Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 15
Amyotrophic Lateral Sclerosis Masquerading as Multiple System Atrophy with Parkinsonism and Anxiety as Initial Manifestations
Received 1 April 2025
Accepted for publication 2 September 2025
Published 10 September 2025 Volume 2025:15 Pages 95—100
DOI https://doi.org/10.2147/DNND.S531647
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Haiyan Tang,1 Jianping Yao,2 Zhuang Wang1
1Department of Neurology, Huzhou Central Hospital, The Fifth School of Clinical Medicine of Zhejiang Chinese Medical University, The Affiliated Central Hospital of Huzhou University, Huzhou, Zhejiang, People’s Republic of China; 2Department of Endocrinology, Huzhou Central Hospital, The Fifth School of Clinical Medicine of Zhejiang Chinese Medical University, The Affiliated Central Hospital of Huzhou University, Huzhou, Zhejiang, People’s Republic of China
Correspondence: Zhuang Wang, Department of Neurology, Huzhou Central Hospital, The Fifth School of Clinical Medicine of Zhejiang Chinese Medical University, The Affiliated Central Hospital of Huzhou University, No.1558 Sanhuan Road, Huzhou, Zhejiang, 313000, People’s Republic of China, Email [email protected]
Introduction: Amyotrophic lateral sclerosis (ALS) and multiple system atrophy (MSA) are both neurodegenerative disorders. While ALS may present with clinical features resembling Parkinsonism, there have been no definitive reports of ALS mimicking MSA, only cases of Primary lateral sclerosis (PLS) mimicking Parkinsonism.
Methods: This article reports a case of ALS presenting with Parkinsonism and anxiety as the initial symptoms. Five years after the initial diagnosis of MSA, the patient developed signs of lower motor neuron involvement, including fasciculations and muscle atrophy, ultimately leading to a revised diagnosis of ALS. This study combines literature analysis to explore the reasons for misdiagnosis and identifies key differentiating features.
Results: Specifically, muscle rigidity in ALS is characterized by a velocity-dependent increase in muscle tone caused by damage to the upper motor neurons. This symptom tends to be more pronounced in the lower limbs than in the upper limbs and is often accompanied by spastic gait. Objective examinations may reveal early atrophy of the frontal and temporal lobes of the cerebrum on head magnetic resonance (MR) imaging, whereas 18F-FDG brain positron emission tomography (PET) may reveal reduced metabolism in the frontal and parietal lobes of the cerebrum with normal basal ganglial function, distinguishing ALS from basal ganglial metabolic decline in MSA.
Discussion: To our knowledge, this is the first case of ALS misdiagnosed as MSA. Clinically, patients with parkinsonism who do not respond to dopaminergic drugs should be cautious about atypical ALS. Muscle rigidity manifesting as upper motor neuron damage, and MR and 18F-FDG brain PET imaging can provide early differential diagnosis indicators.
Keywords: amyotrophic lateral sclerosis, multiple system atrophy, motor neuron disease, Parkinsonism, upper motor neuron
Background
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder1 and represents the most common form of motor neuron disease (MND). It is clinically characterized by progressive skeletal muscle weakness, muscular atrophy, fasciculations, bulbar paralysis, and pyramidal tract signs.2 Multiple system atrophy (MSA), another neurodegenerative disorder, is characterized by autonomic dysfunction as its core clinical feature and may present with concomitant parkinsonism and/or cerebellar syndrome.3 The early clinical manifestations of ALS are variable, and may include extrapyramidal and autonomic symptoms. Initial symptoms of bulbar paralysis can be easily mistaken for cerebellar ataxia, often leading to misdiagnosis as MSA. Here, we present a case of ALS initially presenting with Parkinsonian features and anxiety, which was misdiagnosed as MSA in the early stages, and analyze the reasons of misdiagnosis. This study proposes an approach for the early identification, diagnosis, and management of ALS in the absence of specific biomarkers.
Case Presentation
History
In 2017, a 54-year-old male patient presented with generalized fatigue and anxiety. In 2018, he developed bradykinesia and postural tremor in the right hand, along with symptoms consistent with rapid eye movement sleep behavior disorder (RBD). He also experienced stool incontinence and urinary urgency, but no olfactory loss, hallucinations, or cognitive impairment were reported. In 2020, the patient’s symptoms worsened, with occasional falls, dysarthria, and urinary incontinence. Physical examination showed normal limb muscle strength, slightly increased muscle tone in the right limb, more pronounced in the lower limbs than the upper limbs, and hyperactive tendon reflexes. Cranial MR revealed mild atrophy of the bilateral frontotemporal lobes (Figure 1). Cervical and lumbar vertebral spinal MR showed no significant abnormalities, and electromyography (EMG) results were within normal limits. In October 2020, positron emission tomography (PET) demonstrated a normal distribution of vesicular monoamine transporter 2 (VMAT2) in the bilateral striatal vesicles, decreased 18F-FDG metabolism in the left parietal lobe, and increased FDG metabolism in the left posterior putamen (Figure 2). The patient was initially diagnosed with MSA and anxiety. However, levodopa treatment showed no therapeutic effect. In October 2022, the experienced progressive worsening of gait rigidity, accompanied by recurrent falls, dysphagia, and muscle weakness with fasciculations. He was readmitted and ultimately diagnosed with ALS (see event timeline, Figure 3).
 |
Figure 1 MR imaging of the brain 3 years after symptom onset. (a–d) T2 axial view: bilateral frontotemporal lobe atrophy without cerebellar atrophy and hot cross bun sign, (e) T1 axial view: bilateral frontotemporal lobe atrophy. |
 |
Figure 2 PET-CT (a and b) there were normal distribution of VMAT2 in caudate nucleus and front or back of the putamen bilateral; (c and d) 18F-FDG revealed that metabolism in the left parietal lobe reduced, whereas that in the left back side of the putamen increased. |
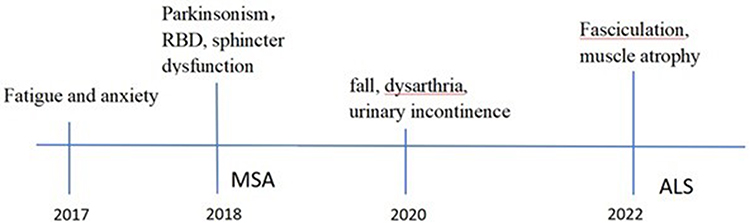 |
Figure 3 History and diagnosis. |
Neurological examination revealed no orthostatic hypotension. Tongue atrophy with fibrillation, dysarthria, normal eye movements without nystagmus were noted. Muscle tone was increased in the lower limbs compared to the upper limbs, accompanied by muscle atrophy and hyperactive tendon reflexes in the upper limbs, as well as deltoid muscle atrophy with fasciculations. Distal upper limbs muscle strength was grade 3–4, while lower limb strength was grade 4+. Bilateral Hoffman’s signs and ankle clonus were present. The patient exhibited leg stiffness and a wide-based freezing gait during ambulation. The Romberg and pull-back tests were both positive.
Laboratory test results, including autoantibody profiles and other immune markers, were within normal ranges. The patient tested negative for syphilis and HIV, and his muscle enzyme levels and vitamin concentrations were also normal. Cognitive function assessments, including the Chinese Mini Mental Status (CMMS) and Montreal Cognitive Assessment (MoCA), yielded normal scores. Ultrasonography showed a residual urine volume of 48 mL. EMG revealed neurogenic changes involving the upper and lower limbs as well as the sternocleidomastoid muscles, with fibrillation or fasciculation potentials observed in the medulla, cervical, thoracic, and lumbar regions. Prolonged motor unit action potential (MUAP) duration, increased amplitude, and elevated polyphasic wave percentage were also noted. EMG findings indicated coexistence of acute denervation and chronic neurogenic lesions, while sensory nerve conduction studies were normal. Genetic testing did not identify pathogenic mutations in genes such as SOD-1, TDP-43, or C9orf72.
Following the diagnosis of ALS, riluzole was initiated to slow disease progression. By October 2023, no significant structural changes were observed in the cerebellar brainstem region (Figure 4). The patient was followed up until June 2024 and exhibited increased difficulty in walking, absence of olfactory loss, and improvement in RBD. He remained capable of independent ambulation and showed no signs of orthostatic hypotension.
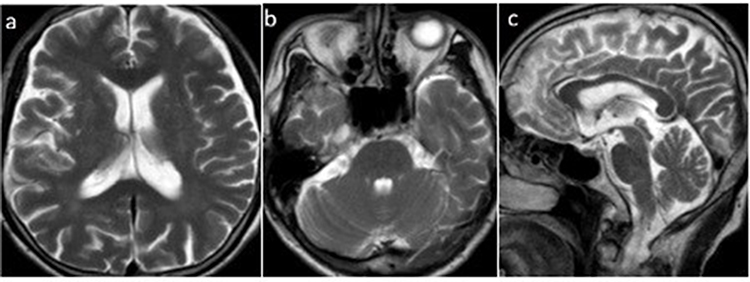 |
Figure 4 MR imaging of the brain 6 years after symptom onset. (a and b) T2 axial view: bilateral frontotemporal lobe atrophy, (b and c) T2 axial and sagittal views: no cerebellar atrophy and hot cross bun signs. |
Discussion and Conclusions
The patient exhibited motor symptoms one year after initially presenting with fatigue and anxiety, characterized by unilateral bradykinesia and tremor. Parkinson’s disease was excluded due to the normal dopamine transporter function indicated by VMAT2 imaging.4 However, a clinically possible diagnosis of MSA could be considered based on the presence of typical RBD, unexplained urinary urgency, and Parkinsonian features, in addition to levodopa insensitivity and lack of characteristic brainstem cross and putaminal fissure signs.3 As the disease progressed, the patient developed typical signs of dysarthria, dysphagia, and fasciculation in the later stages, EMG revealed both upper and lower motor neuron lesions affecting the limbs and sternocleidomastoid muscles, ultimately leading to a diagnosis of ALS.1,5
The review of the patient’s medical history revealed that his early Parkinsonian features and negative EMG results contributed to the neurologist’s misdiagnosis. Patients with anxiety precursors before motor symptoms are easily misdiagnosed with Parkinson’s syndrome. However, Ibrahim et al6 pointed out that emotional instability is a common feature in all patients with MND who are misdiagnosed with Parkinsonism. Considering the broad basis of spastic gait, a combination with dysarthria can easily be mistaken for cerebellar ataxia. In addition, given that the patient exhibited RBD and urinary incontinence in the early stages, he was diagnosed with MSA at several hospitals until he presented with classic fasciculation and muscular atrophy of ALS, as confirmed by EMG. Furthermore, patients with ALS who exhibit prominent pyramidal tract signs may experience sphincter dysfunction.1 Moreover, bladder symptoms are common in primary lateral sclerosis, which is a type of MND.7
Although RBD is a typical non-motor symptom of α-synuclein disease, Zhang et al8 reported that some ALS patients still have sleep disorders.9 Lo et al10 also found RBD in of 2/41 patients with ALS and in of 0/26 healthy controls. The cause of RBD in ALS is unknown, but some patients with ALS11 have been confirmed to contain Lewy bodies in their basal glial cells. In addition, both ALS and MSA have pathogenic factors such as mitochondrial dysfunction, neuroinflammation, and oxidative stress.12 Furthermore, both types of degenerative diseases exhibit abnormal protein folding, which is manifested as alpha-synuclein (α-syn) in MSA, superoxide dismutase 1 (SOD1) and + TAR DNA binding protein (TDP-43) in ALS.12 Moreover, researchers had identified degenerative pathological changes in the cricoarytenoid muscle fibers of patients with MSA that are similar to those observed in ALS. These changes include abnormalities in synaptic contacts, as well as the loss of synaptic vesicles and mitochondria.13,14 Perhaps the similarity in the pathological mechanisms of the two neurodegenerative disorders may lead to comparable clinical manifestations.
If the motor and nonmotor symptoms of this patient mimicked MSA, then why did no cerebellar brainstem atrophy or putaminal fissure signs appear in his brain MR images from the onset to 6 years of follow-up? The first and last MR scans revealed signs of bilateral frontotemporal lobe atrophy. Although no signs of frontotemporal dementia were found in this patient, decreased frontotemporal cortex thickness in ALS could explain this sign.15 In addition, brain PET imaging revealed that 18F-FDG metabolism in the left parietal lobe of the patient decreased, whereas that in the left posterior putamen increased. This finding contradicts the results of Devrome M.16 regarding the low metabolism of the striatum in patients with atypical Parkinson’s disease. In addition, Mabuchi et al17 reported that 18F-FDG brain PET images in three MND cases suggested widespread frontal hypometabolism with basal ganglial retention.
Although objective examinations can effectively aid in diagnosis, their widespread use in early stages is limited due to the high cost of PET and its unavailability in basic healthcare facilities. In some patients with ALS, MR T2-weighted imaging (T2 WI), fluid-attenuated inversion recovery (FLAIR), and diffusion-weighted imaging (DWI) sequences can reveal highly symmetric signals along the pyramidal tracts, suggesting upper motor neuron involvement18 but are unnecessary for diagnosis. Elevated levels of cerebrospinal fluid light chains and serum neurofilaments may also indicate upper motor neuronopathy in ALS but lack diagnostic specificity.5 Nevertheless, MR remains a valuable tool if it can detect subtle frontotemporal lobe atrophy in the early stages of the disease. Clinicians should also be aware that rigidity observed in ALS originates from pyramidal tract dysfunction. This type of rigidity manifests more as spasticity rather than the lead-pipe or cogwheel rigidity typically seen in parkinsonism, and is generally more pronounced in the lower limbs than in the upper limbs. As illustrated in this case, it may be accompanied by tendon hyperreflexia, which could serve as an early distinguishing feature between ALS and MSA.
In conclusion, not all MNDs are purely confined to the motor system. When they present with extrapyramidal symptoms or Sphincter dysfunction, they are classified as atypical MNDs. Atypical ALS is characterized by a relatively prolonged disease course, and when Parkinsonism and sphincter dysfunction appear in the early stages, it can be easily misdiagnosed as MSA. Although previous studies have documented cases of Primary lateral sclerosis (PLS) mimicking Parkinsonism, there are no published reports describing ALS presenting with symptoms resembling Parkinsonism. Furthermore, in this particular case, the misdiagnosis pertains specifically to MSA, rather than Parkinsonism discussed in earlier literature.6,17
This study particularly highlights that patients with ALS exhibit high velocity–dependent muscle tension, which is more pronounced in the lower limbs compared to the upper limbs. Early imaging changes, such as mild atrophy of the frontotemporal cortex observed on early MRI and decreased frontoparietal metabolism with preserved basal ganglia function shown on 18F-FDG PET, can serve as key criteria for differentiating ALS from MSA and preventing long-term misdiagnosis.
We anticipate that the development of highly feasible imaging techniques and other biomarkers for ALS will provide robust evidence to support early diagnosis. Consequently, timely treatment can be initiated to improve patients’ quality of life and extend their survival.
Data Sharing Statement
Data is provided within the paper.
Ethics Approval and Consent to Participate
This study has been approved and permitted for publication by the Medical Ethics Committee of Huzhou Central Hospital, and the patient consented to participate.
Consent for Publication
The patient gave written informed consent for his personal or clinical details along with any identifying images to be published in this study.
Acknowledgment
We thank the patients for participating in this study.
Disclosure
The authors report no conflicts of interest.
References
1. Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293–299. doi:10.1080/146608200300079536
2. Urushitani M, Warita H, Atsuta N, et al. The clinical practice guideline for the management of amyotrophic lateral sclerosis in Japan-update 2023. Rinsho Shinkeigaku. 2024;64(4):252–271. doi:10.5692/clinicalneurol.cn-001946
3. Wenning GK, Stankovic I, Vignatelli L, et al. The movement disorder society criteria for the diagnosis of multiple system atrophy. Mov Disord. 2022;37(6):1131–1148. doi:10.1002/mds.29005
4. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30(12):1591–1601. doi:10.1002/mds.26424
5. Shefner JM, Al-Chalabi A, Baker MR, et al. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020;131(8):1975–1978. doi:10.1016/j.clinph.2020.04.005
6. Norlinah IM, Bhatia KP, Ostergaard K, Howard R, Arabia G, Quinn NP. Primary lateral sclerosis mimicking atypical parkinsonism. Mov Disord. 2007;22(14):2057–2062. doi:10.1002/mds.21645
7. Turner MR, Barohn RJ, Corcia P, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020;91(4):373–377. doi:10.1136/jnnp-2019-322541
8. Zhang F, Niu L, Liu X, et al. Rapid eye movement sleep behavior disorder and neurodegenerative diseases: an update. Aging Dis. 2020;11(2):315–326. doi:10.14336/AD.2019.0324
9. Anghel L, Ciubara A, Nechita A, et al. Sleep disorders associated with neurodegenerative diseases. Diagnostics. 2023;13(18).
10. Lo Coco D, Puligheddu M, Mattaliano P, et al. REM sleep behavior disorder and periodic leg movements during sleep in ALS. Acta Neurol Scand. 2017;135(2):219–224. doi:10.1111/ane.12593
11. Desai J, Swash M. Extrapyramidal involvement in amyotrophic lateral sclerosis: backward falls and retropulsion. J Neurol Neurosurg Psychiatry. 1999;67(2):214–216. doi:10.1136/jnnp.67.2.214
12. Tyler SEB, Tyler LDK. Pathways to healing: plants with therapeutic potential for neurodegenerative diseases. IBRO Neurosci Rep. 2023;14:210–234. doi:10.1016/j.ibneur.2023.01.006
13. Yoshihara T, Yamamura Y, Kaneko F, Abo N, Nomoto M. Neuromuscular junctions of the posterior cricoarytenoid muscle in multiple system atrophy: a case study. Acta Otolaryngol Suppl. 2009;129(562):115–119. doi:10.1080/00016480902911987
14. Yoshihara T, Ishii T, Iwata M, Nomoto M. Ultrastructural and histochemical study of the motor end plates of the intrinsic laryngeal muscles in amyotrophic lateral sclerosis. Ultrastruct Pathol. 1998;22(2):121–126. doi:10.3109/01913129809032266
15. Chio A, Pagani M, Agosta F, Calvo A, Cistaro A, Filippi M. Neuroimaging in amyotrophic lateral sclerosis: insights into structural and functional changes. Lancet Neurol. 2014;13(12):1228–1240. doi:10.1016/S1474-4422(14)70167-X
16. Devrome M, Van Weehaeghe D, De Vocht J, Van Damme P, Van Laere K, Koole M. Glucose metabolic brain patterns to discriminate amyotrophic lateral sclerosis from Parkinson plus syndromes. EJNMMI Res. 2018;8(1):110. doi:10.1186/s13550-018-0458-5
17. Mabuchi N, Watanabe H, Atsuta N, et al. Primary lateral sclerosis presenting parkinsonian symptoms without nigrostriatal involvement. J Neurol Neurosurg Psychiatry. 2004;75(12):1768–1771. doi:10.1136/jnnp.2003.035212
18. Filippi M, Agosta F, Grosskreutz J, et al. Progress towards a neuroimaging biomarker for amyotrophic lateral sclerosis. Lancet Neurol. 2015;14(8):786–788. doi:10.1016/S1474-4422(15)00134-9
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Safety and Efficacy of Mesenchymal Stem Cell Therapy in Multiple System Atrophy: Systematic Review
Elimam N, Albarari SS, Shaalan Y, Elsheikh SM, Alzamari AA, Elmekkawi N, Mogahed R, Alghuweiri RH
Biologics: Targets and Therapy 2026, 20:593367
Published Date: 31 March 2026