Back to Journals » Infection and Drug Resistance » Volume 16
Alterations in the Fecal Microbiota Composition in Pediatric Acute Diarrhea: A Cross-Sectional and Comparative Study of Viral and Bacterial Enteritis
Authors Xiao Q ![]() , Chen B
, Chen B ![]() , Zhu Z, Yang T, Tao E, Hu C, Zheng W
, Zhu Z, Yang T, Tao E, Hu C, Zheng W ![]() , Tang W, Shu X, Jiang M
, Tang W, Shu X, Jiang M
Received 23 March 2023
Accepted for publication 12 August 2023
Published 21 August 2023 Volume 2023:16 Pages 5473—5483
DOI https://doi.org/10.2147/IDR.S410720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Qiulin Xiao,1,2 Bo Chen,1 Zhenya Zhu,1 Ting Yang,1 Enfu Tao,1 Chenmin Hu,1 Wei Zheng,3 Weihong Tang,2 Xiaoli Shu,1 Mizu Jiang1,3
1Gastrointestinal Laboratory, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, National Children’s Regional Medical Center, Hangzhou, Zhejiang, 310052, People’s Republic of China; 2Department of Gastroenterology, Hangzhou Children’s Hospital, Hangzhou, Zhejiang, 310014, People’s Republic of China; 3Department of Gastroenterology, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, National Children’s Regional Medical Center, Hangzhou, Zhejiang, 310052, People’s Republic of China
Correspondence: Mizu Jiang, Department of Gastroenterology, Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, National Children’s Regional Medical Center, 3333 Bin Sheng Road, Hangzhou, Zhejiang, 310052, People’s Republic of China, Tel +86-571-87061007, Fax +86-571-86658672, Email [email protected]
Objective: To examine the association between the fecal microbiota of acute diarrhea in children and provide gut microbiota information related the acute diarrhea with rotavirus.
Patients and Methods: Children with acute diarrhea aged 3– 60 months were selected for the study. Routine stool examination was performed, and stool samples were collected and stored at − 80 °C until further analysis. Fecal microbial DNA was extracted, and DNA concentration and quality were detected. PCR amplification and 16S rDNA high-throughput sequencing analysis using the Illumina MiSeq platform were performed, and intestinal flora was statistically analyzed.
Results: Children with acute diarrhea exhibited gut microbial dysbiosis. Lower microbial diversity and richness were observed in the viral enteritis and bacterial enteritis groups than in the control group. Composition of the microbiota in acute diarrhea differed from that in the control group. The Bacteroidetes/Firmicutes dramatically decreased in the viral enteritis and bacterial enteritis groups. However, the relative abundance of Proteobacteria and Fusobacteria increased, especially in the bacterial enteritis group. In addition, the relative abundance of Actinobacteria had dramatically increased in the viral enteritis group. According to the Kyoto Encyclopedia of Genes and Genomes map analysis, the membrane transport dysfunction was caused by rotavirus infection, while the membrane transport dysfunction was more evident in bacterial infection.
Conclusion: Acute diarrhea infections cause fecal microbiota dysbiosis in children. Changes in fecal microflora in children suggest that the regulation of intestinal flora in children with acute diarrhea should be strengthened.
Keywords: acute diarrhea, microbiome, Fusobacteria, Firmicutes, Bacteroidetes, 16S rDNA sequencing, rotavirus
Introduction
Acute diarrhea is the second leading cause of preventable mortality and morbidity in children worldwide after pneumonia.1 About half a million children die from diarrhea every year.2 The causative agents of diarrhea includes bacteria and viruses, including rotaviruses, adenoviruses, and astroviruses.3 Symptoms of bacterial diarrhea are more severe than viral diarrhea, because the former can cause high fever or cenesthesia.4 With the development of the economy and the improvement of medical and health levels, a large number of antibiotics have been developed for the treatment of bacterial diarrhea. However, Antibiotics also as major disruptors of gut microbiota while treating pathogenic bacteria.5–7 Several studies have shown that bacterial community dysbiosis can lead to intestinal diseases, including cholera,8 inflammatory bowel disease, and autoimmune diseases.9 On the other hand, the pathogenesis of viral diarrhea remains unclear. Rotavirus, a major etiological agent of acute diarrhea in children worldwide10 and there are no specific drugs to date. Therefore, this study aimed to understand the fecal microbiota of children with acute diarrhea, analyze the similarities and differences in fecal microbiota changes between rotavirus enteritis and bacterial enteritis, elucidate the pathogenesis of acute diarrhea, and provide more evidence of gut microbiome data for the treatment of diarrhea in children with beneficial intestinal bacteria, so that clinicians can better implement effective and more targeted treatment for children.
Materials and Methods
Subjects
Based on the clinical symptoms, stool routine, and common enterovirus test results, 55 of 76 children with diarrhea who were treated (without antibiotics prior to enrollment) in the outpatient clinic of a children’s hospital affiliated to Zhejiang University School of Medicine from November 2018 to March 2019 were enrolled in this study. They were aged from 18–60 months. The children were divided into three groups: rotavirus enteritis (R) group (rotavirus antigen test positive), bacterial enteritis (B) group, and a control group with no symptoms (C). This study was approved by the Ethics Committee of Zhejiang University (2018-IRB-104). Written informed consent was obtained from all parents of children. We certify that the study was performed in accordance with the Declaration of Helsinki.
Inclusion Criteria and Exclusion Criteria
The inclusion criteria were as follows: i) Acute diarrhea, watery stool, or mucous blood stool; ii) The course of illness within 14 days; iii) Age from 3–60 months old; iv) Met the diagnostic criteria of rotavirus enteritis or bacterial enteritis as follows: viral enteritis, clinical manifestations of diarrhea, mainly watery stool, and positive in fecal rotavirus; bacterial enteritis, clinical manifestations of diarrhea, mainly stool to mucus blood, routine fecal detection of white/purulent cells in each high magnification field and above, and negative in common enterovirus detection. The exclusion criteria were as follows: i) Patients who had been treated with antibiotics in the last two weeks; ii) Prebiotics have been used in the last two weeks; iii) The course of illness exceeding 14 days; iv) Milk protein allergy in children diagnosed with bacterial enteritis.
Sample and Data Collection
Stool samples from children suffering from acute diarrhea were collected through anal swabbing. The samples collected at convenience. Regular fecal inspection and rapid diarrhea virus antigen detection were performed. Information including age, sex, duration of diarrhea, number of stools per day, stool characteristics, associated clinical symptoms (fever, cough, dehydration, vomiting, abdominal pain, etc.), feeding status, and recent medication were obtained through a questionnaire survey.
DNA Extraction, PCR Amplification, and High-Throughput Sequencing
DNA was extracted from different samples according to the manufacturer’s instructions using a QIAamp®DNA fecal mini-kit. Reagents used to find DNA from trace samples have been shown to be effective in preparing DNA for most bacteria. Buffer A. TE was used as the blank control. The total DNA was eluted in 50 µL elution buffer and stored at −80 °C until amplification using polymerase chain reaction (PCR) by Hangzhou Lianchuan Biotechnology Co., LTD., Zhejiang.
The variable region of 16S rRNA (V3–V4 regions) was amplified using slightly modified primers 338F (5′ -ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACGGGTTCTAAT-3′). PCR products were detected using 2% agarose gel electrophoresis, and the target fragments were recovered. The Axy Prep PCR Cleanup Kit was used for the recovery. For the purified PCR products, the Quant-it PicoGreen dsDNA Assay Kit was used for the quantitative assay of libraries in the Qbit fluorescence quantitative system. The qualified library concentration was above 2 nM. After gradient dilution of the qualified index sequence library (non-repeatable sequence), the PCR product was prepared according to the corresponding proportion of the required sequencing volume and denatured into a single chain by NaOH for on-board sequencing. The MiSeq Reagent Kit was used for 2×300bp double-end sequencing.
The FLASH (V1.2.8) software was used to merge the sequences to grow tags according to the overlap of the double-ended sequences. Barcode and primer sequences introduced by the built database on the sequences were removed, and then Vsearch (V2.3.4) was used to filter the chimera. Clean data uses Vsearch to identify clean tags with a sequence similarity of more than 97% as operational taxonomic units (OTUs) and select the best centroids (located in the geometric center) sequence as the representative sequence of the OTU. QIIME (V1.8.0) was used to analyze the α and β diversity. The β diversity among the three groups was assessed using Principal Component Analysis and Principal Coordinate Analysis. BLAST was used for sequence alignment, and OTU proxy sequences were annotated using the Ribosomal Database Project and NCBI database for species of each representative sequence. Other images were generated using the R package (V3.2.5).
Statistical Analysis
One-way analysis of variance (ANOVA) was used to analyze the differences in intestinal flora among the three groups. We state that H0: there are no significant differences in intestinal flora among the three groups; and H1: there are significant differences in intestinal flora among the three groups. By conducting a one-way analysis of variance and obtaining a P-value less than 0.05, the null hypothesis (H0) would be rejected, indicating that there are significant differences in intestinal flora among the three groups, then the overall mean of the three groups were different, indicating the differences in intestinal flora in children with acute diarrhea caused by different pathogenic factors.
Results
Comparison of Basic Information and Clinical Symptoms Among the Three Groups
The basic information and clinical symptoms of the three groups are shown in Table 1. There were no significant differences in age or sex among the three groups (P > 0.05). In the R group and B group, the disease course was approximately within three days. There were more cases of dehydration in the R group than in the B group, and vomiting symptoms were more obvious in the R group than in the B group. The positive rate of fecal occult blood in B group was higher than that in R group.
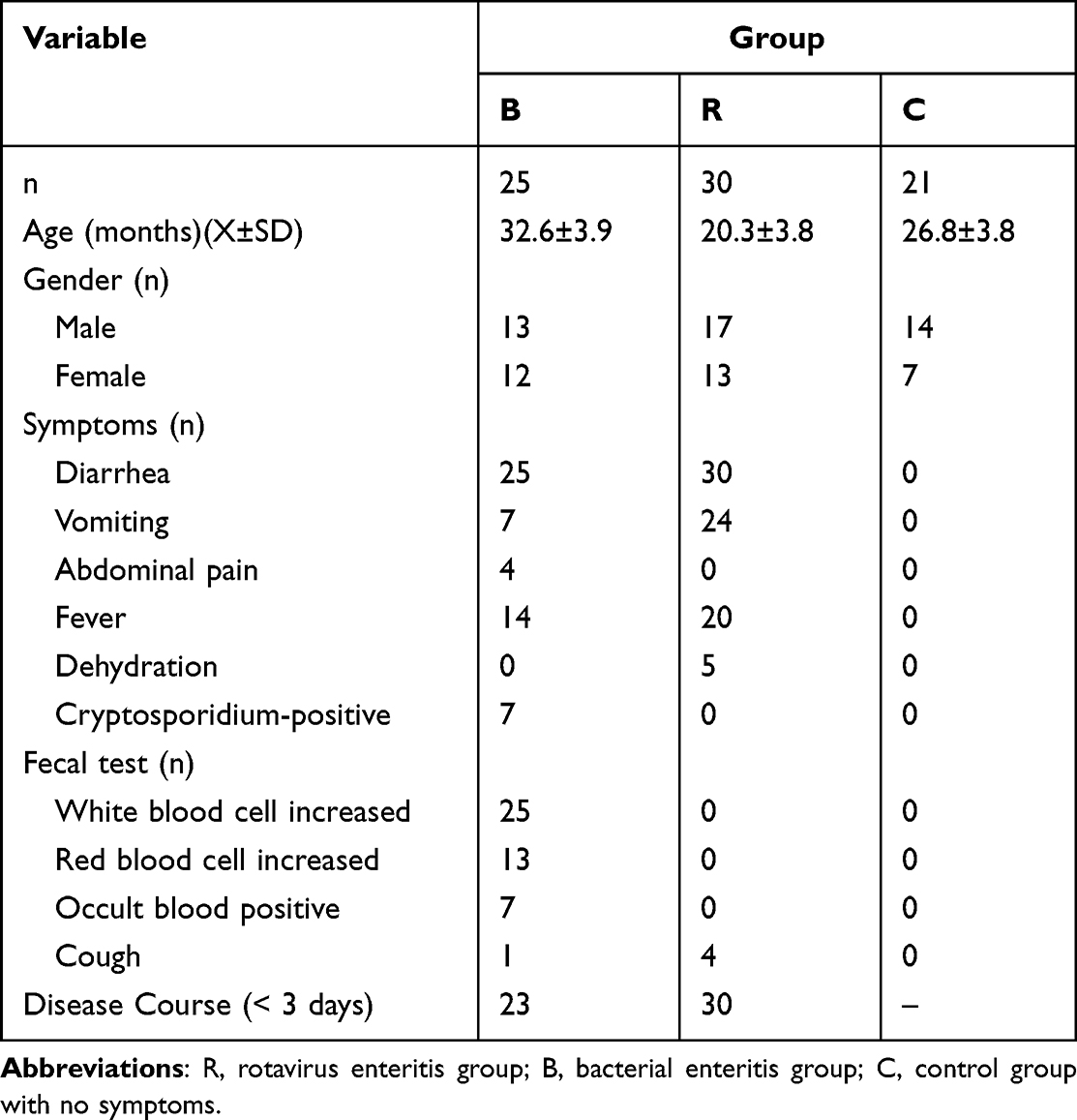 |
Table 1 Comparison of Basic Information and Clinical Symptoms Among Three Groups |
Diversity of Gut Microbiota Among Three Groups
As shown in Figure 1, the closer the distance between the points, the better the biological repeatability of the sample or the similar composition of the same sample. Analysis of similarities was performed, and the results revealed that the R group was weighted and unweighted. The structure of the intestinal microflora in the B group was significantly different from that in the C group. The richness (Goods_coverage rare) and diversity (Shannon) of intestinal flora in the three groups were significantly different, indicating that the sample had sufficient sequencing depth (Figure 2). Furthermore, the microbial diversity and richness of the R and B groups were lower than that of the C group. Similarity analysis showed that the composition of acute diarrhea flora was also different from that of the C group. Therefore, a decrease in microbial diversity was closely related to acute diarrhea.
 |
Figure 1 (A) Principal component analysis diagram; (B) Weighted principal coordinate analysis diagram. Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group; C, control group with no symptom (P<0.05). |
 |
Figure 2 Comparison of ipal coordin diagram of dilution curve results based on OTU level. (A) Shannon index; (B) Goods_coverage. Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group; C, control group with no symptom (P<0.05). |
Overall Distribution of High-Throughput Sequencing Data Sets and Bacterial Taxa in the Three Groups
After filtering-out low-quality reads, an average of 25,136.092 high-quality sequences (total: 1,910,343.0, minimum: 13,688.0, maximum: 42,569.0) were obtained for each sample, and the approximate coverage rate of all samples was ≥ 97%. Therefore, 25,136.092 sequences and 2133 OTUs sequences from each sample were utilized for subsequent analyses.
At the phylum level, Bacteroidetes was dominant in the intestinal microbiome in the three groups (mean: 33.44%; B: 10.50%; R: 34.46%; C: 55.36%), followed by Firmicutes (mean: 28.4%; B 21.18%; R 31.58%; C 32.44%) and Proteobacteria (mean: 32.40%; B: 63.27%; R: 23.65%; C: 10.28%). Compared with the C group, the Bacteroidetes/Firmicutes ratio in the R and B groups were significantly decreased, especially in the B group. Meanwhile, the relative abundance of Proteus/Clostridium was significantly increased in the B group. In addition, the relative abundance of Actinomycetes was significantly increased in the R group (Figure 3). At the genus level, Bacteroides (mean: 27.51%; B: 9.46%; R: 27.81%; C: 45.28%), Faecalibacterium (mean: 7.29%; B: 4.44%; R: 5.54%; C: 11.89%), and Escherichia coli (mean: 22.26%; B: 47.43%; R: 17.41%; C: 1.95%) were the most abundant. E. coli and Fusobacteria increased in the B and R groups compared to the C group, while Bacteroides and Faecalibacterium decreased (Figure 3B). In addition, the three groups shared 1157 OTUs (Figure 3C). There were 133, 42, and 267 unique OTUs for the B, R, and C groups, respectively.
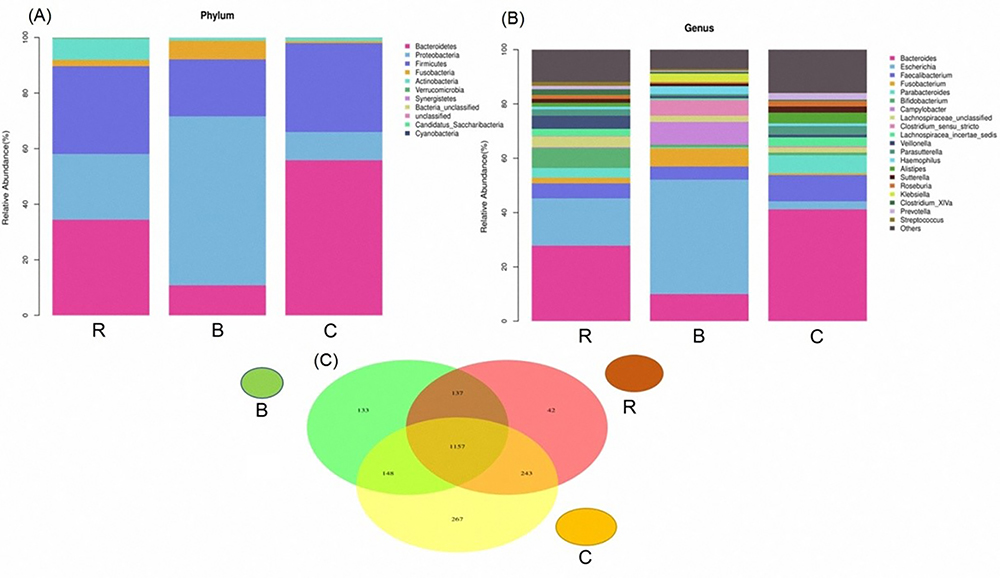 |
Figure 3 (A) Histogram of the relative abundance of species in the three groups at the phylum level; (B) Histogram of the relative abundance of species in the three groups at the genus level; (C) Venn diagram (Represents the number of OTUs shared and unique to each group). Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group; C, control group with no symptom (P<0.05). |
Differential Taxonomic Abundance in Three Groups of Children
We applied LEfSe analysis to further identify the significantly different taxonomic abundance between the three groups and between the R and B groups. Results showed that there were 108 taxa, and the intestinal microbial communities were divided into R and C, B and C, and R and B using a linear discriminant analysis score greater than 2 points (Figure 4). To visualize the impact of acute diarrhea on the intestinal microflora of children, the dominant taxa and the structure of the microflora in each group were represented by a branching diagram. The fecal microbiota of R and B groups were analyzed. In the B group, Proteobacteria and E. coli were more abundant, whereas Bacteroides/Firmicutes ratio was lower compared to the R group (Figure 4). At the family and genus levels, the abundances of Enterobacteriaceae and Campylobacter in B were higher than that in the R group, while Bifidobacteriaceae, Wrigidaceae, and Trichoderma were lower than that in R (Figure 4). In addition, the intestinal microflora composition was compared between groups B and C. In the C group, Bacteroidetes, Firmicutes, Ruminococcaceae, and Clostridium were more abundant than that in the B group. Meanwhile, Enterobacteriaceae, Proteobacteria, E. coli, Campylobacter, and Klebsiella were more abundant in the B group. The composition and changes in the intestinal flora of the R and C groups were compared and analyzed, and there were significant differences in LDA from the phylum to the genus level. The number of Enterobacteriaceae, Bifidobacteriaceae, and E. coli increased significantly in the R group (Figure 5).
 |
Figure 4 The enriched taxa of fecal microbiota in the three groups were presented as branch maps; LDA score was higher than 2 points, which was generated by LefSe analysis; the center point represented the root (bacteria) of the tree, and each ring represented the next lower classification level (p, phylum; c, class; o, order; family; g, genus). Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group; C, control group with no symptom (P<0.05). |
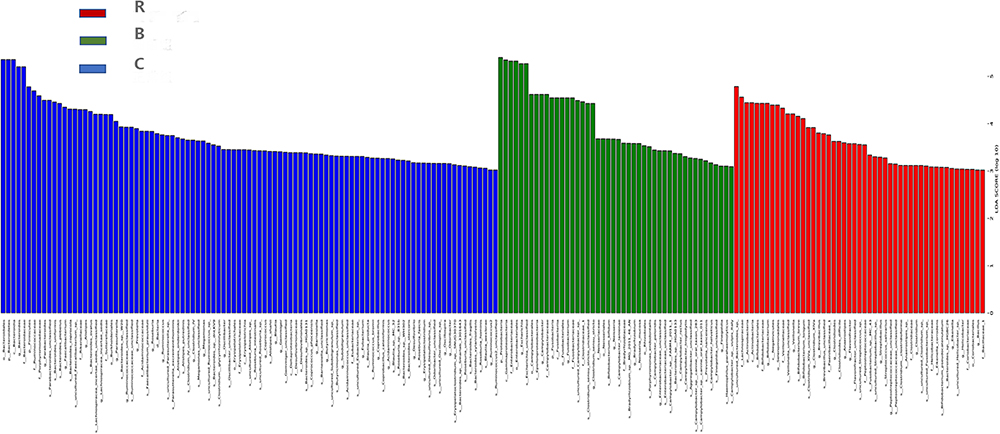 |
Figure 5 Classification histogram of the three groups with the most varied microbial community composition. The analysis showed that the number of Enterobacteriaceae, Escherichia coli and Bifidobacterium increased significantly in the rotavirus group. Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group; C, control group with no symptom (P<0.05). |
Predictive Function of the Three Groups of Children Using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)
PICRUSt results revealed higher stability, generally complementing the classification level of variation. The Kruskal–Wallis test showed statistically significant differences in the metabolism of other amino acids at the KEGG level 2 in a relatively healthy state. The same test was also significant at the KEGG level 3, since we observed differences between the three groups: pentose phosphate pathway, amino acid biosynthesis, ether lipid metabolism, and other ion-coupled transporters. Compared with the R group, the swimming and signal transfer genes of the B group were abundant, and the bacterial groups of these two genes were higher than those of the R group (Figure 6). Compared with the C group, the R group exhibited high energy concentration and membrane transport protein expression, while the membrane transport protein in the R group was lower than that in C group (Figure 7). These results suggest that viral infection leads to membrane transport dysfunction, while bacterial infection leads to more serious membrane transport dysfunction.
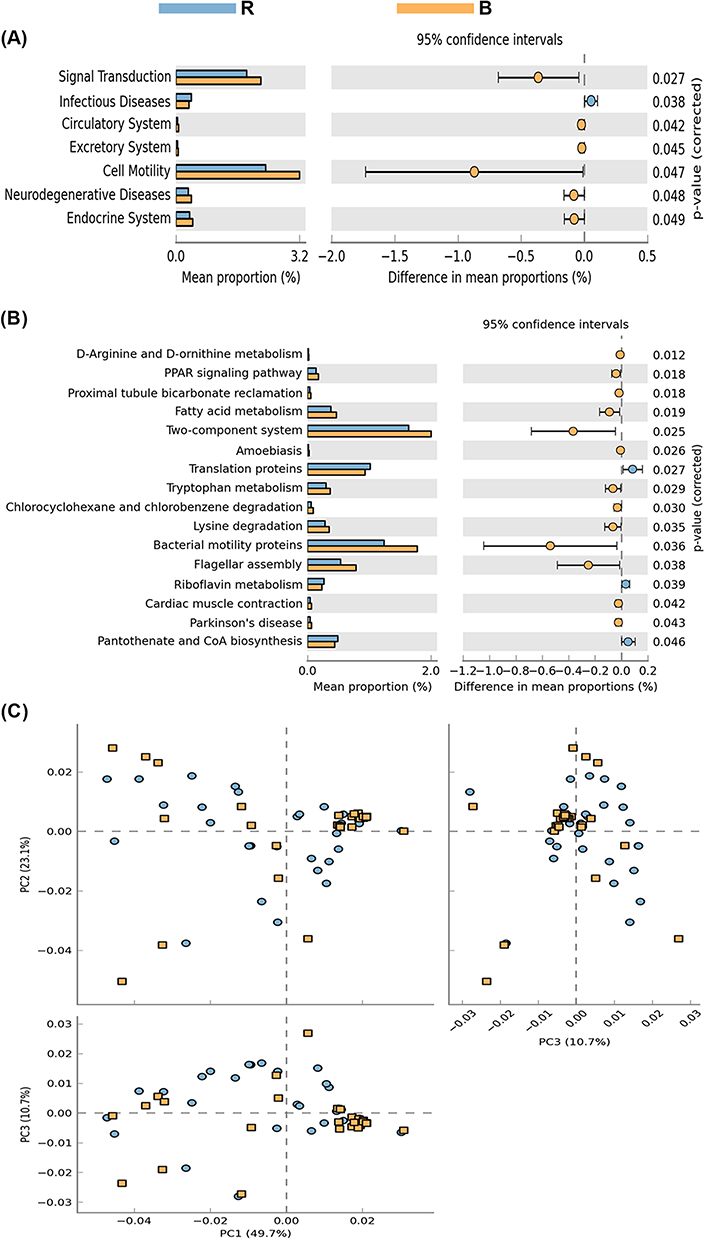 |
Figure 6 KEGG of rotavirus and normal groups. PICRUST is based on the Greengenes full-length sequence database of 16S rRNA genes, and the “closed” reference OTU is used to divide the sequencing results of bacteria. (A) The function of intestinal flora was predicted, and signal transduction and bacterial mobility in rotavirus group were lower than those in bacterial infection group (P < 0.05); (B) Two-component system, bacterial swimming gene, and bacterial infection group were higher than the viral infection group (P < 0.05); (C) Clustering analysis showed that the two groups of functional genes were significantly different at the KEGG level 2 and level 3. Abbreviations: R, rotavirus enteritis group; B, bacterial enteritis group. |
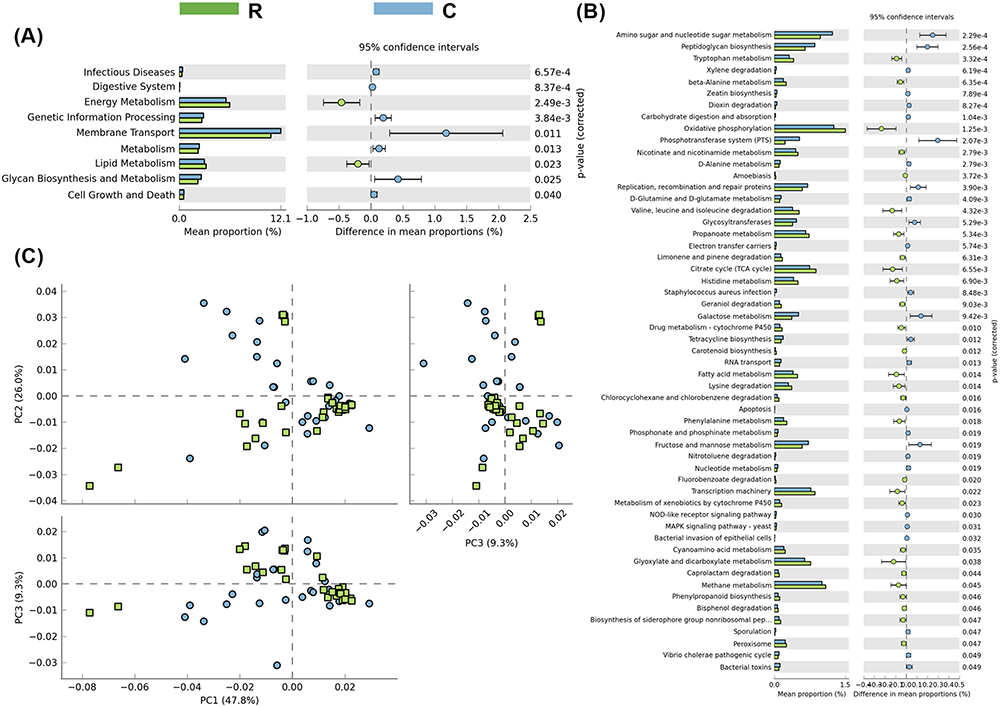 |
Figure 7 KEGG of rotavirus and normal groups. The function of intestinal flora was predicted, (A and B) Energy enrichment and membrane transporter in rotavirus group were lower than those in normal control group (P < 0.05). (C) Cluster analysis showed that except for the above two genes, the difference of other functional genes in rotavirus group and normal group was not significantly separated. Abbreviations: R, rotavirus enteritis group; C, control group with no symptom. |
Discussion
In 2007, the World Health Organization reported that, 30 years ago, there were more than 700 million cases of diarrhea among children under 5 years of age.11 Acute diarrhea is a major public health problem worldwide with more than 2 million deaths each year, mainly affecting children under the age of five in developing countries.12 This disease is particularly common in developing countries. Underlying diseases, such as malnutrition, which can increase the risk of diarrhea are also common in these countries. These factors can have a significant disease burden and economic impact due to direct medical costs, unemployment, declining quality of life, and mortality. In a healthy intestinal environment, the bacterial community can be divided into dominant and subdominant flora. Among all microorganisms, the dominant flora is the most abundant.13 However, the proportion of subdominant bacteria, including aerobes and facultative anaerobes, was relatively small. The number of these two flora remained relatively stable. Once this balance is disrupted, it leads intestinal problems.14 Therefore, further understanding of the structure of acute diarrhea flora and timely and correct intervention are critical, but this depends on the understanding of the mechanism of acute diarrhea to persistent pathogenesis, which is still not well understood. Here, we predictive function of the three groups of children using PICRUSt. The PICRUSt analysis revealed differences in metabolic pathways and gene expression related to amino acid biosynthesis, lipid metabolism, and membrane transporters. The B group exhibited higher abundance of swimming and signal transfer genes, while the R group showed higher energy concentration and membrane transport protein expression. The composition of the microbiota in acute diarrhea differed significantly from that in the control group. Specifically, the Bacteroidetes/Firmicutes ratio dramatically decreased in both viral enteritis and bacterial enteritis groups, indicating an imbalance in these bacterial phyla. Moreover, the relative abundance of Proteobacteria and Fusobacteria increased, especially in the bacterial enteritis group. These findings suggest that specific microbial shifts occur during acute diarrhea. Further analysis at the genus level revealed that certain bacteria showed distinct patterns in abundance. For example, the relative abundance of Actinobacteria significantly increased in the viral enteritis group. Meanwhile, the B and R groups exhibited higher levels of Proteus/Clostridium and E. coli, while Bacteroides and Faecalibacterium decreased compared to the control group. These changes in bacterial abundance further support the notion of dysbiosis in pediatric acute diarrhea.
We also employed KEGG mapping analysis to investigate the functional changes in the gut microbiota during acute diarrhea. The results indicated that rotavirus infection led to membrane transport dysfunction, while bacterial infection resulted in more pronounced membrane transport dysfunction. This suggests that different pathogens may have distinct effects on the gut microbiota’s functional capabilities.
Our findings highlight the presence of gut microbial dysbiosis in pediatric acute diarrhea. The alterations in microbial composition and functional capabilities provide insights into the potential mechanisms underlying the disease. Understanding these changes may contribute to the development of targeted interventions to manage and prevent acute diarrhea in children. However, further research is needed to elucidate the specific mechanisms and explore potential therapeutic approaches.
Some studies have identified intestinal aggregations of enteroaggregative E. coli, but they may be associated with antibiotic use prior to acute diarrhea. Epidemiologists have identified antibiotics as major disruptors of gut microbiota15–17 Analysis of changes in intestinal flora in acute diarrhea may provide guidance for the prevention of persistent diarrhea.
In this study, the intestinal microbiota of 76 enrolled samples was compared, and intestinal microbiota abnormalities were found in children with acute diarrhea. Compared with the C group, the microbial diversity and richness of the R and B groups were lower. Compared with the B group, more bacterial genera were observed in the R group, which is in contrast to a 2013 study.18 According to the study, rotavirus infection resulted in a decrease in microbial community diversity compared to previously unreported bacterial infections. In addition, studies have shown that in some cases, antibiotics are administered prior to confirmation of viral infection,19 which may significantly alter the intestinal microbiota. The abundance of Clostridiaceae and Streptococcaceae varied in children infected with rotavirus and bacteria alone, compared to the intestinal microbiota of healthy controls. At the phylum level, Bacteroidetes, Firmicutes, and Proteobacteria were the most abundant taxa in the three groups of subjects in this study. Similarity analysis showed that the composition of acute diarrhea flora was also different from that of the control group. Our results exhibited that diarrhea causes a large change in the intestinal flora, which has little impact on bacteria adhering to the host, but increases its relative abundance.
As for the differences in the overall relative abundance of specific populations in each group, the present study showed that the populations of Prevotella and Ruminococcus increased significantly in the R group compared to the B group. However, a recent study has clearly indicated that the abundance of Ruminococcus is related to the decreased immunoglobulin A titer targeting bacteria and rotavirus.20 The presence of these bacteria in patients with rotavirus infection may also depend on other factors, including the secretory cell status of the host and rotavirus genotypes, such as mutations in rotavirus type P (P[6] and P[8]).21 On the other hand, population-based studies have shown that Prevotella and Lactococcus have diagnostic value.22
The study might have included participants from a specific geographical region or age group, limiting the generalizability of the findings. Future studies could aim to include a diverse range of participants from different regions or age groups. The current study might have focused on a cross-sectional analysis of the fecal microbiota during acute diarrhea. Conducting longitudinal studies that track the microbiota composition over time can help identify temporal patterns and provide a better understanding of the dynamics of the microbiota during recovery.
Conclusion
We observed that the diversity and abundance of intestinal microorganisms in the rotavirus enteritis group decreased significantly, especially the Bacteroides/Firmicutes ratio, while the relative abundance of Proteobacteria, Fusobacteria, and Actinomycetes increased, which was different from that in the bacterial enteritis group. At the family and genus levels, the number of Enterobacteriaceae, Bifidobacteriaceae, and E. coli increased significantly in the rotavirus enteritis group. Through KEGG map analysis, rotavirus infection leads to membrane transport dysfunction, while bacterial infection leads to more serious membrane transport dysfunction. The changes in intestinal flora in children with rotavirus enteritis suggest that the regulation of intestinal flora in children with acute diarrhea should be strengthened.
Data Sharing Statement
The data presented in this study are available on request from the corresponding author. The data NCBI project SRA number is PRJNA786198.
Acknowledgments
This study was funded by grants from the National Natural Science Foundation of China (No. 81270459), and key research and development project of Zhejiang Province (No. 2021C03064).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no competing interests in this work.
References
1. Toro Monjaraz EM, Ignorosa Arellano KR, Loredo Mayer A, Palacios-González B, Cervantes Bustamante R, Ramírez Mayans JA. Gut microbiota in Mexican children with acute diarrhea: an Observational Study. Pediatr Infect Dis J. 2021;40(8):704–709. doi:10.1097/INF.0000000000003128
2. Yang S, He Y, Zhang J, et al. Viral metagenomics reveals diverse viruses in the fecal samples of children with diarrhea. Virol Sin. 2022;37(1):82–93. doi:10.1016/j.virs.2022.01.012
3. Yin X, Gu X, Yin T, Wen H, Gao X, Zheng X. Study of enteropathogenic bacteria in children with acute diarrhoea aged from 7 to 10 years in Xuzhou, China. Microb Pathog. 2016;91:41–45. doi:10.1016/j.micpath.2015.11.027
4. Iyoha O, Abiodun PO. Human rotavirus genotypes causing acute watery diarrhea among under-five children in Benin City, Nigeria. Niger J Clin Pract. 2015;18(1):48–51. doi:10.4103/1119-3077.146978
5. Wu Z, Shen J, Xu Q, et al. Epigallocatechin-3-gallate improves intestinal gut microbiota homeostasis and ameliorates Clostridioides difficile infection. Nutrients. 2022;14(18):3756. doi:10.3390/nu14183756
6. Guo Y, Yang X, Qi Y, et al. Long-term use of ceftriaxone sodium induced changes in gut microbiota and immune system. Sci Rep. 2017;7(1):43035. doi:10.1038/srep43035
7. Cheng RY, Li M, Li SS, et al. Vancomycin and ceftriaxone can damage intestinal microbiota and affect the development of the intestinal tract and immune system to different degrees in neonatal mice. Pathog Dis. 2017;75(8). doi:10.1093/femspd/ftx104
8. Monira S, Nakamura S, Gotoh K, et al. Metagenomic profile of gut microbiota in children during cholera and recovery. Gut Pathog. 2013;5(1):1. doi:10.1186/1757-4749-5-1
9. Marietta E, Mangalam AK, Taneja V, Murray JA. Intestinal dysbiosis in, and enteral bacterial therapies for, systemic autoimmune diseases. Front Immunol. 2020;11:573079. doi:10.3389/fimmu.2020.573079
10. Gómez-Rial J, Rivero-Calle I, Salas A, Martinón-Torres F. Rotavirus and autoimmunity. J Infect. 2020;81(2):183–189. doi:10.1016/j.jinf.2020.04.041
11. Elliott EJ. Acute gastroenteritis in children. BMJ. 2007;334(7583):35–40. doi:10.1136/bmj.39036.406169.80
12. Jafari F, Garcia-Gil LJ, Salmanzadeh-Ahrabi S, et al. Diagnosis and prevalence of enteropathogenic bacteria in children less than 5 years of age with acute diarrhea in Tehran children’s hospitals. J Infect. 2009;58(1):21–27. doi:10.1016/j.jinf.2008.10.013
13. Christensen EG, Licht TR, Leser TD, Bahl MI. Dietary xylo-oligosaccharide stimulates intestinal bifidobacteria and lactobacilli but has limited effect on intestinal integrity in rats. BMC Res Notes. 2014;7(1):660. doi:10.1186/1756-0500-7-660
14. Qu M, Deng Y, Zhang X, et al. Etiology of acute diarrhea due to enteropathogenic bacteria in Beijing, China. J Infect. 2012;65(3):214–222. doi:10.1016/j.jinf.2012.04.010
15. Mathew S, Smatti MK, Al Ansari K, Nasrallah GK, Al Thani AA, Yassine HM. Mixed viral-bacterial infections and their effects on gut microbiota and clinical illnesses in children. Sci Rep. 2019;9(1):865. doi:10.1038/s41598-018-37162-w
16. Ramirez J, Guarner F, Bustos Fernandez L, Maruy A, Sdepanian VL, Cohen H. Antibiotics as major disruptors of gut microbiota. Front Cell Infect Microbiol. 2020;10:572912. doi:10.3389/fcimb.2020.572912
17. Gough EK. The impact of mass drug administration of antibiotics on the gut microbiota of target populations. Infect Dis Poverty. 2022;11(1):76. doi:10.1186/s40249-022-00999-5
18. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol. 2013;79(17):5112–5120. doi:10.1128/AEM.01043-13
19. Shankar V, Reo NV, Paliy O. Simultaneous fecal microbial and metabolite profiling enables accurate classification of pediatric irritable bowel syndrome. Microbiome. 2015;3(1):73. doi:10.1186/s40168-015-0139-9
20. Rodríguez-Díaz J, García-Mantrana I, Vila-Vicent S, et al. Relevance of secretor status genotype and microbiota composition in susceptibility to rotavirus and norovirus infections in humans. Sci Rep. 2017;7(1):45559. doi:10.1038/srep45559
21. Johan N, Sumit S, Filemon B, et al. Both Lewis and secretor status mediate susceptibility to rotavirus infections in a rotavirus genotype–dependent manner. Clin Infect Dis. 2014;11:1567–1573.
22. Gorvitovskaia A, Holmes SP, Huse SM. Interpreting prevotella and bacteroides as biomarkers of diet and lifestyle. Microbiome. 2016;4(1):1–12. doi:10.1186/s40168-016-0160-7
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Significance of Gut Microbiota on Graves’ Disease
Chen H, Cao J, Zhang F, Xiong W
International Journal of General Medicine 2024, 17:3967-3974
Published Date: 11 September 2024