119–2126. doi:10.1164/ajrccm.152.6.8520784
32. de Serres FJ, Blanco I, Fernández-Bustillo E. Estimated numbers and prevalence of PI*S and PI*Z deficiency alleles of α1-antitrypsin deficiency in Asia. Eur Respir J. 2006;28(6):1091–1099. doi:10.1183/09031936.00029806
33. Denden S, Lakhdar R, Keskes NB, Hamdaoui MH, Chibani JB, Khelil AH. PCR-based screening for the most prevalent alpha 1 antitrypsin deficiency mutations (PI S, Z, and Mmalton) in COPD patients from Eastern Tunisia. Biochem Genet. 2013;51(9–10):677–685. doi:10.1007/s10528-013-9597-6
34. Seixas S, Marques PI. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl Clin Genet. 2021;14:173–194. doi:10.2147/tacg.S257511
35. Presotto MA, Veith M, Trinkmann F, et al. Clinical characterization of a novel alpha1-antitrypsin null variant: piQ0(Heidelberg). Respir Med Case Rep. 2022;35:101570. doi:10.1016/j.rmcr.2021.101570
36. Gadek JE, Klein HG, Holland PV, Crystal RG. Replacement therapy of alpha 1-antitrypsin deficiency. Reversal of protease-antiprotease imbalance within the alveolar structures of PiZ subjects. J Clin Invest. 1981;68(5):1158–1165. doi:10.1172/jci110360
37. Acquavella J, Vágó E, Sorensen HT, Horváth-Puhó E, Hess GP. Registry-based cohort study of alpha-1 antitrypsin deficiency prevalence, incidence and mortality in Denmark 2000-2018. BMJ Open Respir Res. 2022;9(1). doi:10.1136/bmjresp-2022-001281
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Development and Validation of a Multivariable Prediction Model to Identify Acute Exacerbation of COPD and Its Severity for COPD Management in China (DETECT Study): A Multicenter, Observational, Cross-Sectional Study
Yin Y, Xu J, Cai S, Chen Y, Chen Y, Li M, Zhang Z, Kang J
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2093-2106
Published Date: 5 September 2022
Clinical Diagnostic Value of Serum GABA, NE, ET-1, and VEGF in Chronic Obstructive Pulmonary Disease with Pulmonary Hypertension
Yan J, Duan Y, Cheng M
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:1803-1813
Published Date: 19 August 2023
Integrating Mendelian Randomization and Machine Learning to Identify Hypoxia-Related Diagnostic Biomarkers and Causal Relationship in COPD
Fu W, Liu Y, Li R, Jin H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:3187-3202
Published Date: 12 September 2025