Back to Journals » International Journal of Nanomedicine » Volume 17
An Acid-Sensitive Nanofiber Conjugate Based on a Short Aromatic Peptide for Targeted Delivery of Doxorubicin in Liver Cancer
Authors Liang J, Guo R, Xuan M, Sun Q, Wu W
Received 26 January 2022
Accepted for publication 26 June 2022
Published 5 July 2022 Volume 2022:17 Pages 2961—2973
DOI https://doi.org/10.2147/IJN.S359642
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Ju Liang,1 Runfa Guo,1 Maosong Xuan,1 Qiankun Sun,1 Wenlan Wu2
1School of Chemical Engineer and Pharmacy, Henan University of Science and Technology, Luoyang, People’s Republic of China; 2School of Basic Medical Sciences, Henan University of Science and Technology, Luoyang, People’s Republic of China
Correspondence: Ju Liang, School of Chemical Engineer and Pharmacy, Henan University of Science and Technology, Luoyang, 471023, People’s Republic of China, Email [email protected]
Purpose: This study aimed to construct a DOX conjugate with liver tumor targeting and acid sensitivity based on a short aromatic peptide FFYEE, which could amplify the tumor inhibition efficacy of DOX and alleviate tissue toxicity.
Methods: A novel DOX-peptide conjugate, D-gal-FFYEE-hyd-DOX, was constructed by linking DOX to the side chain of FFYEE with acid-sensitive hydrazone bond and by modifying the C-terminal of peptide with α-D-galactosamine (D-gal) as targeting ligand. The structure of D-gal-FFYEE-hyd-DOX was characterized by mass spectrometry, infrared spectroscopy (IR), and UV-Vis spectroscopy (UV-Vis). The assembly characteristics of pentapeptide FFYEE and D-gal-FFYEE-hyd-DOX were observed by transmission electron microscope (TEM). In vitro drug release, cytotoxicity, endocytosis, in vivo antitumor experiment and histopathology analysis were investigated.
Results: Peptide FFYEE endowed the D-gal-FFYEE-hyd-DOX with self-assembly performance and improved biocompatibility. D-gal-FFYEE-hyd-DOX can self-assemble into nanofibers with a diameter of ∼ 40 nm in neutral aqueous solution and significantly reduced the cytotoxicity of free DOX to L02 cells. In vitro drug release results showed that D-gal-FFYEE-hyd-DOX had acid sensitivity and controlled release characteristics. The cytotoxicity and endocytosis investigations confirmed that D-gal-FFYEE-hyd-DOX enhanced the cellular uptake of DOX and inhibition effect on HepG2 cells. In vivo antitumor experiment indicated that D-gal-FFYEE-hyd-DOX could significantly inhibit the growth of liver tumor in mice and reduce the side effects of DOX.
Conclusion: The conjugate D-gal-FFYEE-hyd-DOX with liver tumor targeting and acid sensitivity has the characteristics of strong tumor inhibition and low toxicity, hinting the great clinical application potential for targeted delivery of DOX in cancer treatment.
Keywords: targeted delivery, doxorubicin, short aromatic peptide, nanofiber, biocompatibility
Introduction
Doxorubicin (DOX) is an anthracycline antitumor drug with superiority and broad spectrum of therapeutic efficacy.1,2 After uptake by cells, DOX exerts its efficacy mainly by noncovalent intercalation with double-stranded DNA, resulting in the suppression of DNA replication in fast-proliferating tumors.3 However, similar to other small-molecule chemotherapeutic agents, nonspecific tissue distribution of DOX commonly causes systemic toxicity, such as cardiotoxicity,4,5 bone marrow suppression, nausea and vomiting, which greatly limits its clinical application.6 In order to improve the antitumor effect of DOX and reduce its systemic toxicity, a variety of specific DOX delivery systems are being studied and developed.7,8
The metabolic curve of tumor cells is quite different from that of normal cells, leading to the slightly acidic pH value (pH 6.5–7.0) of the tumor microenvironment (TME).9 Based on the particular acidic TME, researchers are committed to design ideal drug carriers with acid sensitivity to specifically accumulate antitumor drugs in tumor tissues. Such carriers possess good stability at pH 7.4 and rapid drug-release under acidic TME. Acid-sensitive chemical bonds such as hydrazone are often used to connect carriers and drugs to prepare intelligent drug carriers. After cellular uptake, acid hydrolysis occurs to release the drugs in the endosome or lysosome, achieving site-specific delivery.10,11 Kataoka group designed a acid-sensitive polymer, poly(ethylene glycol)-b-Poly(hydrazone asparagine) with hydrazone bond. The self-assembled micelles from such polymer could effectively deliver proteasome inhibitor, MG132, to tumor in situ and induce drug release through acid triggering.12 Recently, Li research group connected DOX and oleic acid via hydrazone bond to prepare nanostructured lipid carrier (NLC). Such acid-sensitive DOX/NLC nanoparticles could accumulate DOX in the tumor site and reduce systemic toxicity, resulting in effective inhibition of breast cancer.13
Besides, drug delivery with ligand-receptor-mediated targeting based on specific surface receptors of tumor cells could further increase the therapeutic accuracy and therapeutic effect. Typical targeting ligands mainly include antibodies, folic acid, galactose/lactose and some oligopeptides.14–16 The galactosyl receptor, asialoglycoprotein receptor (ASGPR), is abundant on the surface of liver tumor cells. Therefore, the carrier modified with galactose could introduce active targeting capability and convenient internalization through endocytosis of liver tumor cells.17–19
In this article, as shown in Figure 1, a novel DOX conjugate (D-gal-FFYEE-hyd-DOX) was synthesized based on an aromatic pentapeptide we previously prepared,20 modified with D-gal through amidation reaction and connected with DOX via hydrazone bond. Observation of transmission electron microscope (TEM) displayed that D-gal-FFYEE-hyd-DOX could self-assemble into nanofibrous network structure in aqueous solution. The data of drug release under different pH conditions in vitro represented rapid controlled release of DOX in pH 5.5 with the cumulative release of over 90%. The investigations of cytotoxicity on HepG2, L02 and EC109 cells hinted improved antitumor effect and reduced toxic side effect. Moreover, in vivo experiments on mouse liver tumor model showed the specific tumor tissue targeting with ideal therapeutic effect and satisfied biosafety. Therefore, combination of the special properties of D-gal and hydrazone bond could make the D-gal-FFYEE-hyd-DOX and the analogues the promising delivery systems for liver tumor treatment.
 |
Figure 1 The schematic diagram for the process of D-gal-FFYEE-hyd-DOX synthesis, assembly, endocytosis and intracellular drug release. |
Experiments and Methods
Reagents and Materials
Doxorubicin hydrochloride (DOX•HCl, purity >98%) was purchased from Aladdin Reagent Co., Ltd. (Shanghai, China). Fully protected FFYEE (Phe-Phe-Tyr-Asp-Asp) (purity >99%) was purchased from Zhejiang Ontores Biotechnologies Co., Ltd. (Zhejiang, China). α-D-galactosamine (D-gal) and hydrazone hydrate were purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). N,N’-dicyclohexylcarbodiimide (DCC), N,N’-diisopropyl ethylamine (DiEA) and N-hydroxysuccinimide (NHS) were provided by Aladdin Reagent Co., Ltd. (Shanghai, China). Tetrahydrofuran (THF) and trifluoroacetic acid (TFA) were purchased from Tianjin Yongda Chemical Reagent Co., Ltd. (China). Dichloromethane (DCM), 2-(7-Azabenzotriazole)-N,N,N’,N’-Tetramethylurea hexafluorophosphate (HATU), hexafluorophosphate triethylamine (TEA) and N,N’-dimethylformamide (DMF) were purchased from Tianjin Fengchuan Chemical Reagent Co., Ltd. (China) and distilled prior to use. RPMI1640 medium and trypsin EDTA digestive solution were purchased from Beijing Solarbio Science & Technology Co., Ltd. (China). High-quality fetal bovine serum (FBS) was purchased from Hangzhou Sijiqing bioengineering Materials Co., Ltd. (Zhejiang, China). DAPI was purchased from Shanghai Aladdin Reagent Co., Ltd. (China). CCK-8 and hematoxylin and eosin staining kit (H&E) were purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Liquid paraffin was purchased from Zhejiang Ontores Biotechnologies Co., Ltd (Jiangsu, China) and used after preheating. All other reagents and solvents were of analytical grade and used directly. EC109, HepG2, L02 and H22 cells were purchased from Procell Life Science & Technology Co., Ltd (China).
Synthesis of D-gal-FFYEE-hyd-DOX
Fully protected peptide FFYEE (labelled as compound 1) (1.0 eq), DCC (1.2 eq) and NHS (1.2 eq) were dissolved in 10 mL of anhydrous THF, and stirred in an ice bath for 4 h. The precipitation was removed by centrifugation and the liquid supernatant was collected. D-gal (1.5 eq) was dissolved in 9 mL of anhydrous DMF, and the solution was then added to the previously collected supernatant. Thereafter, DiEA (4 eq) was added, and the mixed solution was stirred at room temperature for 24 h. Thereafter, the solution was concentrated with iced ether to precipitate the product. After washing with ether for three times, the product was collected, dried overnight and labelled as compound 2.
The compound 2 was dissolved in a mixed solution of DCM and TFA (V:V=1:1). After stirring at room temperature 25°C for 60 min, the solution was filtered and collected. Another 4 mL of DCM and TFA (V:V=1:1) was added to the filtrate. The solution was stirred at room temperature for 20 min, and filtered. The obtained filtrate was distilled under reduced pressure to remove TFA as much as possible. Then, a large amount of iced ether was added for precipitation. The compound 3 was collected after washing and freeze-dried.
The compound 3 (1.0 eq), HATU (0.6 eq) and TEA (2.3 eq) were dissolved in DCM (10 mL). Then, hydrazone hydrate (9.8 eq) was added to the solution at 25 oC before stirring overnight. The mixture was dissolved in an aqueous solution and extracted with DCM. Subsequently, the DCM was dehydrated with anhydrous sodium sulfate. The obtained compound 4 was dried in vacuum.
DOX•HCL and TEA were dissolved in DMSO to obtain desalted doxorubicin (DOX). Compound 4 (1.0 eq), DOX (1.2 eq) and small amount of glacial acetic acid were dissolved in methanol in the dark for 12 h. Further, the solution was concentrated to remove the solvent to obtain nanofiber conjugate, D-gal-FFYEE-hyd-DOX, compound 5. The molecular weight of FFYEE, three intermediates, and the final product D-gal-FFYEE-hyd-DOX was examined by electrospray ionization-mass spectrometry (ESI-MS) and all products were characterized by infrared spectroscopy (IR). The changes in absorption peaks of DOX and D-gal-FFYEE-hyd-DOX were detected by UV-Vis spectroscopy (UV-Vis).
Morphology of Self-Assembled D-gal-FFYEE-hyd-DOX
The morphology of assembly pentapeptide FFYEE and D-gal-FFYEE-hyd-DOX was observed by TEM (TECNAI G20 s-twin). Before observation, the FFYEE and D-gal-FFYEE-hyd-DOX were diluted in an aqueous solution and placed drop wise on a carbon-coated copper grid, damp-dried at room temperature.
In vitro Drug Release Behavior
To quantitate the pH-induced release of DOX from D-gal-FFYEE-hyd-DOX, 1 mg of D-gal-FFYEE-hyd-DOX was dissolved in 2 mL of ultrapure water and transferred to a dialysis bag (MWCO 1000 Da). The dialysis bag was placed into a 50 mL centrifuge tube with 10 mL PBS solutions of different pH values (pH 7.4 and pH 5.5) and stirred at 37°C away from light exposure. At predetermined time interval, the PBS solutions were withdrawn and replaced by another 10 mL of PBS after each sampling. The released DOX from the D-gal-FFYEE-hyd-DOX was measured by the fluorescence spectrophotometer at 589 nm. The DOX concentrations of the samples were calculated based on the standard curve (y = 1.0599x - 0.2442, R2=0.9979). The cumulative DOX release was calculated according to the following formula: cumulative release (%) = (Mt/m∞) × 100. Mt stands for the amount of DOX released from the D-gal-FFYEE-hyd-DOX at a specific time t, and m∞ is the total amount of DOX loaded in the D-gal-FFYEE-hyd-DOX.
Cytotoxicity Assay of D-gal-FFYEE-hyd-DOX
EC109, HepG2 and L02 cells were incubated with D-gal-FFYEE-hyd-DOX for cytotoxicity investigation, respectively. The cells were cultured in the incubator at 37°C with 5% of CO2. After trypsin treatment, the cells were seeded in a 96-well culture plate at a density of 5000 cells per well and incubated for 24 h. D-gal-FFYEE-hyd-DOX (containing 5–200 mg/L DOX) and free DOX were dissolved in 100 μL DMEM and incubated with different cells for 6 h and 12 h. Then, CCK8 kit was used to detect cell survival comparing free DOX as the control. 10 μL of CCK8 solution was slowly added to each well, and the 96-well culture plate was incubated for 4 h. The average optical density (OD) was measured at 450 nm with the ELISA microplate reader (Bio-Rad). Viability (%) = (ODtreated/ODcontrol)×100. ODtreated is the absorbance value of D-gal-FFYEE-hyd-DOX or free DOX, and ODcontrol is the absorbance value of the negative control with PBS only.
Cellular Uptake
EC109, HepG2 and L02 cells were used to investigate the cellular uptake of D-gal-FFYEE-hyd-DOX and free DOX. The cells were inoculated into a 12-well plate with coverslips in 2 mL of DMEM medium with 10% FBS, and incubated for 24 h. Subsequently, 40 mg/L D-gal-FFYEE-hyd-DOX was added and incubated for 6 h with an equal amount of free DOX as a control. After paraformaldehyde fixation, the coverslips were washed three times with PBS. Then, DAPI was added to stain the nucleus for 5 min, and the cells were washed three times with PBS. The whole process was carried out in dark to avoid fluorescence quenching. Finally, the fluorescence images were captured with the Carl Zeiss Axio Observer A1 microscope.
In vivo Anti-Tumor Study
Male Kunming mice, 25±5g, provided by School of Basic Medical Sciences, Henan University of Science and Technology. Housing conditions were thermostatically maintained at 22±2oC with constant humidity (55±2%) and a 12-h light–dark cycle, keeping with national standard (Laboratory Animal-Requirements of Environment and Housing Facilities) (GB 14925–2010). The research protocol of the animal experimentation was approved by the Institutional Animal Ethics Committee, Henan University of Science and Technology, Luo Yang, China and the laboratory animal-guideline for ethical review of animal welfare of the People’s Republic of China (ethical approval number: GB/T 35892–2018).
The cryopreserved H22 cells were resuscitated at 37°C and an animal liver cancer model was established by subcutaneously injecting 0.1 mL H22 cells into the underarm area of mice. The mice were randomly divided into 3 groups (n = 12) and injected via the tail vein with 0.1 mL PBS, free DOX solution, D-gal-FFYEE-hyd-DOX solution (5 mg/kg equivalent DOX), respectively. When the tumor mass reached ~500 mm3, mice were administered once every two days for 7 consecutive times in 14 days. After each administration, the mice were weighed and the tumor mass was measured. The volume of the tumor mass was calculated according to the following formula: V = (A×B2)/2, where A was the length and B stands for the width of the tumor.
Histopathology Examination
The mice were euthanized after the end of the administration. Tumor tissues and other major tissues were isolated and fixed in 2.5% formaldehyde. The fixed paraffin-embedded tissues were sliced into 5 μm sections for histopathology analysis. The tissue sections were mounted on glass slides and stained with hematoxylin and eosin kit (H&E). All sections were examined under a fluorescence microscope (Axio Observer).
Results and Discussion
Synthesis and Characterization of D-gal-FFYEE-hyd-DOX
The synthesis route of D-gal-FFYEE-hyd-DOX is illustrated in Figure 2. The amide moiety was introduced through the reaction of the carboxyl-terminal of FFYEE and amino group of D-gal, which was based on our previous work.20 Thereafter, the tert-butyl protective group in peptide side chain was removed. Then, DOX was connected by hydrazone bond and D-gal-FFYEE-hyd-DOX was prepared finally. As shown in Figure 3A, the molecular weight of full protected FFYEE found in ESI-MS was 944.65 (calcd Mw: 944.14). The molecular weights of three intermediates (compound 2, 3, 4) presented in Figure 3B–D were 1105.30 (calcd Mw: 1105.29), 936.55 (calcd Mw: 936.97) and 950.55 (calcd Mw: 951.00), respectively. As shown in Figure 3E, the molecular weight of the final product D-gal-FFYEE-hyd-DOX found in ESI-MS was 1477.20 (calcd Mw: 1476.51). According to the results of mass spectrometry, the products after each step were successfully obtained. The infrared spectrum of the above compounds is shown in Figure 4. As shown in Figure 4A, the stretching vibration peak at 1650–1680 cm−1 and 1420 cm−1-1400 cm−1 corresponded to the C=O and C-N formed by amide reaction, which preliminarily indicated that D-gal was successfully connected to the pentapeptide FFYEE. As shown in Figure 4B, the new stretching vibration peak at 1500–1630 cm−1 (arrow 3) corresponded to the vibration of C=N in hydrazone bond, indicating that D-gal-FFYEE-hyd-DOX was prepared successfully. As shown in Figure 5, the red shift of the UV characteristic peaks of DOX in the range of 250–300 nm confirmed the above results.
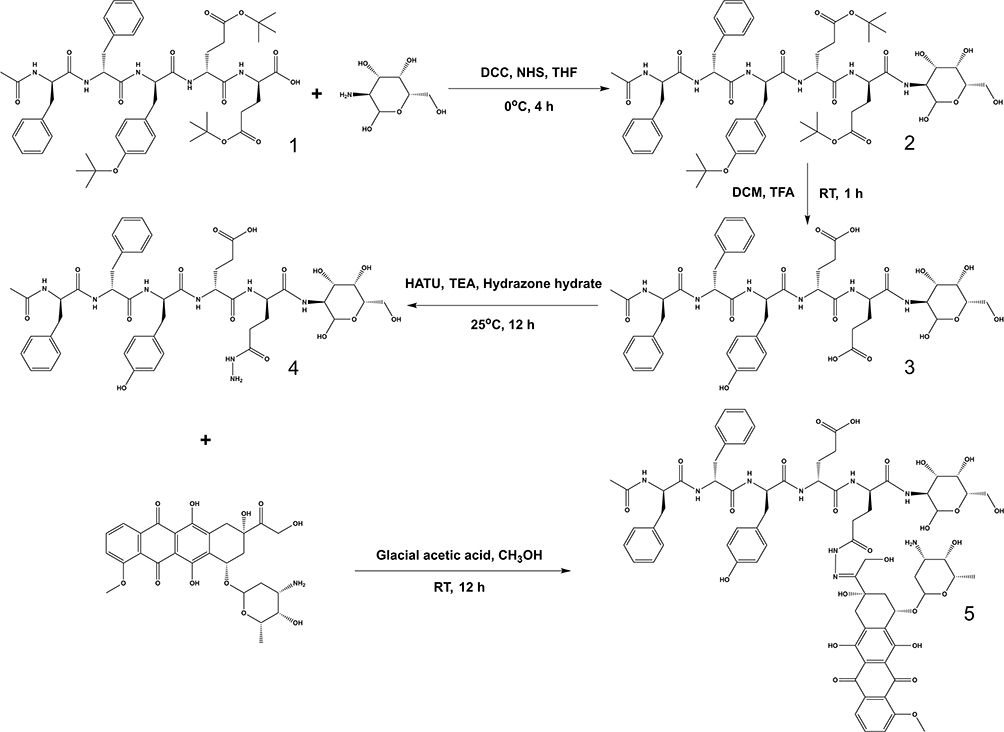 |
Figure 2 Graphical synthesis route of D-gal-FFYEE-hyd-DOX. FFYEE (1), D-gal-FFYEE (2), D-gal-FFYEE of removing the tert-butyl protective group (3), D-gal-FFYEE connected with hydrazide group (4), and D-gal-FFYEE-hyd-DOX (5). |
 |
Figure 3 Mass spectrograms of peptide FFYEE (A), compound 2 (B), compound 3 (C), compound 4 (D) and the final product 5 (E). |
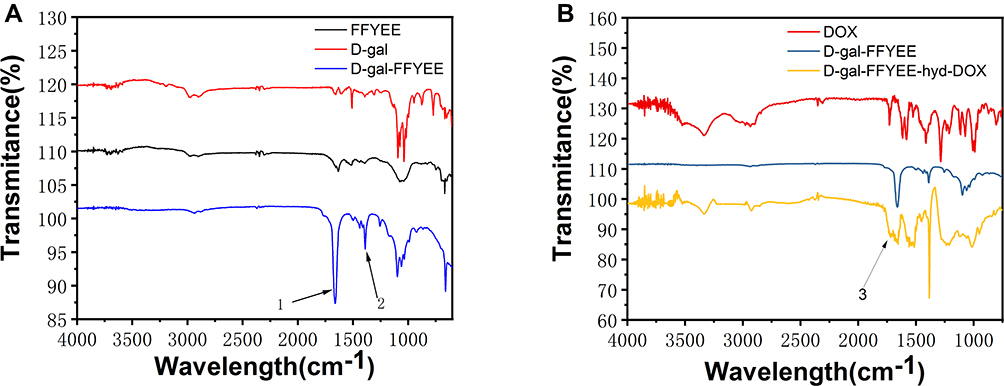 |
Figure 4 The infrared spectrum of the D-gal-FFYEE connected D-gal (A), and the final product D-gal-FFYEE-hyd-DOX (B). |
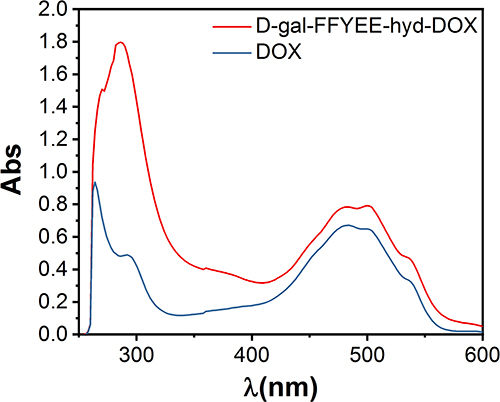 |
Figure 5 UV-Vis spectrum of D-gal-FFYEE-hyd-DOX and DOX. |
Morphology Investigation
Through TEM analysis, both aromatic pentapeptide FFYEE and D-gal-FFYEE-hyd-DOX can self-assemble into nanofibrous structures in neutral aqueous solution. As shown in Figure 6, the diameter of the fibrous structure assembled by FFYEE was ~30 nm. After D-gal and DOX connections, the fibrous structure still existed with significantly increased diameter of ~195 nm.
 |
Figure 6 Transmission electron microscopy (TEM) micrographs of the FFYEE fibers (A) and D-gal-FFYEE-hyd-DOX fibers (B). |
In vitro Drug Release
The extracellular pH values of TME or endosomes are more acidic (pH <6) than that of normal microenvironment (pH 7.4).21 Theoretically, the D-gal-FFYEE-hyd-DOX with acid-sensitive hydrazone bond could exhibit controlled release behavior upon external pH change. Therefore, the DOX release curves of self-assembled nanofiber conjugate in PBS of pH 7.4 and simulative TME were recorded for comparative investigation. From the release profile in Figure 7, the D-gal-FFYEE-hyd-DOX was proved to maintain architecture stability in pH 7.4 PBS because of only about ~20% of DOX released within 72 h. By contrast, decreasing the PBS pH to a value of 5.5, due to the rupture of hydrazone bond causing rapid dissociation of the self-assembled nanofiber conjugate, there is a burst release phenomenon of DOX in acidic dialysis medium. The cumulative release of DOX promptly increased from ~20% to ~70% within 7 h, and exceeded 90% within 24 h. All these data strongly demonstrate that D-gal-FFYEE-hyd-DOX present the features of acid sensitivity and controlled release, suggesting the potential application for tumor therapy.
 |
Figure 7 In vitro DOX release of D-gal-FFYEE-hyd-DOX in PBS (pH 7.4) or PBS (pH 5.5). |
In vitro Cytotoxicity and Intracellular Uptake
The cytotoxicity of D-gal-FFYEE-hyd-DOX and free DOX against EC109 (human esophageal squamous carcinoma cell line), HepG2 (human hepatoma cell line) and L02 cells (human normal liver cell line) was evaluated. As displayed in Figure 8A, the cytotoxicity of D-gal-FFYEE-hyd-DOX and free DOX on HepG2 cells at different incubation times was studied as exhibited. After incubation for 12 h, the D-gal-FFYEE-hyd-DOX revealed stronger killing effect than 6 h at all concentrations. When the equivalent concentration was 200 mg/L, the viability of HepG2 cells in D-gal-FFYEE-hyd-DOX group was 12.9% and 18.8% after 12 h and 6 h of incubation, indicating that the toxicity of D-gal-FFYEE-hyd-DOX to HepG2 cells increased with the increase of incubation time. At the same time, it could be seen that at 6 h, the inhibition of D-gal-FFYEE-hyd-DOX on HepG2 cells was significantly stronger than that of DOX at all drug concentration. Based on the results displayed in Figure 8B, the cell viability of EC109 was significantly higher than that of HepG2 after 12 h incubation with D-gal-FFYEE-hyd-DOX. At the concentration of 200 mg/L, the viability of HepG2 and EC109 cells after 12 h of incubation was 17.5% and 43.9%, respectively, indicating that the killing ability of D-gal-FFYEE-hyd-DOX on HepG2 cells was stronger than that of EC109 cells. Therefore, we can speculate that D-gal-FFYEE-hyd-DOX could deliver more DOX to HepG2 cells rich in ASGPR receptors on the cell surface through the targeting effect of D-galactose. Meanwhile, as shown in Figure 8B, the cell survival rate of L02 cells treated with D-gal-FFYEE-hyd-DOX for 12 h was 63.6%, which suggested that D-gal-FFYEE-hyd-DOX showed lower cytotoxicity to L02 cells compared with HepG2 and EC109 cells.
 |
Figure 8 Cell viability of HepG2 cells incubated with D-gal-FFYEE-hyd-DOX and free DOX for 6 h and 12 h (A). Cell viability of EC109, L02 and HepG2 cells incubated with D-gal-FFYEE-hyd-DOX for 12 h (B). Data are presented as the average (SD, n = 3). |
Moreover, we observed the intracellular DOX uptake of D-gal-FFYEE-hyd-DOX or free DOX after incubation with EC109, HepG2 and L02 cells for 6 h, respectively. It can be seen from Figure 9 that after cells were incubated by D-gal-FFYEE-hyd-DOX, only a small amount of DOX was uptaken by EC109 cells and a large amount of DOX was showing in HepG2 cells. The data indicated that HepG2 cells have a higher uptake rate of D-gal-FFYEE-hyd-DOX than EC109 cells. Such results confirmed that the D-gal moiety of D-gal-FFYEE-hyd-DOX promoted the cellular uptake for the overexpressed ASGPR receptor on HepG2 cells. In addition, compared with HepG2 cells, there was only a small amount of DOX after L02 cells was incubated with D-gal-FFYEE-hyd-DOX, matching the above cytotoxicity results. The difference of cellular uptake rates further showed that the D-gal-FFYEE-hyd-DOX with D-gal targeting ability and acid sensitivity can specifically deliver DOX to HepG2 cells. This enhanced endocytosis facilitated the enrichment of DOX in liver tumors and reduced the systemic toxicity on normal tissues.
 |
Figure 9 Fluorescence inverted microscope image of L02, EC109 and HepG2 cells that were incubated with free DOX and D-gal-FFYEE-hyd-DOX for 6 h, respectively. Blue and red dots stand for the fluorescence of DAPI and DOX, respectively. The scale bar is 50 µm. |
In vivo Anti-Tumor Assay
The anti-tumor assay of D-gal-FFYEE-hyd-DOX in vivo was evaluated by H22 liver tumor-bearing Kunming mice model. As shown in Figure 10A, after 14 days of administration, the tumor volume significantly reduced to 160 mm3 in D-gal-FFYEE-hyd-DOX group comparing the initial volume of 546 mm3. Accordingly, in free DOX group, the tumor volume decreased to approximately 507 mm3. However, the tumor volume increased to 1861 mm3 in mice of PBS group. As shown in Figure 10B, the results of different groups showed that the body weight of D-gal-FFYEE-hyd-DOX and PBS groups did not decrease significantly. On the contrary, the weight of free DOX group decreased significantly, which may be related to the systemic toxicity of free DOX. The data indicated the biocompatibility of D-gal-FFYEE-hyd-DOX in vivo. The representative tumor tissue images are shown in Figure 10C. Obviously, the excised tumor mass volume of DOX and D-gal-FFYEE-hyd-DOX groups exhibited anti-tumor effects of different degrees. The free DOX presented a relatively less antitumor effect, and D-gal-FFYEE-hyd-DOX revealed a much greater inhibition effect on tumor growth. In conclusion, the D-gal-FFYEE-hyd-DOX nanofiber represented remarkable anti-tumor effect and biocompatibility comparing free DOX.
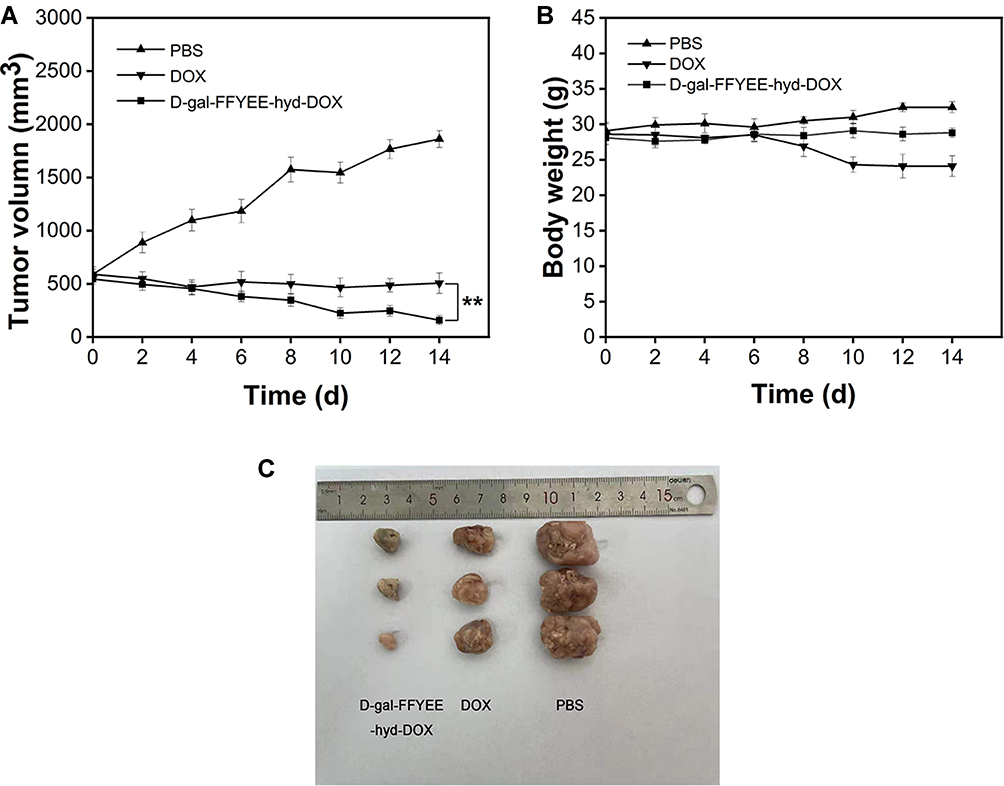 |
Figure 10 In vivo anti-tumor effect of D-gal-FFYEE-hyd-DOX. (A) Tumor growth curves, (B) body weight change curves, (C) representative tumor tissue image in male mice bearing H22 tumor after intravenous administration. The data are presented as the mean ± SD (n = 5). **P < 0.05 DOX vs D-gal-FFYEE-hyd-DOX. |
Histopathology Analysis
The therapeutic effect and tissue toxicity of D-gal-FFYEE-hyd-DOX was evaluated by H&E staining of tumor tissues and other major organs. From Figure 11, tumors treated with PBS were mainly filled with abundant tumor cells without obvious damage, which was characterized by high cell density, many and large nuclei and increased stroma. By contrast, there were a large number of apoptotic or necrosis cells without nuclei in tumor tissues after treatment with D-gal-FFYEE-hyd-DOX or free DOX. Clearly, D-gal-FFYEE-hyd-DOX displayed a significant antitumor effect, even better than free DOX. Compared with free DOX, targeting D-gal-FFYEE-hyd-DOX presented no obvious subacute toxicity to other normal tissues. Moreover, the morphologies of the cells in the heart, liver and spleen were analogous to those of the cells in the PBS group, indicating no apparent toxicity to these tissues. In the group of free DOX, there were obvious heart, liver and spleen damages, such as extensive irregular arrangement and shrinkage of the cells. However, there is no significant toxicity in the lung or kidney tissues. In all, D-gal-FFYEE-hyd-DOX exhibited significant inhibition of tumor growth in vivo without systemic toxicity. In previous similar work, Zhang and coworkers reported an activatable cell-penetrating peptide-conjugated DOX (DOX-CR8G3PK6-DMA) for tumor targeted drug delivery.22 In vivo results showed that this peptidic prodrug could significantly inhibit tumor growth. DOX-ACPP-DMA exhibited significant inhibition and reduced cardiotoxicity and hepatotoxicity of tumor growth in vivo significantly. Neither DOX-ACPP-DMA nor free DOX had significant splenic toxicity. Different from this work, DOX showed some spleen toxicity in our experiment, while conjugate D-gal-FFYEE-hyd-DOX did not show corresponding spleen toxicity. However, it can be seen that the design of peptide-based conjugates has obvious advantages in reducing the systemic toxicity of DOX.
 |
Figure 11 H&E staining of tumor tissue and other tissues isolated from male mice bearing H22 tumor after administration of D-gal-FFYEE-hyd-DOX, DOX and PBS. |
Conclusion
In summary, a novel conjugate D-gal-FFYEE-hyd-DOX with targeting and acid-sensitive drug release characteristics was constructed based on the aromatic pentapeptide FFYEE. D-gal-FFYEE-hyd-DOX can be self-assembled into a nanofiber structure with a diameter of ~200 nm in neutral aqueous solution. In vitro and in vivo experiments showed that D-gal-FFYE-HYD-DOX was an excellent DOX delivery system with increased antitumor activity and reduced side effects. The primary goal of drug delivery systems in cancer therapy is to transport sufficient drugs to disease sites while minimizing the toxicity on major tissues. Compared with other drug delivery systems, the drug precursor or conjugate could minimize the premature drug release and interaction with macromoleculars in vivo, which can reduce the metabolic burden of patients and improve the therapeutic effect.23 In order to increase the tumor-specific inhibition effect of DOX and reduce its toxicity, several prodrugs or conjugates have been developed rapidly in recent years.24,25 Peptide-based conjugates possess better biocompatibility, degradability and more convenient preparation processes.26–28 The conjugated D-gal-FFYEE-hyd-DOX designed in this paper is biodegradable and will not cause unnecessary immunogenic reactions.29,30 Meanwhile, the nanofibers formed by the conjugates sustained stable nanostructures in the water, which could decompose by the stimulation of the specific TME, and release the active drug. Therefore, D-gal-FFYEE-hyd-DOX nanofiber may become an efficient and safe delivery system for cancer therapy in the future.
Acknowledgment
We are grateful for the financial support of the National Natural Science Foundation of China (51403055, U1704150).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Goncalves M, Mignani S, Rodrigues J, Tomas H. A glance over doxorubicin based-nanotherapeutics: from proof-of-concept studies to solutions in the market. J Controlled Release. 2020;317:347–374. doi:10.1016/j.jconrel.2019.11.016
2. Liu C, Ma XR, Zhuang J, Liu LJ, Sun CG. Cardiotoxicity of doxorubicin-based cancer treatment: what is the protective cognition that phytochemicals provide us? Pharmacol Res. 2020;160:105062. doi:10.1016/j.phrs.2020.105062
3. Agudelo D, Bourassa P, Berube G, Tajmir-Riahi HA. Review on the binding of anticancer drug doxorubicin with DNA and tRNA: structural models and antitumor activity. J Photochem Photobiol B-Biol. 2016;158:274–279. doi:10.1016/j.jphotobiol.2016.02.032
4. Li D, Yang Y, Wang S. Role of acetylation in doxorubicin-induced cardiotoxicity. Redox Biol. 2021;46:102089. doi:10.1016/j.redox.2021.102089
5. Hu C, Zhang X, Wei WY, et al. Matrine attenuates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via maintaining AMPK alpha/UCP2 pathway. Acta Pharmaceutica Sinica B. 2019;9(4):690–701. doi:10.1016/j.apsb.2019.03.003
6. Cui H, Huan ML, Ye WL, et al. Mitochondria and nucleus dual delivery system to overcome DOX resistance. Mol Pharm. 2017;14(3):746–756. doi:10.1021/acs.molpharmaceut.6b01016
7. Duan HX, Liu YH, Gao ZG, Huang W. Recent advances in drug delivery systems for targeting cancer stem cells. Acta Pharmaceutica Sinica B. 2021;11(1):55–70. doi:10.1016/j.apsb.2020.09.016
8. He YM, Lei L, Cao J, et al. A combinational chemo-immune therapy using an enzyme-sensitive nanoplatform for dual-drug delivery to specific sites by cascade targeting. Sci Adv. 2021;7(6). doi:10.1126/sciadv.aba0776
9. Liu J, Chen Q, Feng L, Liu Z. Nanomedicine for tumor microenvironment modulation and cancer treatment enhancement. Nano Today. 2018;21:55–73. doi:10.1016/j.nantod.2018.06.008
10. Zhang YJ, Sun T, Jiang C. Biomacromolecules as carriers in drug delivery and tissue engineering. Acta Pharmaceutica Sinica B. 2018;8(1):34–50. doi:10.1016/j.apsb.2017.11.005
11. Takano S, Islam W, Nakazawa K, Maeda H, Sakurai K, Fujii S. Phosphorylcholine-grafted molecular bottlebrush-doxorubicin conjugates: high structural stability, long circulation in blood, and efficient anticancer activity. Biomacromolecules. 2021;22(3):1186–1196. doi:10.1021/acs.biomac.0c01704
12. Quader S, Cabral H, Mochida Y, et al. Selective intracellular delivery of proteasome inhibitors through pH-sensitive polymeric micelles directed to efficient antitumor therapy. J Controlled Release. 2014;188:67–77. doi:10.1016/j.jconrel.2014.05.048
13. Li W, Fu J, Ding Y, Liu D, Jia N. Low density lipoprotein-inspired nanostructured lipid nanoparticles containing pro-doxorubicin to enhance tumor-targeted therapeutic efficiency. Acta Biomaterialia. 2019;96:456–467. doi:10.1016/j.actbio.2019.06.051
14. Guo FY, Yu N, Jiao YL, et al. Star polyester-based folate acid-targeting nanoparticles for doxorubicin and curcumin co-delivery to combat multidrug-resistant breast cancer. Drug Deliv. 2021;28(1):1709–1721. doi:10.1080/10717544.2021.1960926
15. Toy R, Bauer L, Hoimes C, Ghaghada KB, Karathanasis E. Targeted nanotechnology for cancer imaging. Adv Drug Deliv Rev. 2014;76:79–97. doi:10.1016/j.addr.2014.08.002
16. Petrov RA, Mefedova SR, Yamansarov EY, et al. New small-molecule glycoconjugates of docetaxel and GalNAc for targeted delivery to hepatocellular carcinoma. Mol Pharm. 2021;18(1):461–468. doi:10.1021/acs.molpharmaceut.0c00980
17. D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting - strategies and applications. J Controlled Release. 2015;203:126–139. doi:10.1016/j.jconrel.2015.02.022
18. Sharma R, Porterfield JE, An HT, et al. Rationally designed galactose dendrimer for hepatocyte-specific targeting and intracellular drug delivery for the treatment of liver disorders. Biomacromolecules. 2021;22(8):3574–3589. doi:10.1021/acs.biomac.1c00649
19. Naguib MJ, Kamel AM, Negmeldin AT, Elshafeey AH, Elsayed I. Molecular docking and statistical optimization of taurocholate-stabilized galactose anchored bilosomes for the enhancement of sofosbuvir absorption and hepatic relative targeting efficiency. Drug Deliv. 2020;27(1):996–1009. doi:10.1080/10717544.2020.1787557
20. Liang J, Xuan M, Wu W, Li J. GSH-responsive nanofibrous prodrug formed by a short naphthylacetic acid-terminated peptide for 6-mercaptopurine delivery. J Drug Deliv Sci Technol. 2021;65:102691. doi:10.1016/j.jddst.2021.102691
21. Ovais M, Mukherjee S, Pramanik A, et al. Designing stimuli-responsive upconversion nanoparticles that exploit the tumor microenvironment. Adv Mater. 2020;32(22):2000055. doi:10.1002/adma.202000055
22. Cheng H, Zhu J, Xu X, et al. Activable cell-penetrating peptide conjugated prodrug for tumor targeted drug delivery. ACS Appl Mater Interfaces. 2015;7:16061. doi:10.1016/j.addr.2016.06.015
23. Mozaffari S, Salehi D, Mahdipoor P, et al. Design and application of hybrid cyclic-linear peptide-doxorubicin conjugates as a strategy to overcome doxorubicin resistance and toxicity. Eur J Med Chem. 2021;226:113836. doi:10.1016/j.ejmech.2021.113836
24. Alam Khan S, Jawaid Akhtar M. Structural modification and strategies for the enhanced doxorubicin drug delivery. Bioorg Chem. 2022;120:105599. doi:10.1016/j.bioorg.2022.105599
25. Sohail M, Sun Z, Li Y, Gu X, Xu H. Research progress in strategies to improve the efficacy and safety of doxorubicin for cancer chemotherapy. Expert Rev Anticancer Ther. 2021;21(12):1385–1398. doi:10.1080/14737140.2021.1991316
26. Alas M, Saghaeidehkordi A, Kaur K. Peptide-drug conjugates with different linkers for cancer therapy. J Med Chem. 2021;64(1):216–232. doi:10.1021/acs.jmedchem.0c01530
27. Hao T, Fu Y, Yang Y, et al. Tumor vasculature-targeting PEGylated peptide-drug conjugate prodrug nanoparticles improve chemotherapy and prevent tumor metastasis. Eur J Med Chem. 2021;219:113430. doi:10.1016/j.ejmech.2021.113430
28. Deng X, Mai R, Zhang C, et al. Discovery of novel cell-penetrating and tumor-targeting peptide-drug conjugate (PDC) for programmable delivery of paclitaxel and cancer treatment. Eur J Med Chem. 2021;213:113050. doi:10.1016/j.ejmech.2020.113050
29. Fang Y, Wang H. Molecular engineering of peptide-drug conjugates for therapeutics. Pharmaceutics. 2022;14(1):212. doi:10.3390/pharmaceutics14010212
30. Zhang Y, He P, Zhang P, Yi X, Xiao C, Chen X. Polypeptides-drug conjugates for anticancer therapy. Adv Healthc Mater. 2021;10(11):e2001974. doi:10.1002/adhm.202001974
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.